Perinatal Undernutrition, Metabolic Hormones, and Lung Development


Abstract
1. Introduction
2. Effect of Metabolic Hormones in Lung Development
2.1. Ghrelin
2.2. Leptin
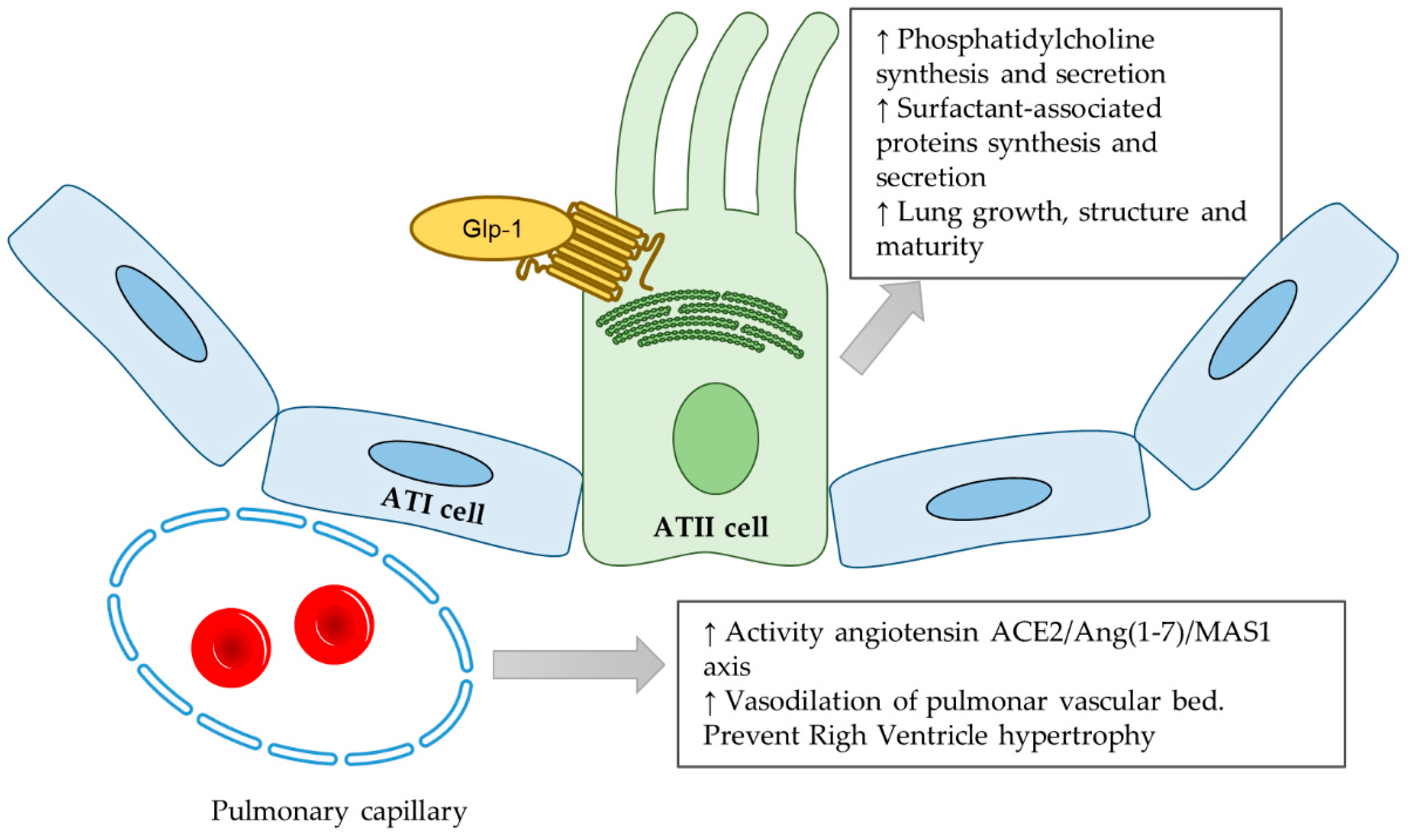
2.3. GLP-1
2.4. Retinoids
2.5. Cholecalciferol
3. Effect of Undernutrition on Lung Development and Adult Lung Function
- (1)
- Severe nausea and vomiting period that persists more than the first trimester [115];
- (2)
- The “Maternal Depletion Syndrome,” a product of a short inter-pregnancy interval, not allowing sufficient time to replenish energy reserves and recovery of mothers, which promotes a depletion of both macro- and micronutrients [116];
- (3)
- Teenager pregnancy, where the mother, who may still be growing, competes with the fetus for resources [117];
- (4)
- Use and abuse of tobacco [118]; and
- (5)
- Alcohol/drugs [119], which may promote placenta under-function and reduced nutrient supply to fetus and/or maternal undernutrition.
