Lipid Accumulation and Chronic Kidney Disease

 ,
,
Abstract
1. Introduction
2. Epidemiological Studies
3. The Mechanism of Lipid Transport and Metabolism in the Kidneys
3.1. Lipid Uptake by CD36 in the Kidney
3.2. Lipid Uptake by Other Transporters
3.3. Regulation of Lipogenesis in the Kidney
4. Lipid-Induced Renal Damage
5. Treatment
5.1. Bile Acid Receptor Agonists
5.2. PPAR Agonists
5.3. C/EBPα and C/EBPβ Inhibition
5.4. CD36 Antagonists
5.5. SREBP-1 Inhibitors
5.6. Other Targets
5.7. Non-Targeted Therapies
5.8. Lifestyle Modification
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am. J. Kidney Dis. 2002, 39 (Suppl. 1), S1–S266. [Google Scholar]
- GBD Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Collins, A.J.; Foley, R.N.; Chavers, B.; Gilbertson, D.; Herzog, C.; Johansen, K.; Kasiske, B.; Kutner, N.; Liu, J.; St Peter, W.; et al. United States Renal Data System 2011 Annual Data Report: Atlas of chronic kidney disease & end-stage renal disease in the United States. Am. J. Kidney Dis. 2012, 59 (Suppl. 1), e1–e420. [Google Scholar]
- Kramer, H.; Luke, A.; Bidani, A.; Cao, G.; Cooper, R.; McGee, D. Obesity and prevalent and incident CKD: The Hypertension Detection and Follow-Up Program. Am. J. Kidney Dis. 2005, 46, 587–594. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. New Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V.D.; Chagnac, A.; de Vries, A.P.; Levi, M.; Porrini, E.; Herman-Edelstein, M.; Praga, M. Obesity-related glomerulopathy: Clinical and pathologic characteristics and pathogenesis. Nat. Rev. Nephrol. 2016, 12, 453–471. [Google Scholar] [CrossRef]
- Kuwahara, S.; Hosojima, M.; Kaneko, R.; Aoki, H.; Nakano, D.; Sasagawa, T.; Kabasawa, H.; Kaseda, R.; Yasukawa, R.; Ishikawa, T.; et al. Megalin-Mediated Tubuloglomerular Alterations in High-Fat Diet-Induced Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1996–2008. [Google Scholar] [CrossRef]
- Gelber, R.P.; Kurth, T.; Kausz, A.T.; Manson, J.E.; Buring, J.E.; Levey, A.S.; Gaziano, J.M. Association between body mass index and CKD in apparently healthy men. Am. J. Kidney Dis. 2005, 46, 871–880. [Google Scholar] [CrossRef]
- Munkhaugen, J.; Lydersen, S.; Wideroe, T.E.; Hallan, S. Prehypertension, obesity, and risk of kidney disease: 20-year follow-up of the HUNT I study in Norway. Am. J. Kidney Dis. 2009, 54, 638–646. [Google Scholar] [CrossRef]
- Felizardo, R.J.; da Silva, M.B.; Aguiar, C.F.; Camara, N.O. Obesity in kidney disease: A heavyweight opponent. World J. Nephrol. 2014, 3, 50–63. [Google Scholar] [CrossRef]
- Krikken, J.A.; Lely, A.T.; Bakker, S.J.; Navis, G. The effect of a shift in sodium intake on renal hemodynamics is determined by body mass index in healthy young men. Kidney Int. 2007, 71, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Sheng, Z.; Yao, L. Obesity-related glomerulopathy: Pathogenesis, pathologic, clinical characteristics and treatment. Front. Med. 2017, 11, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Li, S.J.; Chen, H.P.; Wang, Q.W.; Li, L.S.; Liu, Z.H. Obesity-related glomerulopathy in China: A case series of 90 patients. Am. J. Kidney Dis. 2008, 52, 58–65. [Google Scholar] [CrossRef]
- Dai, D.; Chang, Y.; Chen, Y.; Chen, S.; Yu, S.; Guo, X.; Sun, Y. Visceral Adiposity Index and Lipid Accumulation Product Index: Two Alternate Body Indices to Identify Chronic Kidney Disease among the Rural Population in Northeast China. Int. J. Environ. Res. Public Health 2016, 13, 1231. [Google Scholar] [CrossRef] [PubMed]
- Kambham, N.; Markowitz, G.S.; Valeri, A.M.; Lin, J.; D’Agati, V.D. Obesity-related glomerulopathy: An emerging epidemic. Kidney Int. 2001, 59, 1498–1509. [Google Scholar] [CrossRef]
- Yamagata, K.; Ishida, K.; Sairenchi, T.; Takahashi, H.; Ohba, S.; Shiigai, T.; Narita, M.; Koyama, A. Risk factors for chronic kidney disease in a community-based population: A 10-year follow-up study. Kidney Int. 2007, 71, 159–166. [Google Scholar] [CrossRef]
- Takamatsu, N.; Abe, H.; Tominaga, T.; Nakahara, K.; Ito, Y.; Okumoto, Y.; Kim, J.; Kitakaze, M.; Doi, T. Risk factors for chronic kidney disease in Japan: A community-based study. BMC Nephrol. 2009, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.E.; Sarnak, M.J. Managing dyslipidemia in chronic kidney disease. J. Gen. Intern. Med. 2004, 19, 1045–1052. [Google Scholar] [CrossRef]
- Yamada, Y.; Matsui, K.; Takeuchi, I.; Fujimaki, T. Association of genetic variants with dyslipidemia and chronic kidney disease in a longitudinal population-based genetic epidemiological study. Int. J. Mol. Med. 2015, 35, 1290–1300. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef]
- Afshinnia, F.; Rajendiran, T.M.; Soni, T.; Byun, J.; Wernisch, S.; Sas, K.M.; Hawkins, J.; Bellovich, K.; Gipson, D.; Michailidis, G.; et al. Impaired beta-Oxidation and Altered Complex Lipid Fatty Acid Partitioning with Advancing CKD. J. Am. Soc. Nephrol. 2018, 29, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Appel, G.B.; Radhakrishnan, J.; Avram, M.M.; DeFronzo, R.A.; Escobar-Jimenez, F.; Campos, M.M.; Burgess, E.; Hille, D.A.; Dickson, T.Z.; Shahinfar, S.; et al. Analysis of metabolic parameters as predictors of risk in the RENAAL study. Diabetes Care 2003, 26, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Kimura, T.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Matsusaka, T.; et al. High-Fat Diet-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney. J. Am. Soc. Nephrol. 2017, 28, 1534–1551. [Google Scholar] [CrossRef] [PubMed]