4. Undernutrition and Hormones in Lung Development
Author Contributions
Funding
Conflicts of Interest
References
- Kelly, R.W. Nutrition and placental development. Proc. Nutr. Soc. Aust. 1992, 17, 203–211. [Google Scholar]
- McCance, R.A.; Widdowson, E.M. The determinants of growth and form. Proc. R. Soc. Lond. B Biol. Sci. 1974, 185, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Clark, P.M. Fetal undernutrition and disease in later life. Rev. Reprod. 1997, 2, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- DiFiore, J.W.; Wilson, J.W. Lung development. Semin. Pediatr. Surg. 1994, 3, 221–232. [Google Scholar] [PubMed]
- Burri, P.H. Structural aspects of postnatal lung development – alveolar formation and growth. Biol. Neonate 2006, 89, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zosky, G.R. Lung development. Photochem. Photobiol. Sci. 2017, 16, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Stocks, J.; Hislop, A.; Sonnappa, S. Early lung development: Lifelong effect on respiratory health and disease. Lancet Respir. Med. 2013, 9, 728–742. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Wang, G.; Lee, H.M.; Englander, E.; Greeley, G.H., Jr. Ghrelin—not just another stomach hormone. Regul. Pept. 2002, 105, 75–81. [Google Scholar] [CrossRef]
- Wren, A.M.; Small, C.J.; Ward, H.L.; Murphy, K.G.; Dakin, C.L.; Taheri, S.; Kennedy, A.R.; Roberts, G.H.; Morgan, D.G.; Ghatei, M.A.; et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 2000, 141, 4325–4328. [Google Scholar] [CrossRef] [PubMed]
- Cortelazzi, D.; Cappiello, V.; Morpurgo, P.S.; Ronzoni, S.; Nobile De Santis, M.S.; Cetin, I.; Beck-Peccoz, P.; Spada, A. Circulating levels of ghrelin in human fetuses. Eur. J. Endocrinol. 2003, 149, 111–116. [Google Scholar] [CrossRef]
- Volante, M.; Fulcheri, E.; Allìa, E.; Cerrato, M.; Pucci, A.; Papotti, M. Ghrelin expression in fetal, infant and adult human lung. J. Histochem. Cytochem. 2002, 50, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, K.; Nakagawa, M.; Baba, Y.; Sato, M.; Toshinai, K.; Date, Y.; Nakazato, M.; Kojima, M.; Miyazato, M.; Kaiya, H.; et al. Maternal ghrelin plays an important role in rat fetal development during pregnancy. Endocrinology 2006, 147, 1333–1342. [Google Scholar] [CrossRef]
- Nunes, S.; Nogueira-Silva, C.; Dias, E.; Moura, R.S.; Correia-Pinto, J. Ghrelin and obestatin: Different role in fetal lung development? Peptides 2008, 29, 2150–2158. [Google Scholar] [CrossRef]
- Santos, M.; Bastos, P.; Gonzaga, S.; Roriz, J.M.; Baptista, M.J.; Nogueira-Silva, C.; Melo-Rocha, G.; Henriques-Coelho, T.; Roncon-Albuquerque, R., Jr.; Leite-Moreira, A.F.; et al. Ghrelin expression in human and rat fetal lungs and the effect of ghrelin administration in nitrofen-induced congenital diaphragmatic hernia. Pediatr. Res. 2006, 59, 531–537. [Google Scholar] [CrossRef]
- Pereira-Terra, P.; Moura, R.S.; Nogueira-Silva, C.; Correia-Pinto, J. Neuroendocrine factors regulate retinoic acid receptors in normal and hypoplastic lung development. J. Physiol. 2015, 593, 3301–3311. [Google Scholar] [CrossRef]
- Xu, Y.P.; Zhu, J.J.; Cheng, F.; Jiang, K.W.; Gu, W.Z.; Shen, Z.; Wu, Y.D.; Liang, L.; Du, L.Z. Ghrelin ameliorates hypoxia-induced pulmonary hypertension via phosphor-GSK3 β/β-catenin signalling in neonatal rats. J. Mol. Endocrinol. 2011, 47, 33–43. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Malli, F.; Papaiopannou, A.L.; Gourgoulianis, K.L.; Daniil, Z. The role of leptin in the respiratory system: An overview. Respir. Res. 2010, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clarck, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef]
- Gorska, E.; Popko, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Hoggard, N.; Mercer, J.G.; Rayner, D.V.; Moar, K.; Trayhurn, P.; Williams, L.M. Localization of leptin receptor mRNA splice variants in murine peripheral tissues by RT-PCR and in situ hybridization. Biochem. Biophys. Res. Commun. 1997, 232, 383–387. [Google Scholar] [CrossRef]
- Masuzaki, H.; Ogawa, Y.; Sagawa, N.; Hosoda, K.; Matsumoto, T.; Mise, H.; Nishimura, H.; Yoshimasa, Y.; Tanaka, I.; Mori, T.; et al. Nonadipose tissue production of leptin: Leptin as a novel placenta-derived hormone in humans. Nat. Med. 1997, 3, 1023–1033. [Google Scholar] [CrossRef]
- Vernooy, J.H.; Drummen, N.E.; van Suylen, R.J.; Cloots, R.H.; Möller, G.M.; Bracke, K.R.; Zuyderduyn, S.; Dentener, M.A.; Brusselle, G.G.; Hiemstra, P.S.; et al. Enhanced pulmonary leptin expression in patients with severe COPD and asymptomatic smokers. Thorax 2009, 64, 26–32. [Google Scholar] [CrossRef]
- Belmeyer, A.; Martino, J.M.; Chandel, N.S.; Scott Budinger, G.R.; Dean, D.A.; Mutlu, G.M. Leptin resistance protects mice from hyperoxia-induced acute lung injury. Am. J. Respir. Crit. Care Med. 2007, 175, 587–594. [Google Scholar] [CrossRef]
- Torday, J.S.; Sun, H.; Wang, L.; Torres, E.; Sunday, M.E.; Rubin, L.P. Leptin mediates the parathyroid hormone-related protein paracrine stimulation of fetal lung maturation. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L405–L410. [Google Scholar] [CrossRef]
- Hoggard, N.; Hunter, L.; Duncan, J.S.; Williams, L.M.; Trayhurn, P.; Mercer, J.G. Leptin and leptin receptor mRNA and protein expression in the murine fetus and placenta. Proc. Natl. Acad. Sci. USA 1997, 94, 11073–11078. [Google Scholar] [CrossRef]
- Bergen, H.T.; Cherlet, T.C.; Manuel, P.; Scott, J.E. Identification of leptin receptors in lung and isolated fetal type II cells. Am. J. Respir. Cell Mol. Biol. 2002, 27, 71–77. [Google Scholar] [CrossRef]
- Henson, M.C.; Swan, K.F.; Edwards, D.E.; Hoyle, G.W.; Purcell, J.; Castracane, V.D. Leptin receptor expression in fetal lung increases in late gestation in the baboon: A model for human pregnancy. Reproduction 2004, 127, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, J.P.; Huang, H.; Wang, Z.H.; Cheng, R.; Cai, W.B. Leptin promotes fetal lung maturity and upregulats SP-A expression in pulmonary alveoli type-II epithelial cells involving TTF-1 activation. PLoS ONE 2013, 8, e69297. [Google Scholar] [CrossRef]
- Kirwin, S.M.; Bhandari, V.; Dimatteo, D.; Barone, C.; Johnson, L.; Paul, S.; Spitzer, A.R.; Chander, A.; Hassink, S.G.; Funanage, V.L. Leptin enhances lung maturity in the fetal rat. Pediatr. Res. 2006, 60, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Schehr, A.; Ikegami, M. Leptin does not influence surfactant synthesis in fetal sheep and mice lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L498–L505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Blasio, M.J.; Boije, M.; Kempster, S.L.; Smith, G.C.; Charnock-Jones, D.S.; Denyer, A.; Hughes, A.; Wooding, F.B.; Blache, D.; Fowden, A.L.; et al. Leptin Matures Aspects of Lung Structure and Function in the Ovine Fetus. Endocrinology 2016, 157, 395–404. [Google Scholar] [CrossRef]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef]
- Huang, K.; Rabold, R.; Abston, E.; Schofield, B.; Misra, V.; Galdzicka, E.; Lee, H.; Biswal, S.; Mitzner, W.; Tankersley, C.G. Effects of leptin deficiency on postnatal lung development in mice. J. Appl. Physiol. 2008, 105, 249–259. [Google Scholar] [CrossRef]
- Song, Y.; Yu, Y.; Wang, D.; Chai, S.; Liu, D.; Xiao, X.; Huang, Y. Maternal high-fat diet feeding during pregnancy and lactation augments lung inflammation and remodelling in offspring. Respir. Physiol. Neurobiol. 2015, 207, 1–6. [Google Scholar] [CrossRef]
- Khrisanapant, W.; Sengmeuang, P.; Kukongviriyapan, U.; Pasurivong, O.; Pakdeechotel, P. Plasma leptin levels and a restrictive lung in obese thai children and adolescents. Southeast Asian J. Trop. Med. Public Health 2015, 46, 116–124. [Google Scholar]
- Holst, J.J. From the Incretin Concept and the Discovery of GLP-1 to Today’s Diabetes Therapy. Front. Endocrinol. 2019, 10, 260. [Google Scholar] [CrossRef]
- Bullock, B.P.; Heller, R.S.; Habener, J.F. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology 1996, 137, 2968–2978. [Google Scholar] [CrossRef] [PubMed]
- Romaní-Pérez, M.; Outeiriño-Iglesias, V.; Gil-Lozano, M.; González-Matías, L.C.; Mallo, F.; Vigo, E. Pulmonary GLP-1 receptor increases at birth and exogenous GLP-1 Receptor agonists augmented surfactant-protein levels in litters from normal and nitrofen-treated pregnant rats. Endocrinology 2013, 154, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Benito, E.; Blazquez, E.; Bosch, M.A. Glucagon-like peptide-1-(7–36) amide increases pulmonary surfactant secretion through a cyclic adenosine 3’, 5’-monophosphate-dependent protein kinase mechanism in rat type II pneumocytes. Endocrinology 1998, 139, 2363–2368. [Google Scholar] [CrossRef] [PubMed]
- Vara, E.; Arias-Díaz, J.; Garcia, C.; Balibrea, J.L.; Blázquez, E. Glucagon-like peptide-1 (7–36) amide stimulates surfactant secretion in human type II pneumocytes. Am. J. Respir. Crit. Care Med. 2001, 163, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, M.P.; Kampmeijer, A.; Jimenez, J.; Zia, S.; Vanbree, R.; Verbist, G.; Toelen, J.; Deprest, J.A. The Effect of Transplacental Administration of Glucagon-Like Peptide-1 on Fetal Lung Development in the Rabbit Model of Congenital Diaphragmatic Hernia. Fetal Diagn. Ther. 2016, 39, 125–133. [Google Scholar] [CrossRef]
- Fandiño, J.; Vaz, A.A.; Toba, L.; Romaní-Pérez, M.; González-Matías, L.C.; Mallo, F.; Diz-Chaves, Y. Liraglutide Enhances the Activity of the ACE/Ang(1–7)/Mas Receptor Pathway in Lungs of Male Pups from Food-Restricted Mothers and Prevents the Reduction of SP-A. Int. J. Endocrinol. 2018, 2018, 6920620. [Google Scholar] [CrossRef]
- Romaní-Pérez, M.; Outeiriño-Iglesias, V.; Moya, C.M.; Santisteban, P.; González-Matías, L.C.; Vigo, E.; Mallo, F. Activation of the GLP-1 Receptor by Liraglutide Increases ACE2 Expression, Reversing Right Ventricle Hypertrophy, and Improving the Production of SP-A and SP-B in the Lungs of Type 1 Diabetes Rats. Endocrinology 2015, 156, 3559–3569. [Google Scholar] [CrossRef]
- Ross, A.C.; Ternus, M.E. Vitamin A as a hormone: Recent advances in understanding the actions of retinol, retinoic acid, and beta carotene. J. Am. Diet. Assoc. 1993, 93, 1285–1290. [Google Scholar] [CrossRef]
- Conaway, H.H.; Henning, P.; Lerner, U.H. Vitamin a metabolism, action, and role in skeletal homeostasis. Endocr. Rev. 2013, 34, 766–797. [Google Scholar] [CrossRef]