- Wahl, P.; Ducasa, G.M.; Fornoni, A. Systemic and renal lipids in kidney disease development and progression. Am. J. Physiol. Ren. Physiol. 2016, 310, F433–F445. [Google Scholar] [CrossRef] [PubMed]
- Moorhead, J.F.; Chan, M.K.; El-Nahas, M.; Varghese, Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 1982, 2, 1309–1311. [Google Scholar] [CrossRef]
- Armesilla, A.L.; Vega, M.A. Structural organization of the gene for human CD36 glycoprotein. J. Biol. Chem. 1994, 269, 18985–18991. [Google Scholar]
- Nicholson, A.C.; Febbraio, M.; Han, J.; Silverstein, R.L.; Hajjar, D.P. CD36 in atherosclerosis. The role of a class B macrophage scavenger receptor. Ann. N. Y. Acad. Sci. 2000, 902, 128–131, discussion 131–133. [Google Scholar] [CrossRef]
- Susztak, K.; Ciccone, E.; McCue, P.; Sharma, K.; Bottinger, E.P. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005, 2, e45. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Chen, Y.; Huang, W.; Viterna, J.; Liu, J.; Westfall, K.; Tian, J.; Bartlett, D.J.; Tang, W.H.; Xie, Z.; et al. CD36 and Na/K-ATPase-alpha1 form a proinflammatory signaling loop in kidney. Hypertension 2013, 61, 216–224. [Google Scholar] [CrossRef]
- Okamura, D.M.; Lopez-Guisa, J.M.; Koelsch, K.; Collins, S.; Eddy, A.A. Atherogenic scavenger receptor modulation in the tubulointerstitium in response to chronic renal injury. Am. J. Physiol. Ren. Physiol. 2007, 293, F575–F585. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.Z.; Varghese, Z.; Powis, S.H.; Moorhead, J.F. Human mesangial cells express inducible macrophage scavenger receptor. Kidney Int. 1999, 56, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Xiao, Y.; Luo, X.; Zhao, Y.; Zhao, L.; Wang, Y.; Wu, T.; Wei, L.; Chen, Y. Inflammatory stress promotes the development of obesity-related chronic kidney disease via CD36 in mice. J. Lipid Res. 2017, 58, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef]
- Baranova, I.N.; Vishnyakova, T.G.; Bocharov, A.V.; Kurlander, R.; Chen, Z.; Kimelman, M.L.; Remaley, A.T.; Csako, G.; Thomas, F.; Eggerman, T.L.; et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J. Biol. Chem. 2005, 280, 8031–8040. [Google Scholar] [CrossRef] [PubMed]
- Okamura, D.M.; Pennathur, S.; Pasichnyk, K.; Lopez-Guisa, J.M.; Collins, S.; Febbraio, M.; Heinecke, J.; Eddy, A.A. CD36 regulates oxidative stress and inflammation in hypercholesterolemic CKD. J. Am. Soc. Nephrol. 2009, 20, 495–505. [Google Scholar] [CrossRef]
- Bige, N.; Shweke, N.; Benhassine, S.; Jouanneau, C.; Vandermeersch, S.; Dussaule, J.C.; Chatziantoniou, C.; Ronco, P.; Boffa, J.J. Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction. Kidney Int. 2012, 81, 1226–1238. [Google Scholar] [CrossRef]
- Cui, W.; Maimaitiyiming, H.; Zhou, Q.; Norman, H.; Zhou, C.; Wang, S. Interaction of thrombospondin1 and CD36 contributes to obesity-associated podocytopathy. Biochim. Biophys. Acta 2015, 1852, 1323–1333. [Google Scholar] [CrossRef]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Kuniyasu, A.; Horiuchi, S.; Nakayama, H. Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J. Biol. Chem. 2001, 276, 3195–3202. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Chanda, D.; Nabben, M.; Neumann, D.; Glatz, J.F. Post-translational modifications of CD36 (SR-B2): Implications for regulation of myocellular fatty acid uptake. Biochim. Biophys. Acta 2016, 1862, 2253–2258. [Google Scholar] [CrossRef]
- Wang, C.; Hu, L.; Zhao, L.; Yang, P.; Moorhead, J.F.; Varghese, Z.; Chen, Y.; Ruan, X.Z. Inflammatory stress increases hepatic CD36 translational efficiency via activation of the mTOR signalling pathway. PLoS ONE 2014, 9, e103071. [Google Scholar] [CrossRef]
- Thorne, R.F.; Ralston, K.J.; de Bock, C.E.; Mhaidat, N.M.; Zhang, X.D.; Boyd, A.W.; Burns, G.F. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim. Biophys. Acta 2010, 1803, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Guthmann, F.; Maehl, P.; Preiss, J.; Kolleck, I.; Rustow, B. Ectoprotein kinase-mediated phosphorylation of FAT/CD36 regulates palmitate uptake by human platelets. Cell. Mol. Life Sci. CMLS 2002, 59, 1999–2003. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.; Narisawa, S.; Millan, J.L.; Widmaier, E.P. Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1738–R1747. [Google Scholar] [CrossRef] [PubMed]
- Coe, N.R.; Bernlohr, D.A. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim. Biophys. Acta 1998, 1391, 287–306. [Google Scholar] [CrossRef]
- Schaffer, J.E.; Lodish, H.F. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 1994, 79, 427–436. [Google Scholar] [CrossRef]
- Anderson, C.M.; Stahl, A. SLC27 fatty acid transport proteins. Mol. Asp. Med. 2013, 34, 516–528. [Google Scholar] [CrossRef]
- Gertow, K.; Skoglund-Andersson, C.; Eriksson, P.; Boquist, S.; Orth-Gomer, K.; Schenck-Gustafsson, K.; Hamsten, A.; Fisher, R.M. A common polymorphism in the fatty acid transport protein-1 gene associated with elevated post-prandial lipaemia and alterations in LDL particle size distribution. Atherosclerosis 2003, 167, 265–273. [Google Scholar] [CrossRef]