- Marill, J.; Idres, N.; Capron, C.C.; Nguyen, E.; Chabot, G.G. Retinoica cid metabolism and mechanism of action: A review. Curr. Drug Metab. 2003, 4, 1–10. [Google Scholar] [CrossRef]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the reanscription of retinoid-target genes. Gene 2004, 328, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; McCaffery, P.J.; Drager, U.C.; De Luca, L.M. Retinoids in embryonal development. Physiol. Rev. 2000, 80, 1021–1054. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Roth, C.B.; Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin a deficiency. Effects of restoration of vitamin A at various times during gestation. Am. J. Anat. 1953, 92, 189–217. [Google Scholar] [CrossRef]
- Lammer, E.J.; Chen, D.T.; Hoar, R.M.; Agnish, N.D.; Benke, P.J.; Braun, J.T.; Curry, C.T.; Fernhoff, P.M.; Grix, A.W., Jr.; Lott, I.T.; et al. Retinoid acid embryopathy. N. Engl. J. Med. 1985, 313, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Mollard, R.; Viville, S.; Ward, S.J.; Décimo, D.; Chambon, P.; Dollé, P. Tissue-specific expression of retinoic acid receptor isoform transcripts in the mouse embryo. Mech. Dev. 2000, 94, 223–232. [Google Scholar] [CrossRef]
- Malpel, S.; Mendelsohn, C.; Cardoso, W.V. Regulation of retinoic acid signalling during lung morphogenesis. Development 2000, 127, 3057–3067. [Google Scholar]
- Chazaud, C.; Dollé, P.; Rossant, J.; Mollard, R. Retinoic acid signalling regulates murine bronchial tubule. Mech. Dev. 2003, 120, 691–700. [Google Scholar] [CrossRef]
- Timoneda, J.; Rodríguez-Fernández, L.; Zaragozá, R.; Marín, M.P.; Cabezuelo, M.T.; Torres, L.; Viña, J.R.; Barber, T. Vitamin A Deficiency and the Lung. Nutrients 2018, 10, 1132. [Google Scholar] [CrossRef]
- Mendelsohn, C.; Lohnes, D.; Décimo, D.; Lufkin, T.; LeMeur, M.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar]
- Geevarghese, S.K.; Chytil, F. Depletion of retinyl esters in the lungs coincides with lung prenatal morphological maturation. Biochem. Biophys. Res. Commun. 1994, 200, 529–535. [Google Scholar] [CrossRef]
- Hind, M.; Corcoran, J.; Maden, M. Temporal/spatial expression of retinoid binding proteins and RAR isoforms in the postnatal lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L468–L476. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.W.; Kong, X.Y.; Zhu, X.X.; Zhu, G.Q.; Ma, J.S.; Liu, X.X. Retinoic acid promotes primary fetal alveolar epithelial type II cell proliferation and differentiation to alveolar epithelial type I cells. In Vitro Cell. Dev. Biol. Anim. 2015, 51, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Grubor, B.; Meyerholtz, D.K.; Lazic, T.; DeMacedo, M.M.; Derscheid, R.J.; Hostetter, J.M.; Gallup, J.M.; DeMartini, J.C.; Ackermann, M.R. Regulation of surfactant protein and defensin mRNA expression in cultured ovine type II pneumocytes by all-trans retinoic acid and VEGF. Int. J. Exp. Pathol. 2006, 87, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Bohn, D.; Tamura, M.; Perrin, D.; Barker, G.; Rabinovitch, M. Ventilatory predictors of pulmonary hypoplasia in congenital diaphragmatic hernia, confirmed by morphologic assessment. J. Pediatr. 1987, 11, 423–431. [Google Scholar] [CrossRef]
- Greer, J.J.; Babiuk, R.P.; Thebaud, B. Etiology of congenital diaphragmatic hernia: The retinoid hypothesis. Pediatr. Res. 2003, 53, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.H. Incidence of congenital diaphragmatic hernia in the young of rats bred on a diet deficient in Vitamin A. Am. J. Dis. Child. 1941, 62, 888–889. [Google Scholar]
- Major, D.; Cadenas, M.; Fournier, L.; Leclerc, S.; Lefebvre, M.; Cloutier, R. Retinol status of newborn infants with congenital diaphragmatic hernia. Pediatr. Surg. Int. 1998, 13, 547–549. [Google Scholar] [CrossRef]
- Nardiello, C.; Miziková, I.; Silva, D.M.; Ruiz-Camp, J.; Mayer, K.; Vadász, I.; Herold, S.; Seeger, W.; Morty, R.E. Standardisation of oxygen exposure in the development of mouse models for brochopulmonary dysplasia. Dis. Model. Mech. 2017, 10, 185–196. [Google Scholar] [CrossRef]
- Veness-Meehan, K.A.; Botone, F.G., Jr.; Stiles, A.D. Effects of retinoic acid on airspace development and lung collagen in hyperoxia-exposed newborn rats. Pediatr. Res. 2000, 48, 434–444. [Google Scholar] [CrossRef]
- Veness-Meehan, K.A.; Pierce, R.A.; Moats-Staats, B.M.; Stiles, A.D. Retinoic acid attenuates O2-induced inhibition of lung septation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L971–L980. [Google Scholar] [CrossRef]
- Ozer, E.A.; Kumral, A.; Ozer, E.; Duman, N.; Yilmaz, O.; Ozkal, S.; Ozkan, H. Effect of retinoic acid on oxygen-induced lung injury in the newborn rat. Pediatr. Pulmonol. 2005, 39, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Londhe, V.A.; Maisonet, T.M.; Lopez, B.; Shin, B.C.; Huynh, J.; Devaskar, S.U. Retinoic acid rescues alveolar hypoplasia in the calorie-restricted developing rat lung. Am. J. Respir. Cell. Mol. Biol. 2013, 48, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, R.B.; Tibboel, D.; Post, M.; Keijzer, R. Congenital diaphragmatic hernia: Comparisons of animal models and relevance to the human situation. Neonatology 2009, 96, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Mey, J.; Babiuk, R.P.; Clugston, R.; Zhang, W.; Greer, J.J. Retinal dehydrogenase-2 is inhibited by compounds that induce congenital diaphragmatic hernias in rodents. Am. J. Pathol. 2003, 162, 673–679. [Google Scholar] [CrossRef]
- Chen, M.H.; MacGowan, A.; Ward, S.; Bavik, C.; Greer, J.J. The activation of the retinoic acid response element is inhibited in an animal model of congenital diaphragmatic hernia. Biol. Neonate 2003, 83, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, N.; Montedonico, S.; Takayasu, H.; Paradisi, F.; Puri, P. Disturbance of retinol transportation causes nitrofen-induced hypoplastic lung. J. Pediatr. Surg. 2007, 42, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Montedonico, S.; Nakazawa, N.; Puri, P. Retinoic acid rescues lung hypoplasia in nitrofen-induced hypoplastic foetal rat lung explants. Pediatr. Surg. Int. 2006, 22, 2–8. [Google Scholar] [CrossRef]
- Thébaud, B.; Tibboel, D.; Rambaud, C.; Mercier, J.C.; Bourbon, J.R.; Dinh-Xuan, A.T.; Archer, S.L. Vitamin A decreases the incidence and severity of nitrofen-induced congenital diaphragmatic hernia in rats. Am. J. Physiol. 1999, 277, L423–L429. [Google Scholar] [CrossRef]
- Thébaud, B.; Barlier-Mur, A.M.; Chailley-Heu, B.; Henrion-Caude, A.; Tibboel, D.; Dinh-Xuan, A.T.; Bourbon, J.R. Restoring effects of vitamin A on surfactant synthesis in nitrofen-induced congenital diaphragmatic hernia in rats. Am. J. Respir. Crit. Care Med. 2001, 164, 1083–1089. [Google Scholar] [CrossRef]
- Montedonico, S.; Sugimoto, K.; Felle, P.; Bannigan, J.; Puri, P. Prenatal treatment with retinoic acid promotes pulmonary alveologenesis in the nitrofen model of congenital diaphragmatic hernia. J. Pediatr. Surg. 2008, 43, 500–507. [Google Scholar] [CrossRef]
- Sugimoto, K.; Takayasu, H.; Nakazawa, N.; Montedonico, S.; Puri, P. Prenatal treatment with retinoic acid accelerates type 1 alveolar cell proliferation of the hypoplastic lung in the nitrofen model of congenital diaphragmatic hernia. J. Pediatr. Surg. 2008, 43, 367–372. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Haussler, C.A.; Hsieh, J.C.; Thompson, P.D.; Selznick, S.H.; Dominguez, C.E.; Jurutka, P.W. The nuclear vitamin D receptor: Biological and molecular regulatory properties revealed. J. Bone Miner. Res. 1998, 13, 325–349. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Guillozo, H.; Garabédian, M.; Balsan, S. Lung as a possible additional target organ for vitamin D during fetal life in the rat. Biol. Neonate 1987, 52, 232–240. [Google Scholar] [CrossRef]
- Marin, L.; Dufour, M.E.; Tordet, C.; Nguyen, M. 1, 25(OH) 2D3 stimulates phospholipid biosynthesis and surfactant release in fetal rat lung explants. Biol. Neonate 1990, 57, 257–260. [Google Scholar] [CrossRef]
- Edelson, J.D.; Chan, S.; Jassal, D.; Post, M.; Tanswell, A.K. Vitamin D stimulates DNA synthesis in alveolar type-II cells. Biochim. Biophys. Acta 1994, 1221, 159–166. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Guillozo, H.; Marin, L.; Tordet, C.; Koite, S.; Garabedian, M. Evidence for a vitamin D paracrine system regulating maturation of developing rat lung epithelium. Am. J. Physiol. 1996, 271, L392–L399. [Google Scholar] [CrossRef]
- Nguyen, M.; Trubert, C.L.; Rizk-Rabin, M.; Rehan, V.K.; Besançon, F.; Cayre, Y.E.; Garabédian, M. 1, 25-Dihydroxyvitamin D3 and fetal lung maturation: Immunogold detection of VDR expression in pneumocytes type II cells and effect on fructose 1, 6 bisphosphatase. J. Steroid. Biochem. Mol. Biol. 2004, 89–90, 93–97. [Google Scholar] [CrossRef]
- Phokela, S.S.; Peleg, S.; Moya, F.R.; Alcorn, J.L. Regulation of human pulmonart surfactant protein gene expression by 1alpha, 25-dihydroxyvitamin D3. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L617–L626. [Google Scholar] [CrossRef]
- Foong, R.E.; Bosco, A.; Jones, A.C.; Gout, A.; Gorman, S.; Hart, P.H.; Zosky, G.R. The effects of in utero vitamin D deficiency on airway smooth muscle mass and lung function. Am. J. Respir. Cell Mol. Biol. 2015, 53, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Zosky, G.R.; Berry, L.J.; Elliot, J.G.; James, A.L.; Gorman, S.; Hart, P.H. Vitamin D deficiency causes deficits in lung function and alters lung structure. Am. J. Respir. Crit. Care Med. 2011, 183, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Lykkedegn, S.; Sorensen, G.L.; Beck-Nielsen, S.S.; Pilecki, B.; Duelund, L.; Marcussen, N.; Christesen, H.T. Vitamin D Depletion in Pregnancy Decreases Survival Time, Oxygen Saturation, Lung Weight and Body Weight in Preterm Rat Offspring. PLoS ONE 2016, 11, e0155203. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.L.; Hollis, B.W. The Implications of Vitamin D Status during Pregnancy on Mother and her Developing Child. Front. Endocrinol. 2018, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Saadoon, A.; Ambalavanan, N.; Zink, K.; Ashraf, A.P.; MacEwen, M.; Nicola, T.; Fanucchi, M.V.; Harris, W.T. Effect of Prenatal versus Postnatal Vitamin D Deficiency on Pulmonary Structure and Function in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 56, 383–392. [Google Scholar] [CrossRef]
- Taylor, S.K.; Sakurai, R.; Sakurai, T.; Rehan, V.K. Inhaled Vitamin D: A Novel Strategy to Enhance Neonatal Lung Maturation. Lung 2016, 194, 931–943. [Google Scholar] [CrossRef]