- Dourlen, P.; Sujkowski, A.; Wessells, R.; Mollereau, B. Fatty acid transport proteins in disease: New insights from invertebrate models. Prog. Lipid Res. 2015, 60, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Evans, J.G.; Pattel, S.; Hirsch, D.; Lodish, H.F. Insulin causes fatty acid transport protein translocation and enhanced fatty acid uptake in adipocytes. Dev. Cell 2002, 2, 477–488. [Google Scholar] [CrossRef]
- Falcon, A.; Doege, H.; Fluitt, A.; Tsang, B.; Watson, N.; Kay, M.A.; Stahl, A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E384–E393. [Google Scholar] [CrossRef] [PubMed]
- Bloksgaard, M.; Neess, D.; Faergeman, N.J.; Mandrup, S. Acyl-CoA binding protein and epidermal barrier function. Biochim. Biophys. Acta 2014, 1841, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Milger, K.; Herrmann, T.; Becker, C.; Gotthardt, D.; Zickwolf, J.; Ehehalt, R.; Watkins, P.A.; Stremmel, W.; Fullekrug, J. Cellular uptake of fatty acids driven by the ER-localized acyl-CoA synthetase FATP4. J. Cell Sci. 2006, 119 Pt 22, 4678–4688. [Google Scholar] [CrossRef]
- Gertow, K.; Rosell, M.; Sjogren, P.; Eriksson, P.; Vessby, B.; de Faire, U.; Hamsten, A.; Hellenius, M.L.; Fisher, R.M. Fatty acid handling protein expression in adipose tissue, fatty acid composition of adipose tissue and serum, and markers of insulin resistance. Eur. J. Clin. Nutr. 2006, 60, 1406–1413. [Google Scholar] [CrossRef]
- Stremmel, W.; Strohmeyer, G.; Borchard, F.; Kochwa, S.; Berk, P.D. Isolation and partial characterization of a fatty acid binding protein in rat liver plasma membranes. Proc. Natl. Acad. Sci. USA 1985, 82, 4–8. [Google Scholar] [CrossRef]
- Haunerland, N.H.; Spener, F. Fatty acid-binding proteins--insights from genetic manipulations. Prog. Lipid Res. 2004, 43, 328–349. [Google Scholar] [CrossRef]
- Murphy, E.J. Sterol carrier protein-2: Not just for cholesterol any more. Mol. Cell. Biochem. 2002, 239, 87–93. [Google Scholar] [CrossRef]
- Petrescu, A.D.; Huang, H.; Martin, G.G.; McIntosh, A.L.; Storey, S.M.; Landrock, D.; Kier, A.B.; Schroeder, F. Impact of L-FABP and glucose on polyunsaturated fatty acid induction of PPARalpha-regulated beta-oxidative enzymes. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G241–G256. [Google Scholar] [CrossRef]
- Hostetler, H.A.; McIntosh, A.L.; Atshaves, B.P.; Storey, S.M.; Payne, H.R.; Kier, A.B.; Schroeder, F. L-FABP directly interacts with PPARalpha in cultured primary hepatocytes. J. Lipid Res. 2009, 50, 1663–1675. [Google Scholar] [CrossRef]
- Issemann, I.; Prince, R.; Tugwood, J.; Green, S. A role for fatty acids and liver fatty acid binding protein in peroxisome proliferation? Biochem. Soc. Trans. 1992, 20, 824–827. [Google Scholar] [CrossRef]
- Wolfrum, C.; Borrmann, C.M.; Borchers, T.; Spener, F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha- and gamma-mediated gene expression via liver fatty acid binding protein: A signaling path to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 2323–2328. [Google Scholar] [CrossRef] [PubMed]
- Weisiger, R.A. Mechanisms of intracellular fatty acid transport: Role of cytoplasmic-binding proteins. J. Mol. Neurosci. 2007, 33, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Van Breda, E.; Van der Vusse, G.J. Intracellular transport of fatty acids in muscle. Role of cytoplasmic fatty acid-binding protein. Adv. Exp. Med. Biol. 1998, 441, 207–218. [Google Scholar]
- Watanabe, S.; Ichikawa, D.; Sugaya, T.; Ohata, K.; Inoue, K.; Hoshino, S.; Kimura, K.; Shibagaki, Y.; Kamijo-Ikemori, A. Urinary Level of Liver-Type Fatty Acid Binding Protein Reflects the Degree of Tubulointerstitial Damage in Polycystic Kidney Disease. Kidney Blood Press Res. 2018, 43, 1716–1729. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Rosen, E.D.; Brun, R.; Hauser, S.; Adelmant, G.; Troy, A.E.; McKeon, C.; Darlington, G.J.; Spiegelman, B.M. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 1999, 3, 151–158. [Google Scholar] [CrossRef]
- Matsusue, K.; Gavrilova, O.; Lambert, G.; Brewer, H.B., Jr.; Ward, J.M.; Inoue, Y.; LeRoith, D.; Gonzalez, F.J. Hepatic CCAAT/enhancer binding protein alpha mediates induction of lipogenesis and regulation of glucose homeostasis in leptin-deficient mice. Mol. Endocrinol. 2004, 18, 2751–2764. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Halaihel, N.; Zhang, W.; Rogers, T.; Levi, M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J. Biol. Chem. 2002, 277, 18919–18927. [Google Scholar] [CrossRef]
- Jin, K.; Norris, K.; Vaziri, N.D. Dysregulation of hepatic fatty acid metabolism in chronic kidney disease. Nephrol. Dial. Transplant. 2013, 28, 313–320. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, Y.; Han, J.; Hu, J.; He, T.; Yan, T.; Huang, N.; Zhang, Q.; Mei, H.; Liao, Y.; et al. Atgl deficiency induces podocyte apoptosis and leads to glomerular filtration barrier damage. FEBS J. 2017, 284, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- Majka, S.M.; Barak, Y.; Klemm, D.J. Concise review: Adipocyte origins: Weighing the possibilities. Stem Cells 2011, 29, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet. Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Arici, M.; Brown, J.; Williams, M.; Harris, K.P.; Walls, J.; Brunskill, N.J. Fatty acids carried on albumin modulate proximal tubular cell fibronectin production: A role for protein kinase C. Nephrol. Dial. Transplant. 2002, 17, 1751–1757. [Google Scholar] [CrossRef]