- Zosky, G.R.; Hart, P.H.; Whitehouse, A.J.; Kusel, M.M.; Ang, W.; Foong, R.E.; Chen, L.; Holt, P.G.; Sly, P.D.; Hall, G.L. Vitamin D deficiency at 16 to 20 weeks’ gestation is associated with impaired lung function and asthma at 6 years of age. Ann. Am. Thorac. Soc. 2014, 11, 571–577. [Google Scholar] [CrossRef]
- Ataseven, F.; Aygün, C.; Okuyucu, A.; Bedir, A.; Kücük, Y.; Kücüködük, S. Is vitamin d deficiency a risk factor for respiratory distress syndrome? Int. J. Vitam. Nutr. Res. 2013, 83, 232–237. [Google Scholar] [CrossRef]
- Backström, M.C.; Mäki, R.; Kuusela, A.L.; Sievänen, H.; Koivisto, A.M.; Ikonen, R.S.; Kouri, T.; Mäki, M. Randomised controlled trial of vitamin D supplementation on bone density and biochemical indices in preterm infants. Arch. Dis. Child. Fetal Neonatal Ed. 1999, 80, F161–F166. [Google Scholar] [CrossRef]
- Yurt, M.; Liu, J.; Sakurai, R.; Gong, M.; Husain, S.M.; Siddiqui, M.A.; Husain, M.; Villareal, P.; Akcay, F.; Torday, J.S.; et al. Vitamin D supplementation blocks pulmonary structural and functional changes in a rat model of perinatal vitamin D deficiency. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L859–L867. [Google Scholar] [CrossRef]
- Swanson, A.M.; David, A.L. Animal models of fetal growth restriction: Considerations for translational medicine. Placenta 2015, 36, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Rees, S.; Ng, J.; Dickson, K.; Nicholas, T.; Harding, R. Growth retardation and the development of the respiratory system in fetal sheep. Early Hum. Dev. 1991, 26, 13–27. [Google Scholar] [CrossRef]
- Cock, M.L.; Albuquerque, C.A.; Joyce, B.J.; Hooper, S.B.; Harding, R. Effects of intrauterine growth restriction on lung liquid dynamics and lung development in fetal sheep. Am. J. Obstet. Gynecol. 2001, 184, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Maritz, G.S.; Cock, M.L.; Louey, S.; Joyce, B.J.; Albuquerque, C.A.; Harding, R. Effects of fetal growth restriction on lung development before and after birth: A morphometric analysis. Pediatr. Pulmonol. 2001, 32, 201–210. [Google Scholar] [CrossRef]
- Rozance, P.J.; Seedorf, G.J.; Brown, A.; Roe, G.; O’Meara, M.C.; Gien, J.; Tang, J.R.; Abman, S.H. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L860–L871. [Google Scholar] [CrossRef]
- Maritz, G.S.; Cock, M.L.; Louey, S.; Suzuki, K.; Harding, R. Fetal growth restriction has long-term effects on postnatal lung structure in sheep. Pediatr. Res. 2004, 55, 287–295. [Google Scholar] [CrossRef]
- Cock, M.L.; Joyce, B.J.; Hooper, S.B.; Wallace, M.J.; Gagnon, R.; Brace, R.A.; Louey, S.; Harding, R. Pulmonary elastin synthesis and deposition in developing and mature sheep: Effects of intrauterine growth restriction. Exp. Lung Res. 2004, 30, 405–418. [Google Scholar] [CrossRef]
- Orgeig, S.; Crittenden, T.A.; Marchant, C.; McMillen, I.C.; Morrison, J.L. Intrauterine growth restriction delays surfactant protein maturation in the sheep fetus. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L575–L583. [Google Scholar] [CrossRef]
- Soo, J.Y.; Orgeig, S.; McGillick, E.V.; Zhang, S.; McMillen, I.C.; Morrison, J.L. Normalisation of surfactant protein –A and –B expression in the lungs of low birth weight lambs by 21 days old. PLoS ONE 2017, 12, e0181185. [Google Scholar] [CrossRef]
- Deng, F.T.; Ouyang, W.X.; Ge, L.F.; Zhang, L.; Chai, X.Q. Expression of lung surfactant proteins SP-B and SP-C and their modulating factors in fetal lung of FGT rats. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 122–128. [Google Scholar] [CrossRef]
- Gortner, L.; Hilhendorff, A.; Bähner, T.; Ensen, M.; Reiss, I.; Rudloff, S. Hypoxia-induced intrauterine growth retardation: Effects on pulmonary development and surfactant protein transcription. Biol. Neonate 2005, 88, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Wang, L.F.; Su, B. Effects of maternal undernutrition during late gestation on the lung surfactant system and morphometry in rats. Pediatr. Res. 2004, 56, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Karadag, A.; Sakurai, R.; Wang, Y.; Guo, P.; Desai, M.; Ross, M.G.; Torday, J.S.; Rehan, V.K. Effect of maternal food restriction on fetal rat lung lipid differentiation program. Pediatr. Pulmonol. 2009, 44, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Joss-Moore, L.A.; Wang, Y.; Yu, X.; Campbell, M.S.; Callaway, C.W.; McKnight, R.A.; Wint, A.; Dahl, M.J.; Dull, R.O.; Albertine, K.H.; et al. IUGR decreases elastin mRNA expression in the developing rat lung and alters elastin content and lung compliance in the mature rat lung. Physiol. Genomics 2011, 43, 499–505. [Google Scholar] [CrossRef]
- Snell, L.H.; Haughey, B.P.; Buck, G.; Marecki, M.A. Metabolic crisis: Hyperemesis gravidarum. J. Perinat. Neonatal Nurs. 1998, 12, 26–37. [Google Scholar] [CrossRef]
- Wendt, A.; Gibbs, C.M.; Peters, S.; Hogue, C.J. Impact of increasing inter-pregnancy interval on maternal and infant health. Paediatr. Perinat. Epidemiol. 2012, 26, 239–258. [Google Scholar] [CrossRef]
- Scholl, T.O.; Hediger, M.L. A review of the epidemiology of nutrition and adolescent pregnancy: Maternal growth during pregnancy and its effect on the fetus. J. Am. Coll. Nutr. 1993, 12, 101–107. [Google Scholar] [CrossRef]
- McEwoy, C.T.; Spindel, E.R. Pulmonary Effects of Maternal Smoking on the Fetus and Child: Effects on Lund Development, Respiratory Morbidities, and Life Long Lung Health. Paediatr. Respir. Rev. 2017, 21, 27–33. [Google Scholar] [CrossRef]
- Sebastiani, G.; Borrás-Novell, C.; Casanova, M.A.; Pascual Tutusaus, M.; Ferrero Martínez, S.; Gómez Roig, M.D.; García-Algar, O. The Effects of Alcohol and Drugs of Abuse on Maternal Nutritional Profile during Pregnancy. Nutrients 2018, 10, 1008. [Google Scholar] [CrossRef]
- Sharma, P.; McKay, K.; Rosenkrantz, T.S.; Hussain, N. Comparisons of mortality and pre-discharge respiratory outcomes in small-for-gestational-age and appropriate-for-gestational-age premature infants. BMC Pediatr. 2004, 8, 4–9. [Google Scholar] [CrossRef]
- Torrance, H.L.; Voorbij, H.A.; Wijnberger, L.D.; van Bel, F.; Visser, G.H. Lung maturation in small for gestational age foetuses from pregnancies complicated by placental insufficiency or maternal hypertension. Early Hum. Dev. 2008, 84, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.E.; Kennedy, K.; Broyles, S.; Rosenfeld, C.R. The small for gestational age infant: Accelerated or delayed pulmonary maturation? Increased or decreased survival? Pediatrics 1995, 95, 534–538. [Google Scholar] [PubMed]
- Giapros, V.; Drougia, A.; Krallis, V.; Theocharis, P.; Andronikou, S. Morbidity and mortality patterns in small-for-gestational age infants born preterm. J. Maternal. Fetal Neonatal Med. 2012, 25, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Haglund, B.; Odlind, V.; Altman, M.; Ewald, U.; Kieler, H. Perinatal conditions related to growth restriction and inflammation are associated with an increased risk of bronchopulmonary dysplasia. Acta Paediatr. 2015, 104, 259–263. [Google Scholar] [CrossRef]
- Wjst, M.; Popescu, M.; Trepka, M.J.; Heinrich, J.; Wichmann, H.E. Pulmonary function in children with initial low birth weight. Pediatr. Allergy Immunol. 1998, 9, 80–90. [Google Scholar] [CrossRef]
- Matharu, K.; Ozanne, S.E. The Fetal Origins of Disease and Associations with Low Birthweight. NeoReviews 2004, 5, e522–e526. [Google Scholar] [CrossRef]
- Briana, D.D.; Malamitsi-Puchner, A. Small for gestational age birth weight: Impact on lung structure and function. Paediatr. Respir. Rev. 2013, 14, 256–262. [Google Scholar] [CrossRef]
- Saad, N.J.; Patel, J.; Burney, P.; Minelli, C. Birth Weight and Lung Function in Adulthood: A Systematic Review and Meta-analysis. Ann. Am. Thorac. Soc. 2017, 14, 994–1004. [Google Scholar] [CrossRef]
- Lelijveld, N.; Kerac, M.; Seal, A.; Chimwezi, E.; Wells, J.C.; Heyderman, R.S.; Nyirenda, M.J.; Stocks, J.; Kirkby, J. Long-term effect of severe acute malnutrition on lung function in Malawian children: A cohort study. Eur. Respir. J. 2017, 49, 1601301. [Google Scholar] [CrossRef]
- Lopuhaä, C.E.; Roseboom, T.J.; Osmond, C.; Barker, D.J.; Ravelli, A.C.; Bleker, O.P.; van der Zee, J.S.; van der Meulen, J.H. Atopy, lung function, and obstructive airways disease after prenatal exposure to famine. Thorax 2000, 55, 555–561. [Google Scholar] [CrossRef]
- Wang, Z.; Zou, Z.; Yang, Z.; Dong, Y.; Ma, J. Association between exposure to the Chinese famine during infancy and the risk of self-reported chronic lung diseases in adulthood: A cross-sectional study. BMJ Open 2017, 7, e015476. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.O.; Sterne, J.A.; Montgomery, S.M.; Azima, H. Birth weight, body mass index and asthma in young adults. Thorax 1999, 54, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Hagström, B.; Nyberg, P.; Nilsson, P.M. Asthma in adult life—is there an association with birth weight? Scand. J. Prim. Health Care 1998, 16, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Black, R.E.; Allen, L.H.; Bhutta, Z.A.; Caulfield, L.E.; de Onis, M.; Ezzati, M.; Mathers, C.; Rivera, J. Maternal and child undernutrition: Global and regional exposures and health consequences. Lancet 2008, 371, 243–260. [Google Scholar] [CrossRef]
- Bastos Maia, S.; Rolland Souza, A.S.; Costa Caminha, M.F.; Lins da Silva, S.; Callou Cruz, R.S.B.L.; Carvalho Dos Santos, C.; Batista Filho, M. Vitamin A and Pregnancy: A Narrative Review. Nutrients 2019, 11, 681. [Google Scholar] [CrossRef]
- Ahima, R.S.; Prabakaran, D.; Mantzoros, C.; Qu, D.; Lowell, B.; Maratos-Flier, E.; Flier, J.S. Role of leptin in the neuroendocrine response to fasting. Nature 1996, 382, 250–252. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Toro, A.; Vilariño-García, T.; Maymó, J.; Guadix, P.; Dueñas, J.L.; Fernández-Sánchez, M.; Varone, C.; Sánchez-Margalet, V. Leptin action in normal and pathological pregnancies. J. Cell. Mol. Med. 2018, 22, 716–727. [Google Scholar] [CrossRef]
- Catov, J.M.; Patrick, T.E.; Powers, R.W.; Ness, R.B.; Harger, G.; Roberts, J.M. Maternal leptin across pregnancy in women with small-for-gestational-age-infants. Am. J. Obstet. Gynecol. 2007, 196, 558-e1. [Google Scholar] [CrossRef]
- Laivuori, H.; Gallaher, M.J.; Collura, L.; Crombleholme, W.R.; Markovic, N.; Rajakumar, A.; Hubel, C.A.; Roberts, J.M.; Powers, R.W. Relationships between maternal plasma leptin, placental leptin mRNA and protein in normal pregnancy, pre-eclampsia and intrauterine growth restriction without pre-eclampsia. Mol. Hum. Reprod. 2006, 12, 551–556. [Google Scholar] [CrossRef]