- Gao, X.; Wu, J.; Qian, Y.; Fu, L.; Wu, G.; Xu, C.; Mei, C. Oxidized high-density lipoprotein impairs the function of human renal proximal tubule epithelial cells through CD36. Int. J. Mol. Med. 2014, 34, 564–572. [Google Scholar] [CrossRef]
- Gibbs, P.E.; Lerner-Marmarosh, N.; Poulin, A.; Farah, E.; Maines, M.D. Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor. FASEB J. 2014, 28, 2478–2491. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Mandel, L.J. Metabolic substrates, cellular energy production, and the regulation of proximal tubular transport. Annu. Rev. Physiol. 1985, 47, 85–101. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Gordon, D.M.; Weeks, M.F.; Moore, K.H.; Hall, J.E.; Hinds, T.D., Jr.; Stec, D.E. Loss of biliverdin reductase-A promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 315, F323–F331. [Google Scholar] [CrossRef]
- Decleves, A.E.; Mathew, A.V.; Cunard, R.; Sharma, K. AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 2011, 22, 1846–1855. [Google Scholar] [CrossRef]
- Miyamoto, S.; Hsu, C.C.; Hamm, G.; Darshi, M.; Diamond-Stanic, M.; Decleves, A.E.; Slater, L.; Pennathur, S.; Stauber, J.; Dorrestein, P.C.; et al. Mass Spectrometry Imaging Reveals Elevated Glomerular ATP/AMP in Diabetes/obesity and Identifies Sphingomyelin as a Possible Mediator. EBioMedicine 2016, 7, 121–134. [Google Scholar] [CrossRef]
- Modaresi, A.; Nafar, M.; Sahraei, Z. Oxidative stress in chronic kidney disease. Iran. J. Kidney Dis. 2015, 9, 165–179. [Google Scholar] [PubMed]
- Jiang, T.; Wang, X.X.; Scherzer, P.; Wilson, P.; Tallman, J.; Takahashi, H.; Li, J.; Iwahashi, M.; Sutherland, E.; Arend, L.; et al. Farnesoid X receptor modulates renal lipid metabolism, fibrosis, and diabetic nephropathy. Diabetes 2007, 56, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Jiang, T.; Shen, Y.; Caldas, Y.; Miyazaki-Anzai, S.; Santamaria, H.; Urbanek, C.; Solis, N.; Scherzer, P.; Lewis, L.; et al. Diabetic nephropathy is accelerated by farnesoid X receptor deficiency and inhibited by farnesoid X receptor activation in a type 1 diabetes model. Diabetes 2010, 59, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Chu, L.; Xu, Z.; Song, X.; Sun, D.; Kullak-Ublick, G.A. Farnesoid X receptor activation protects the kidney from ischemia-reperfusion damage. Sci. Rep. 2017, 7, 9815. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Visentin, M.; Gui, T.; Zhao, L.; Thasler, W.E.; Hausler, S.; Hartling, I.; Cremonesi, A.; Hiller, C.; Kullak-Ublick, G.A. Effects of Farnesoid X Receptor Activation on Arachidonic Acid Metabolism, NF-kB Signaling, and Hepatic Inflammation. Mol. Pharmacol. 2018, 94, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Herman-Edelstein, M.; Weinstein, T.; Levi, M. Bile acid receptors and the kidney. Curr. Opin. Nephrol. Hypertens. 2018, 27, 56–62. [Google Scholar] [CrossRef]
- Sato, H.; Genet, C.; Strehle, A.; Thomas, C.; Lobstein, A.; Wagner, A.; Mioskowski, C.; Auwerx, J.; Saladin, R. Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem. Biophys. Res. Commun. 2007, 362, 793–798. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Huang, B.Q.; Radtke, B.N.; Gajdos, G.B.; Splinter, P.L.; Masyuk, T.V.; Gradilone, S.A.; LaRusso, N.F. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1013–G1024. [Google Scholar] [CrossRef]
- Wang, X.X.; Edelstein, M.H.; Gafter, U.; Qiu, L.; Luo, Y.; Dobrinskikh, E.; Lucia, S.; Adorini, L.; D’Agati, V.D.; Levi, J.; et al. G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes. J. Am. Soc. Nephrol. 2016, 27, 1362–1378. [Google Scholar] [CrossRef]
- Wang, X.X.; Wang, D.; Luo, Y.; Myakala, K.; Dobrinskikh, E.; Rosenberg, A.Z.; Levi, J.; Kopp, J.B.; Field, A.; Hill, A.; et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 2018, 29, 118–137. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Schoonjans, K.; Staels, B.; Auwerx, J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J. Lipid Res. 1996, 37, 907–925. [Google Scholar]
- Tanaka, Y.; Kume, S.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sakaguchi, M.; Sugimoto, T.; Koya, D.; Haneda, M.; Kashiwagi, A.; et al. Fenofibrate, a PPARalpha agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 2011, 79, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diabetes Rep. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Auboeuf, D.; Raspe, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef]
- Lemberger, T.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: A nuclear receptor signaling pathway in lipid physiology. Annu. Rev. Cell Dev. Biol. 1996, 12, 335–363. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, O.; Attman, P.O.; Gause-Nilsson, I.; Svensson, M.K.; Alaupovic, P. Dual PPAR alpha/gamma Agonism Normalizes Lipoprotein Profile of Renal Dyslipidemia. PPAR Res. 2013, 2013, 391628. [Google Scholar] [CrossRef]
- Grommes, C.; Landreth, G.E.; Heneka, M.T. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol. 2004, 5, 419–429. [Google Scholar] [CrossRef]
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From orphan receptors to drug discovery. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef]
- Berger, J.; Bailey, P.; Biswas, C.; Cullinan, C.A.; Doebber, T.W.; Hayes, N.S.; Saperstein, R.; Smith, R.G.; Leibowitz, M.D. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: Binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology 1996, 137, 4189–4195. [Google Scholar] [CrossRef]
- Chan, D.T.; Watts, G.F.; Irish, A.B.; Dogra, G.K. Rosiglitazone does not improve vascular function in subjects with chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 3543–3549. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhang, Q.; Liu, L.; Yin, S.; Liu, Z.; Cao, W. Klotho restoration via acetylation of Peroxisome Proliferation-Activated Receptor gamma reduces the progression of chronic kidney disease. Kidney Int. 2017, 92, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Maquigussa, E.; Paterno, J.C.; de Oliveira Pokorny, G.H.; da Silva Perez, M.; Varela, V.A.; da Silva Novaes, A.; Schor, N.; Boim, M.A. Klotho and PPAR Gamma Activation Mediate the Renoprotective Effect of Losartan in the 5/6 Nephrectomy Model. Front. Physiol. 2018, 9, 1033. [Google Scholar] [CrossRef]
- Chao, C.T.; Chen, Y.C.; Chiang, C.K.; Huang, J.W.; Fang, C.C.; Chang, C.C.; Yen, C.J. Interplay between Superoxide Dismutase, Glutathione Peroxidase, and Peroxisome Proliferator Activated Receptor Gamma Polymorphisms on the Risk of End-Stage Renal Disease among Han Chinese Patients. Oxid. Med. Cell. Longev. 2016, 2016, 8516748. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Spiegelman, B.M. Transcriptional activation of adipogenesis. Curr. Opin. Cell Biol. 1999, 11, 689–694. [Google Scholar] [CrossRef]
- Birkenmeier, E.H.; Gwynn, B.; Howard, S.; Jerry, J.; Gordon, J.I.; Landschulz, W.H.; McKnight, S.L. Tissue-specific expression, developmental regulation, and genetic mapping of the gene encoding CCAAT/enhancer binding protein. Genes Dev. 1989, 3, 1146–1156. [Google Scholar] [CrossRef]
- Zhang, D.E.; Zhang, P.; Wang, N.D.; Hetherington, C.J.; Darlington, G.J.; Tenen, D.G. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 569–574. [Google Scholar] [CrossRef]
- Halmos, B.; Huettner, C.S.; Kocher, O.; Ferenczi, K.; Karp, D.D.; Tenen, D.G. Down-regulation and antiproliferative role of C/EBPalpha in lung cancer. Cancer Res. 2002, 62, 528–534. [Google Scholar]
- Friedman, A.D.; Keefer, J.R.; Kummalue, T.; Liu, H.; Wang, Q.F.; Cleaves, R. Regulation of granulocyte and monocyte differentiation by CCAAT/enhancer binding protein alpha. Blood Cells Mol. Dis. 2003, 31, 338–341. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Shimizu, Y.; Takahashi, H.; Yajima, H.; Yokoyama, Y.; Ishigami, K.; Tabeya, T.; Suzuki, C.; Matsui, M.; Naishiro, Y.; et al. CCAAT/enhancer binding protein alpha (C/EBPalpha)(+) M2 macrophages contribute to fibrosis in IgG4-related disease? Mod. Rheumatol. 2015, 25, 484–486. [Google Scholar] [CrossRef] [PubMed]
- De Winther, M.P.; Hofker, M.H. Scavenging new insights into atherogenesis. J. Clin. Investig. 2000, 105, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Li, X.; Qian, S.W.; Guo, L.; Huang, H.Y.; He, Q.; Liu, Y.; Ma, C.G.; Tang, Q.Q. Transcriptional activation of histone H4 by C/EBPbeta during the mitotic clonal expansion of 3T3-L1 adipocyte differentiation. Mol. Biol. Cell 2011, 22, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, X.; Huang, J.X.; Huang, H.Y.; Zhang, Y.Y.; Qian, S.W.; Zhu, H.; Zhang, Y.D.; Liu, Y.; Liu, Y.; et al. Histone demethylase Kdm4b functions as a co-factor of C/EBPbeta to promote mitotic clonal expansion during differentiation of 3T3-L1 preadipocytes. Cell Death Differ. 2012, 19, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.C.; Cao, Z.; Classon, M.; McKnight, S.L. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995, 9, 168–181. [Google Scholar] [CrossRef]
- Souza, A.C.; Bocharov, A.V.; Baranova, I.N.; Vishnyakova, T.G.; Huang, Y.G.; Wilkins, K.J.; Hu, X.; Street, J.M.; Alvarez-Prats, A.; Mullick, A.E.; et al. Antagonism of scavenger receptor CD36 by 5A peptide prevents chronic kidney disease progression in mice independent of blood pressure regulation. Kidney Int. 2016, 89, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mulumba, M.; Ong, H.; Lubell, W.D. Diversity-Oriented Synthesis of Cyclic Azapeptides by A3-Macrocyclization Provides High-Affinity CD36-Modulating Peptidomimetics. Angew. Chem. 2017, 56, 6284–6288. [Google Scholar] [CrossRef]
- Bessi, V.L.; Labbe, S.M.; Huynh, D.N.; Menard, L.; Jossart, C.; Febbraio, M.; Guerin, B.; Bentourkia, M.; Lecomte, R.; Carpentier, A.C.; et al. EP 80317, a selective CD36 ligand, shows cardioprotective effects against post-ischaemic myocardial damage in mice. Cardiovasc. Res. 2012, 96, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, A.V.; Wu, T.; Baranova, I.N.; Birukova, A.A.; Sviridov, D.; Vishnyakova, T.G.; Remaley, A.T.; Eggerman, T.L.; Patterson, A.P.; Birukov, K.G. Synthetic Amphipathic Helical Peptides Targeting CD36 Attenuate Lipopolysaccharide-Induced Inflammation and Acute Lung Injury. J. Immunol. 2016, 197, 611–619. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef]
- Jiang, T.; Liebman, S.E.; Lucia, M.S.; Li, J.; Levi, M. Role of altered renal lipid metabolism and the sterol regulatory element binding proteins in the pathogenesis of age-related renal disease. Kidney Int. 2005, 68, 2608–2620. [Google Scholar] [CrossRef]