- Jaquet, D.; Leger, J.; Tabone, M.D.; Czernichow, P.; Levy-Marchal, C. High serum leptin concentrations during catch-up growth of children born with intrauterine growth retardation. J. Clin. Endocrinol. Metab. 1999, 84, 1949–1953. [Google Scholar] [CrossRef]
- Guler, N.; Kirerleri, E.; Ones, U.; Tamay, Z.; Salmayenli, N.; Darendeliler, F. Leptin: Does it have any role in childhood asthma? J. Allergy Clin. Immunol. 2004, 114, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, G.; Tschöp, M.; Papotti, M.; Deghenghi, R.; Heiman, M.; Ghigo, E. Neuroendocrine and peripheral activities of ghrelin: Implications in metabolism and obesity. Eur. J. Pharmacol. 2002, 440, 235–254. [Google Scholar] [CrossRef]
- Fuglsang, J. Ghrelin in pregnancy and lactation. Vitam. Horm. 2008, 77, 259–284. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.J.; Luque, E.M.; Ponzio, M.F.; Cantarelli, V.; Diez, M.; Figueroa, S.; Vicenti, L.M.; Carlini, V.P.; Martini, A.C. The role of intragestational ghrelin on postnatal development and reproductive programming in mice. Reproduction 2018, 156, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Onal, E.E.; Cinaz, P.; Atalay, Y.; Türkyilmaz, C.; Bideci, A.; Aktürk, A.; Okumus, N.; Unal, S.; Koç, E.; Ergenekon, E. Umbilical cord ghrelin concentrations in small- and appropriate-for-gestational age newborns infants: Relationship to anthropometric markers. J. Endocrinol. 2004, 180, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Yalinbas, E.E.; Binay, C.; Simsek, E.; Aksit, M.A. The Role of Umbilical Cord Blood Concentration of IGF-I, IGF-II, Leptin, Adiponectin, Ghrelin, Resistin, and Visfatin in Fetal Growth. Am. J. Perinatol. 2019, 36, 600–608. [Google Scholar] [CrossRef]
- Otto, B.; Tschöp, M.; Heldwein, W.; Pfeiffer, A.F.; Diederich, S. Endogenous and exogenous glucocorticoids decrease plasma ghrelin in humans. Eur. J. Endocrinol. 2004, 151, 113–117. [Google Scholar] [CrossRef]
- Valsamakis, G.; Margeli, A.; Vitoratos, N.; Boutsiadis, A.; Sakkas, E.G.; Papadimitriou, G.; Al-Daghri, N.M.; Botsis, D.; Kumar, S.; Papassotiriou, I.; et al. The role of maternal gut hormones in normal pregnancy: Fasting plasma active glucagon-like peptide 1 level is a negative predictor of fetal abdomen circumference and maternal weight change. Eur. J. Endocrinol. 2010, 162, 897–903. [Google Scholar] [CrossRef][Green Version]
- Johnson, M.L.; Saffrey, M.J.; Taylor, V.J. Gastrointestinal capacity, gut hormones and appetite change during rat pregnancy and lactation. Reproduction 2019, 157, 431–443. [Google Scholar] [CrossRef]
- Bonde, L.; Vilsboll, T.; Nielsen, T.; Bagger, J.I.; Svare, J.A.; Holst, J.J.; Larsen, S.; Knop, F.K. Reduced postprandial GLP-1 responses in women with gestational diabetes mellitus. Diabetes Obes. Metab. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Sukumar, N.; Bagias, C.; Goljan, I.; Weldeselassie, Y.; Gharanei, S.; Tan, B.K.; Holst, J.J.; Saravanan, P. Reduced GLP-1 Secretion at 30 Minutes After a 75-g Oral Glucose Load Is Observed in Gestational Diabetes Mellitus: A Prospective Cohort Study. Diabetes 2018, 67, 2650–2656. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Hormone | Action in Lung Development | References |
|---|---|---|
| Ghrelin | Fetal lung branching | [15,16] |
| Upregulating RA receptors/ sensitizing RA action | [17] | |
| Leptin | Enhance lung maturity | [28,31,32,33,34] |
| In vitro phosphatidylcholine secretion | [28] | |
| In vitro SFTPs expression | [28,32,33,34] | |
| GLP-1 | In vitro phosphatidylcholine secretion | [43,44] |
| In vivo SFTPs expression | [42,46,47] | |
| Increase ACE2/Ang (1-7)/MasR branch of the renin-angiotensin system | [46,47] | |
| Retinoic acid | Formation of bronchial tubules during pseudoglandular phase | [57] |
| Lung maturation | [62,69,70,72,77,78,79] | |
| In vitro Proliferation of ATII cells and differentiation to ATI cells | [62,81] | |
| In vitro and in vivo SFTPs expression | [62,63] | |
| Cholecalciferol | Branching morphogenesis | [91] |
| In vitro proliferation of ATII cells | [96] | |
| In vitro surfactant phospholipids secretion | [96] | |
| In vitro SFTPs expression | [96] | |
| Lung maturation | [95,96] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fandiño, J.; Toba, L.; González-Matías, L.C.; Diz-Chaves, Y.; Mallo, F. Perinatal Undernutrition, Metabolic Hormones, and Lung Development. Nutrients 2019, 11, 2870. https://doi.org/10.3390/nu11122870
Fandiño J, Toba L, González-Matías LC, Diz-Chaves Y, Mallo F. Perinatal Undernutrition, Metabolic Hormones, and Lung Development. Nutrients. 2019; 11(12):2870. https://doi.org/10.3390/nu11122870
Chicago/Turabian StyleFandiño, Juan, Laura Toba, Lucas C. González-Matías, Yolanda Diz-Chaves, and Federico Mallo. 2019. "Perinatal Undernutrition, Metabolic Hormones, and Lung Development" Nutrients 11, no. 12: 2870. https://doi.org/10.3390/nu11122870
APA StyleFandiño, J., Toba, L., González-Matías, L. C., Diz-Chaves, Y., & Mallo, F. (2019). Perinatal Undernutrition, Metabolic Hormones, and Lung Development. Nutrients, 11(12), 2870. https://doi.org/10.3390/nu11122870