- Cho, H.; Um, J.; Lee, J.H.; Kim, W.H.; Kang, W.S.; Kim, S.H.; Ha, H.H.; Kim, Y.C.; Ahn, Y.K.; Jung, D.W.; et al. ENOblock, a unique small molecule inhibitor of the non-glycolytic functions of enolase, alleviates the symptoms of type 2 diabetes. Sci. Rep. 2017, 7, 44186. [Google Scholar] [CrossRef]
- Brosius, F.C., 3rd; He, J.C. JAK inhibition and progressive kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Goll, G.L.; Kvien, T.K. New-generation JAK inhibitors: How selective can they be? Lancet 2018, 391, 2477–2478. [Google Scholar] [CrossRef]
- Bachelez, H.; van de Kerkhof, P.C.; Strohal, R.; Kubanov, A.; Valenzuela, F.; Lee, J.H.; Yakusevich, V.; Chimenti, S.; Papacharalambous, J.; Proulx, J.; et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: A phase 3 randomised non-inferiority trial. Lancet 2015, 386, 552–561. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 320. [Google Scholar] [CrossRef] [PubMed]
- Bertocchio, J.P.; Warnock, D.G.; Jaisser, F. Mineralocorticoid receptor activation and blockade: An emerging paradigm in chronic kidney disease. Kidney Int. 2011, 79, 1051–1060. [Google Scholar] [CrossRef]
- Xiao, X.; Li, H.; Yang, J.; Qi, X.; Zu, X.; Yang, J.; Zhong, J.; Cao, R.; Liu, J.; Wen, G. Wnt/beta-catenin signaling pathway and lipolysis enzymes participate in methylprednisolone induced fat differential distribution between subcutaneous and visceral adipose tissue. Steroids 2014, 84, 30–35. [Google Scholar] [CrossRef]
- Holdaas, H.; Fellstrom, B.; Jardine, A.G.; Holme, I.; Nyberg, G.; Fauchald, P.; Gronhagen-Riska, C.; Madsen, S.; Neumayer, H.H.; Cole, E.; et al. Effect of fluvastatin on cardiac outcomes in renal transplant recipients: A multicentre, randomised, placebo-controlled trial. Lancet 2003, 361, 2024–2031. [Google Scholar] [CrossRef]
- Baigent, C.; Landray, M.J.; Reith, C.; Emberson, J.; Wheeler, D.C.; Tomson, C.; Wanner, C.; Krane, V.; Cass, A.; Craig, J.; et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): A randomised placebo-controlled trial. Lancet 2011, 377, 2181–2192. [Google Scholar] [CrossRef]
- Hu, P.J.; Wu, M.Y.; Lin, T.C.; Chen, T.T.; Wu, Y.C.; Su, S.L.; Lu, K.C.; Chen, J.S.; Sung, F.C.; Lee, C.T.; et al. Effect of Statins on Renal Function in Chronic Kidney Disease Patients. Sci. Rep. 2018, 8, 16276. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Gui, T.; Hiller, C.; Kullak-Ublick, G.A. Farnesoid X Receptor Protects against Kidney Injury in Uninephrectomized Obese Mice. J. Biol. Chem. 2016, 291, 2397–2411. [Google Scholar] [CrossRef] [PubMed]
- Isnard Bagnis, C.; Deray, G.; Baumelou, A.; Le Quintrec, M.; Vanherweghem, J.L. Herbs and the kidney. Am. J. Kidney Dis. 2004, 44, 1–11. [Google Scholar] [CrossRef]
- Nahin, R.L.; Barnes, P.M.; Stussman, B.J.; Bloom, B. Costs of complementary and alternative medicine (CAM) and frequency of visits to CAM practitioners: United States, 2007. Natl. Health Stat. Rep. 2009, 18, 1–14. [Google Scholar]
- Saravanan, S.; Pari, L. Protective effect of thymol on high fat diet induced diabetic nephropathy in C57BL/6J mice. Chem.-Biol. Interact. 2016, 245, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Jiang, L.Y.; Wu, S.; Jiang, W.J.; Xie, L.; Li, W.; Yang, C.H. Proteomic Analysis Reveals the Renoprotective Effect of Tribulus terrestris against Obesity-Related Glomerulopathy in Rats. Biol. Pharm. Bull. 2018, 41, 1430–1439. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, D.; Lu, F.; Zou, X.; Xu, L.; Wang, K.; Huang, W.; Su, H.; Zhang, C.; Gao, Y.; et al. Coptidis Rhizoma inhibits NLRP3 inflammasome activation and alleviates renal damage in early obesity-related glomerulopathy. Phytomedicine 2018, 49, 52–65. [Google Scholar] [CrossRef]
- Yang, X.; Xu, W.; Huang, K.; Zhang, B.; Wang, H.; Zhang, X.; Gong, L.; Luo, Y.; He, X. Precision toxicology shows that troxerutin alleviates ochratoxin A-induced renal lipotoxicity. FASEB J. 2019, 33, 2212–2227. [Google Scholar] [CrossRef]
- Guan, Y.; Wu, X.X.; Duan, J.L.; Yin, Y.; Guo, C.; Wei, G.; Wang, Y.H.; Zhu, Y.R.; Weng, Y.; Xi, M.M.; et al. Effects and Mechanism of Combination of Rhein and Danshensu in the Treatment of Chronic Kidney Disease. Am. J. Chin. Med. 2015, 43, 1381–1400. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.F.; Chang, H.C.; Huang, S.L.; Chen, C.L.; Chen, W.T.; Yang, C.C. Prescribed Renoprotective Chinese Herbal Medicines Were Associated with a Lower Risk of All-Cause and Disease-Specific Mortality among Patients with Chronic Kidney Disease: A Population-Based Follow-Up Study in Taiwan. Evid.-Based Complement. Altern. Med. 2017, 2017, 5632195. [Google Scholar] [CrossRef]
- Akyol, A.D.; Yildirim, Y.; Toker, E.; Yavuz, B. The use of complementary and alternative medicine among chronic renal failure patients. J. Clin. Nurs. 2011, 20, 1035–1043. [Google Scholar] [CrossRef]
- Stieger, B.; Mahdi, Z.M.; Jager, W. Intestinal and Hepatocellular Transporters: Therapeutic Effects and Drug Interactions of Herbal Supplements. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Visentin, M.; Biason, P.; Toffoli, G. Drug interactions among the epidermal growth factor receptor inhibitors, other biologics and cytotoxic agents. Pharmacol. Ther. 2010, 128, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Cano, N.J.; Budde, K.; Chazot, C.; Kovesdy, C.P.; Mak, R.H.; Mehrotra, R.; Raj, D.S.; Sehgal, A.R.; Stenvinkel, P.; et al. Diets and enteral supplements for improving outcomes in chronic kidney disease. Nat. Rev. Nephrol. 2011, 7, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Kim, D.K.; Han, S.S.; Park, J.H.; Shin, S.J.; Lee, S.H.; Choi, B.S.; Lim, C.S.; Kim, S.; Chin, H.J. Weight loss has an additive effect on the proteinuria reduction of angiotensin II receptor blockers in hypertensive patients with chronic kidney disease. Kidney Res. Clin. Pr. 2018, 37, 49–58. [Google Scholar] [CrossRef]
- Friedman, A.N.; Chambers, M.; Kamendulis, L.M.; Temmerman, J. Short-term changes after a weight reduction intervention in advanced diabetic nephropathy. Clin. J. Am. Soc. Nephrol. 2013, 8, 1892–1898. [Google Scholar] [CrossRef]
- Saiki, A.; Nagayama, D.; Ohhira, M.; Endoh, K.; Ohtsuka, M.; Koide, N.; Oyama, T.; Miyashita, Y.; Shirai, K. Effect of weight loss using formula diet on renal function in obese patients with diabetic nephropathy. Int. J. Obes. 2005, 29, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Vamenta-Morris, H.; Dreisbach, A.; Shoemaker-Moyle, M.; Abdel-Rahman, E.M. Internet claims on dietary and herbal supplements in advanced nephropathy: Truth or myth. Am. J. Nephrol. 2014, 40, 393–398. [Google Scholar] [CrossRef]
- Zeller, K.; Whittaker, E.; Sullivan, L.; Raskin, P.; Jacobson, H.R. Effect of restricting dietary protein on the progression of renal failure in patients with insulin-dependent diabetes mellitus. New Engl. J. Med. 1991, 324, 78–84. [Google Scholar] [CrossRef]
- Marzocco, S.; Dal Piaz, F.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Tartaglia, D.; Autore, G.; Di Iorio, B. Very low protein diet reduces indoxyl sulfate levels in chronic kidney disease. Blood Purif. 2013, 35, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.R.; Di Micco, L.; Marzocco, S.; De Simone, E.; De Blasio, A.; Sirico, M.L.; Nardone, L.; UBI Study Group. Very Low-Protein Diet (VLPD) Reduces Metabolic Acidosis in Subjects with Chronic Kidney Disease: The “Nutritional Light Signal” of the Renal Acid Load. Nutrients 2017, 9, 69. [Google Scholar] [CrossRef]
- Klahr, S.; Levey, A.S.; Beck, G.J.; Caggiula, A.W.; Hunsicker, L.; Kusek, J.W.; Striker, G. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of Diet in Renal Disease Study Group. New Engl. J. Med. 1994, 330, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Kopple, J.D.; Wang, X.; Beck, G.J.; Collins, A.J.; Kusek, J.W.; Greene, T.; Levey, A.S.; Sarnak, M.J. Effect of a very low-protein diet on outcomes: Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) Study. Am. J. Kidney Dis. 2009, 53, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.M.; Muntner, P.; Rizk, D.V.; McClellan, W.M.; Warnock, D.G.; Newby, P.K.; Judd, S.E. Dietary patterns and risk of death and progression to ESRD in individuals with CKD: A cohort study. Am. J. Kidney Dis. 2014, 64, 204–213. [Google Scholar] [CrossRef]
- Azadbakht, L.; Atabak, S.; Esmaillzadeh, A. Soy protein intake, cardiorenal indices, and C-reactive protein in type 2 diabetes with nephropathy: A longitudinal randomized clinical trial. Diabetes Care 2008, 31, 648–654. [Google Scholar] [CrossRef]
- Jing, Z.; Wei-Jie, Y. Effects of soy protein containing isoflavones in patients with chronic kidney disease: A systematic review and meta-analysis. Clin. Nutr. 2016, 35, 117–124. [Google Scholar] [CrossRef]
- Goraya, N.; Wesson, D.E. Dietary interventions to improve outcomes in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 505–510. [Google Scholar] [CrossRef]
- Goraya, N.; Simoni, J.; Jo, C.; Wesson, D.E. Dietary acid reduction with fruits and vegetables or bicarbonate attenuates kidney injury in patients with a moderately reduced glomerular filtration rate due to hypertensive nephropathy. Kidney Int. 2012, 81, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Asghari, G.; Momenan, M.; Yuzbashian, E.; Mirmiran, P.; Azizi, F. Dietary pattern and incidence of chronic kidney disease among adults: A population-based study. Nutr. Metab. 2018, 15, 88. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.W.; Tsai, W.H.; Liu, J.S.; Kuo, K.L. Association of Vegetarian Diet with Chronic Kidney Disease. Nutrients 2019, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Khatri, M.; Moon, Y.P.; Scarmeas, N.; Gu, Y.; Gardener, H.; Cheung, K.; Wright, C.B.; Sacco, R.L.; Nickolas, T.L.; Elkind, M.S. The association between a Mediterranean-style diet and kidney function in the Northern Manhattan Study cohort. Clin. J. Am. Soc. Nephrol. 2014, 9, 1868–1875. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, K.B.; Hyun, Y.Y.; Chang, Y.; Ryu, S.; Choi, Y. DASH dietary pattern and chronic kidney disease in elderly Korean adults. Eur. J. Clin. Nutr. 2017, 71, 755–761. [Google Scholar] [CrossRef]
- Asghari, G.; Yuzbashian, E.; Mirmiran, P.; Azizi, F. The association between Dietary Approaches to Stop Hypertension and incidence of chronic kidney disease in adults: The Tehran Lipid and Glucose Study. Nephrol. Dial. Transplant. 2017, 32 (Suppl. 2), ii224–ii230. [Google Scholar] [CrossRef]
- Asghari, G.; Farhadnejad, H.; Mirmiran, P.; Dizavi, A.; Yuzbashian, E.; Azizi, F. Adherence to the Mediterranean diet is associated with reduced risk of incident chronic kidney diseases among Tehranian adults. Hypertens. Res. 2017, 40, 96–102. [Google Scholar] [CrossRef]
- Mazaraki, A.; Tsioufis, C.; Dimitriadis, K.; Tsiachris, D.; Stefanadi, E.; Zampelas, A.; Richter, D.; Mariolis, A.; Panagiotakos, D.; Tousoulis, D.; et al. Adherence to the Mediterranean diet and albuminuria levels in Greek adolescents: Data from the Leontio Lyceum ALbuminuria (3L study). Eur. J. Clin. Nutr. 2011, 65, 219–225. [Google Scholar] [CrossRef]
- Diaz-Lopez, A.; Bullo, M.; Martinez-Gonzalez, M.A.; Guasch-Ferre, M.; Ros, E.; Basora, J.; Covas, M.I.; del Carmen Lopez-Sabater, M.; Salas-Salvado, J.; Investigators, P.R.S. Effects of Mediterranean diets on kidney function: A report from the PREDIMED trial. Am. J. Kidney Dis. 2012, 60, 380–389. [Google Scholar] [CrossRef]
- Davidson, M.H. Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am. J. Cardiol. 2006, 98, 27i–33i. [Google Scholar] [CrossRef]
- Prichard, S.S. Impact of dyslipidemia in end-stage renal disease. J. Am. Soc. Nephrol. 2003, 14 (Suppl. 4), S315–S320. [Google Scholar] [CrossRef]
- Friedman, A.N. Omega-3 fatty acid supplementation in advanced kidney disease. Semin. Dial. 2010, 23, 396–400. [Google Scholar] [CrossRef]
- Lim, A.K.; Manley, K.J.; Roberts, M.A.; Fraenkel, M.B. Fish oil treatment for kidney transplant recipients: A meta-analysis of randomized controlled trials. Transplantation 2007, 83, 831–838. [Google Scholar] [CrossRef]
- Tatsioni, A.; Chung, M.; Sun, Y.; Kupelnick, B.; Lichtenstein, A.H.; Perrone, R.; Chew, P.; Lau, J.; Bonis, P.A. Effects of fish oil supplementation on kidney transplantation: A systematic review and meta-analysis of randomized, controlled trials. J. Am. Soc. Nephrol. 2005, 16, 2462–2470. [Google Scholar] [CrossRef]
- Bonis, P.A.; Chung, M.; Tatsioni, A.; Sun, Y.; Kupelnick, B.; Lichtenstein, A.; Perrone, R.; Chew, P.; DeVine, D.; Lau, J. Effects of omega-3 fatty acids on organ transplantation. Evid. Rep. Technol. Assess. 2005, 115, 1–11. [Google Scholar]
- Donadio, J.V., Jr.; Grande, J.P.; Bergstralh, E.J.; Dart, R.A.; Larson, T.S.; Spencer, D.C. The long-term outcome of patients with IgA nephropathy treated with fish oil in a controlled trial. Mayo Nephrology Collaborative Group. J. Am. Soc. Nephrol. 1999, 10, 1772–1777. [Google Scholar] [PubMed]
- Zanetti, M.; Gortan Cappellari, G.; Barbetta, D.; Semolic, A.; Barazzoni, R. Omega 3 Polyunsaturated Fatty Acids Improve Endothelial Dysfunction in Chronic Renal Failure: Role of eNOS Activation and of Oxidative Stress. Nutrients 2017, 9, 895. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, P.M.; Ferraccioli, G.F.; Gambaro, G.; Fulignati, P.; Costanzi, S. Combined treatment with renin-angiotensin system blockers and polyunsaturated fatty acids in proteinuric IgA nephropathy: A randomized controlled trial. Nephrol. Dial. Transplant. 2009, 24, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Lee, S.M.; Lee, M.H.; Son, Y.K.; Kim, S.E.; An, W.S. Effect of Omega-3 Fatty Acid on STAMP2 Expression in the Heart and Kidney of 5/6 Nephrectomy Rat Model. Mar. Drugs 2018, 16, 398. [Google Scholar] [CrossRef]
- Miller, E.R., 3rd; Juraschek, S.P.; Anderson, C.A.; Guallar, E.; Henoch-Ryugo, K.; Charleston, J.; Turban, S.; Bennett, M.R.; Appel, L.J. The effects of n-3 long-chain polyunsaturated fatty acid supplementation on biomarkers of kidney injury in adults with diabetes: Results of the GO-FISH trial. Diabetes Care 2013, 36, 1462–1469. [Google Scholar] [CrossRef]



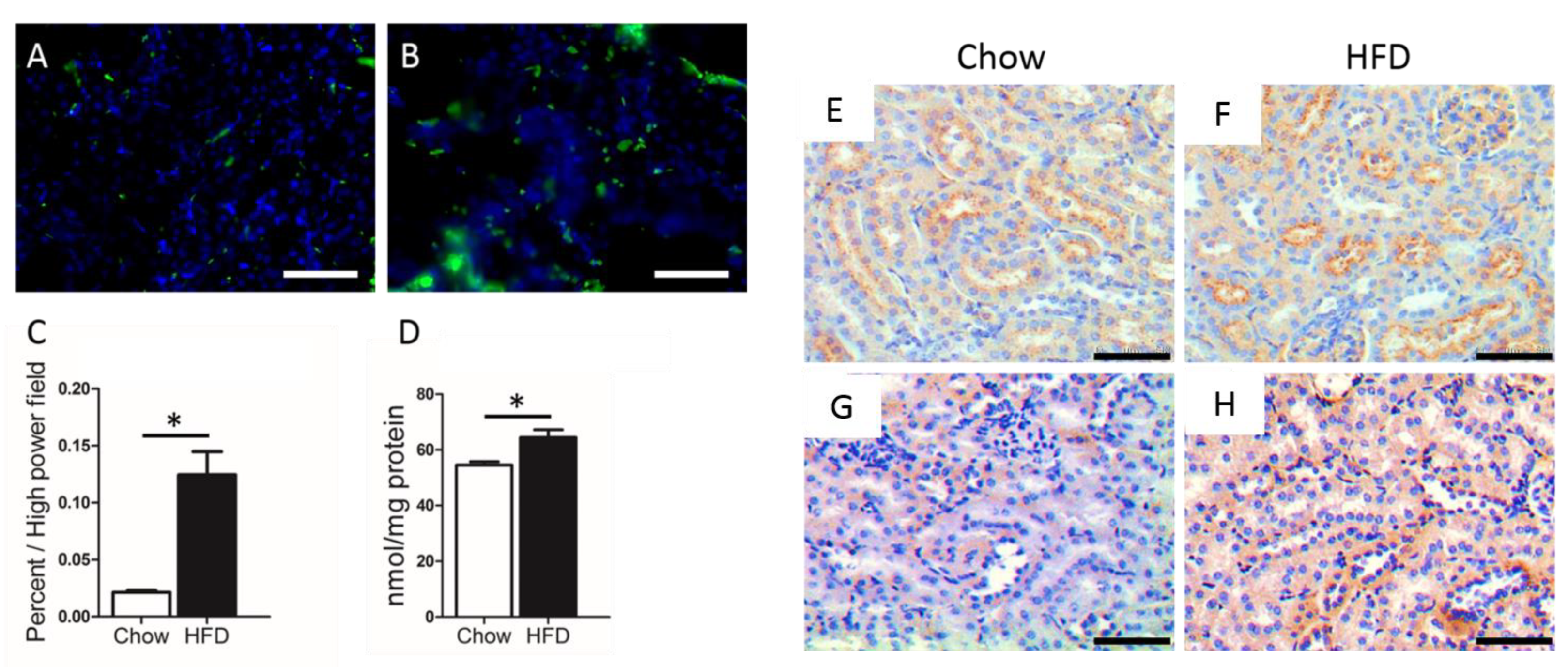{kind=link}
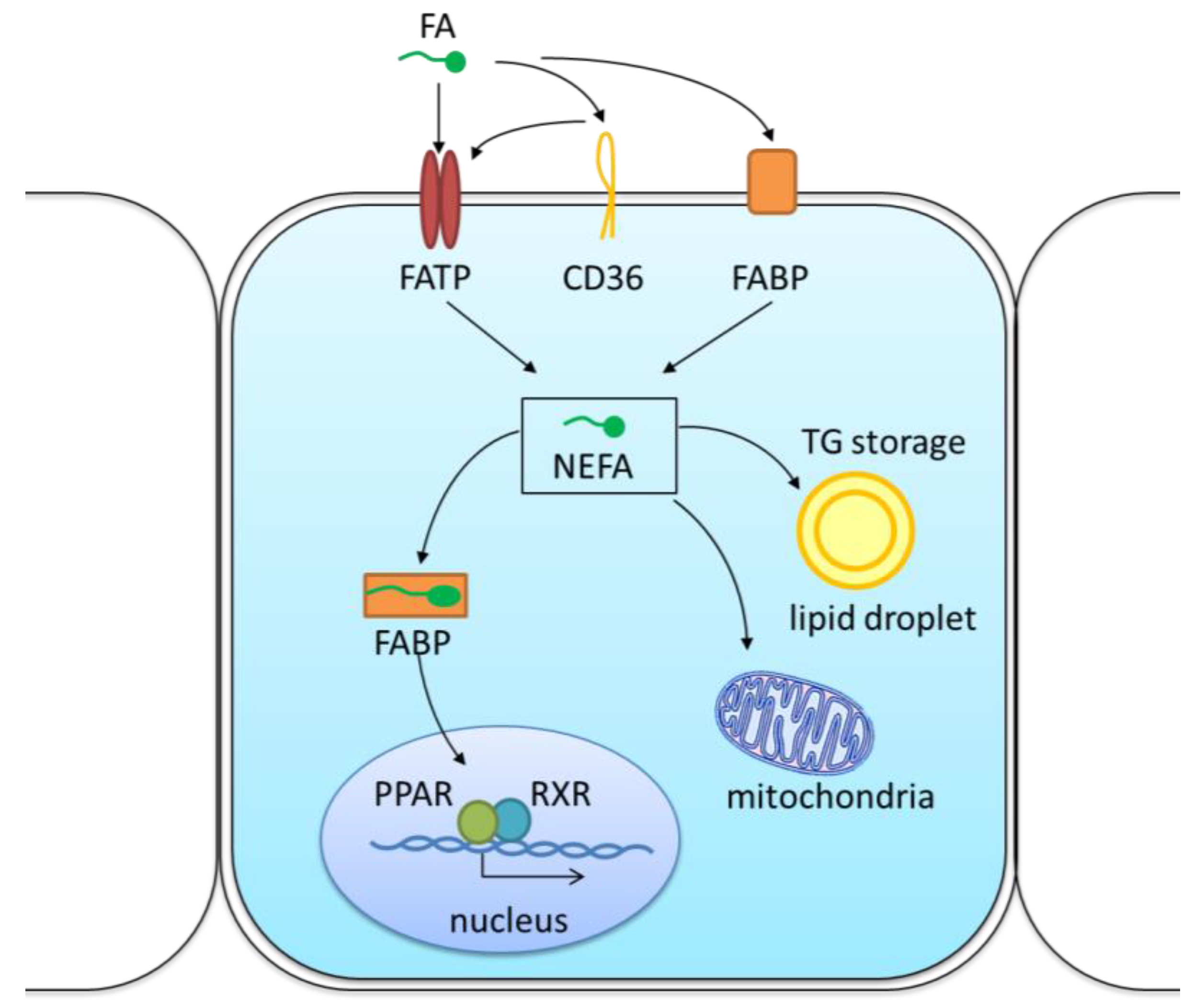{kind=link}
| Compound | Species | Target | Kidney Outcome | Reference |
|---|---|---|---|---|
| GW4064/CA | Mouse | FXR | Glomerulosclerosis ↓ Tubulointerstitial fibrosis ↓ Proteinuria ↓ | [83] |
| OCA | Mouse | FXR | Glomerulosclerosis ↓ Tubulointerstitial fibrosis ↓ Proteinuria ↓ | [84,135] |
| INT-777 | Mouse | TGR5 | Glomerulosclerosis ↓ Tubulointerstitial fibrosis ↓ Proteinuria ↓ | [90] |
| INT-767 | Mouse | FXR/TGR5 | Glomerulosclerosis ↓ Tubulointerstitial fibrosis ↓ Proteinuria ↓ | [91] |
| Fenofibrate | Mouse | PPARα | Glomerulosclerosis ↓ Tubulointerstitial fibrosis ↓ Proteinuria ↓ | [94] |
| Rosiglitazone | Human | PPARɣ | HOMA score ↓ Hs-CRP ↓ Blood pressure = | [102] |
| 5A peptide | Mouse | CD36 | Glomerulosclerosis ↓ Tubulointerstitial fibrosis ↓ Proteinuria ↓ | [118] |
| ENOblock | Mouse | enolase | Inflammation ↓ Tubulointerstitial fibrosis ↓ | [124] |
| Statin | Mouse/Human | HMG-CoA reductase | Proteinuria ↓ | [134] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. https://doi.org/10.3390/nu11040722
Gai Z, Wang T, Visentin M, Kullak-Ublick GA, Fu X, Wang Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients. 2019; 11(4):722. https://doi.org/10.3390/nu11040722
Chicago/Turabian StyleGai, Zhibo, Tianqi Wang, Michele Visentin, Gerd A. Kullak-Ublick, Xianjun Fu, and Zhenguo Wang. 2019. "Lipid Accumulation and Chronic Kidney Disease" Nutrients 11, no. 4: 722. https://doi.org/10.3390/nu11040722
APA StyleGai, Z., Wang, T., Visentin, M., Kullak-Ublick, G. A., Fu, X., & Wang, Z. (2019). Lipid Accumulation and Chronic Kidney Disease. Nutrients, 11(4), 722. https://doi.org/10.3390/nu11040722