The Extra-Virgin Olive Oil Polyphenols Oleocanthal and Oleacein Counteract Inflammation-Related Gene and miRNA Expression in Adipocytes by Attenuating NF-κB Activation

 ,
,  ,
,  ,
,  ,
, 
 ,
,  , ,
, , 
 and
and 
Abstract
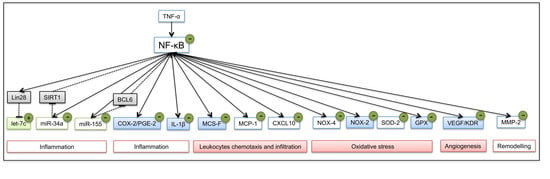
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
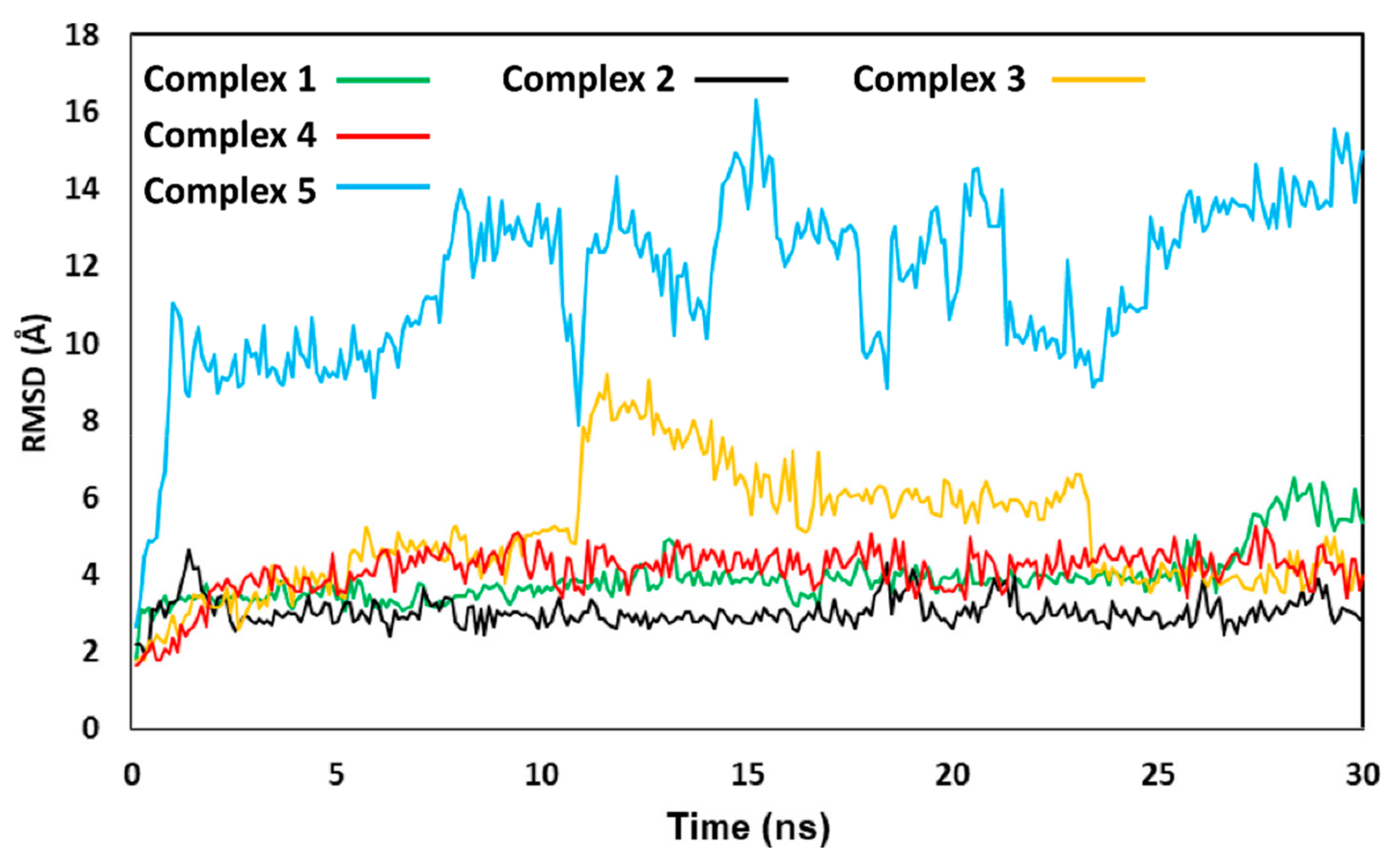{kind=link}
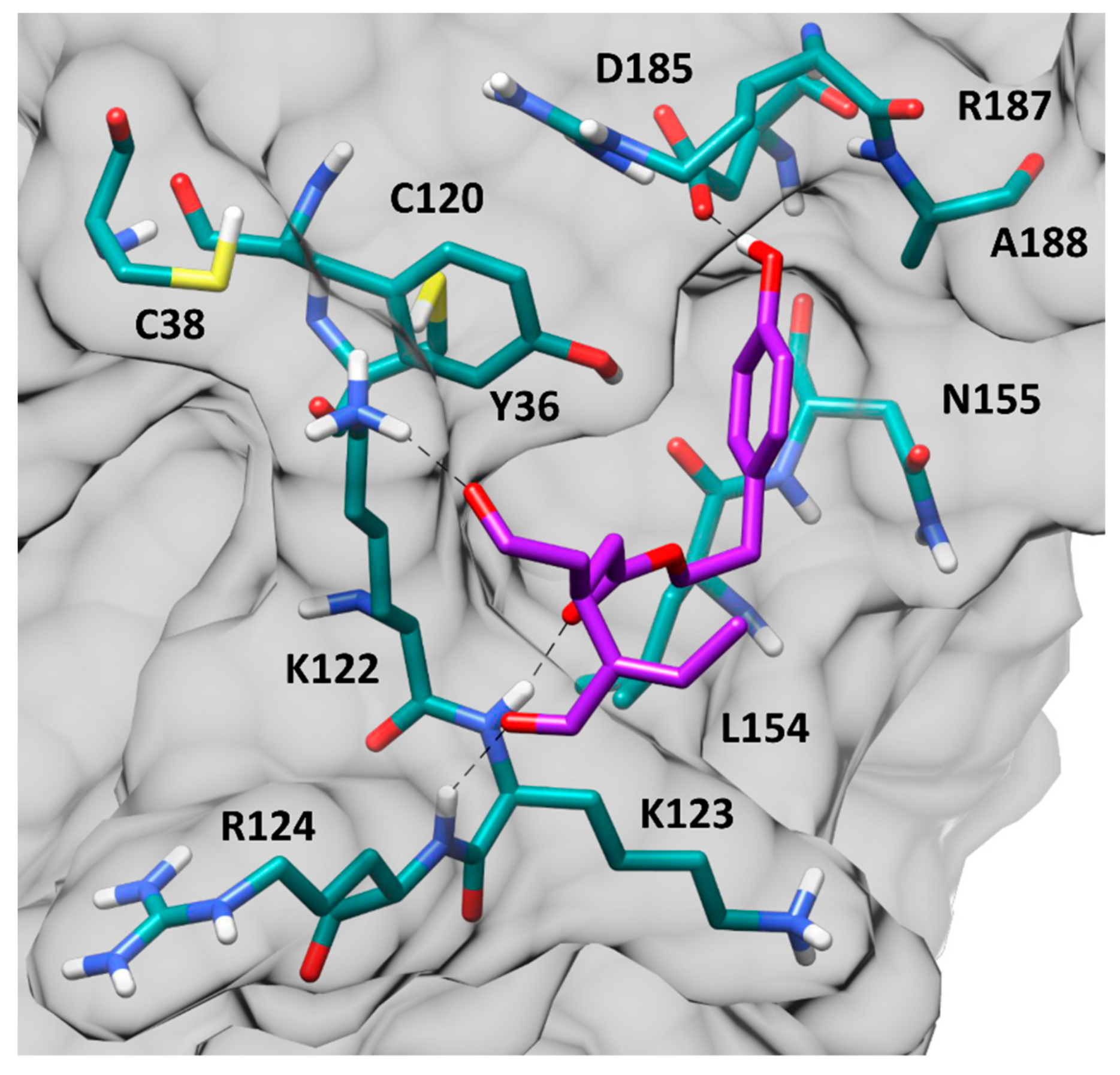{kind=link}
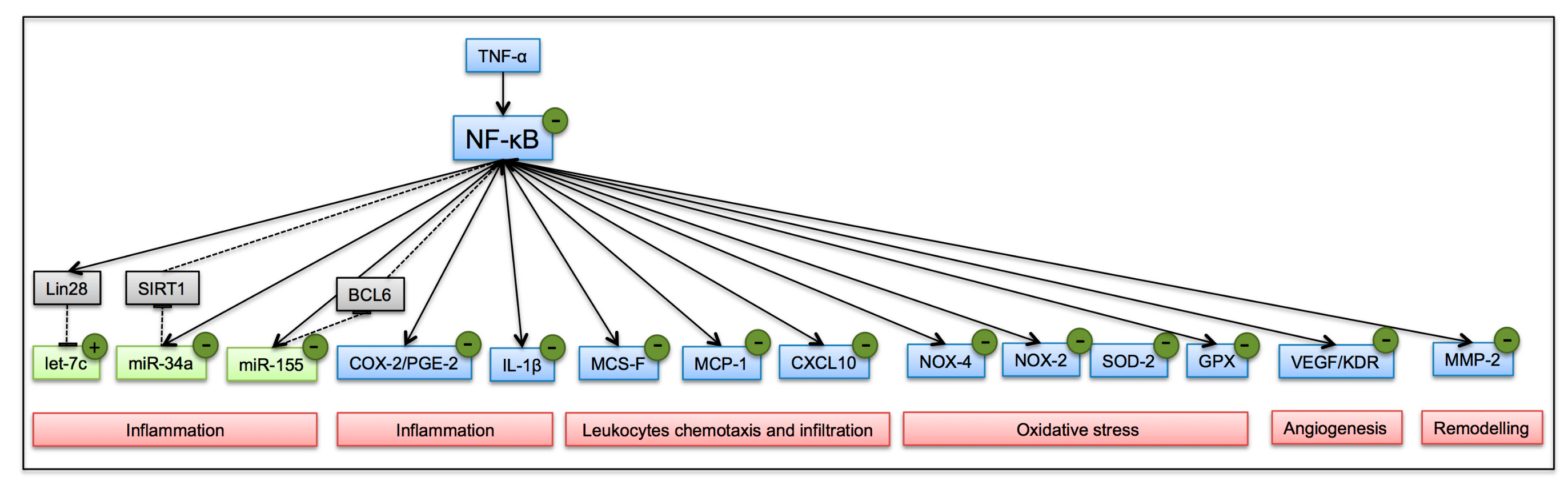{kind=link}
1. Introduction
2. Materials and Methods
2.1. Extraction, Purification, and Characterization of OC and OA
2.2. Cell Culture and Treatments
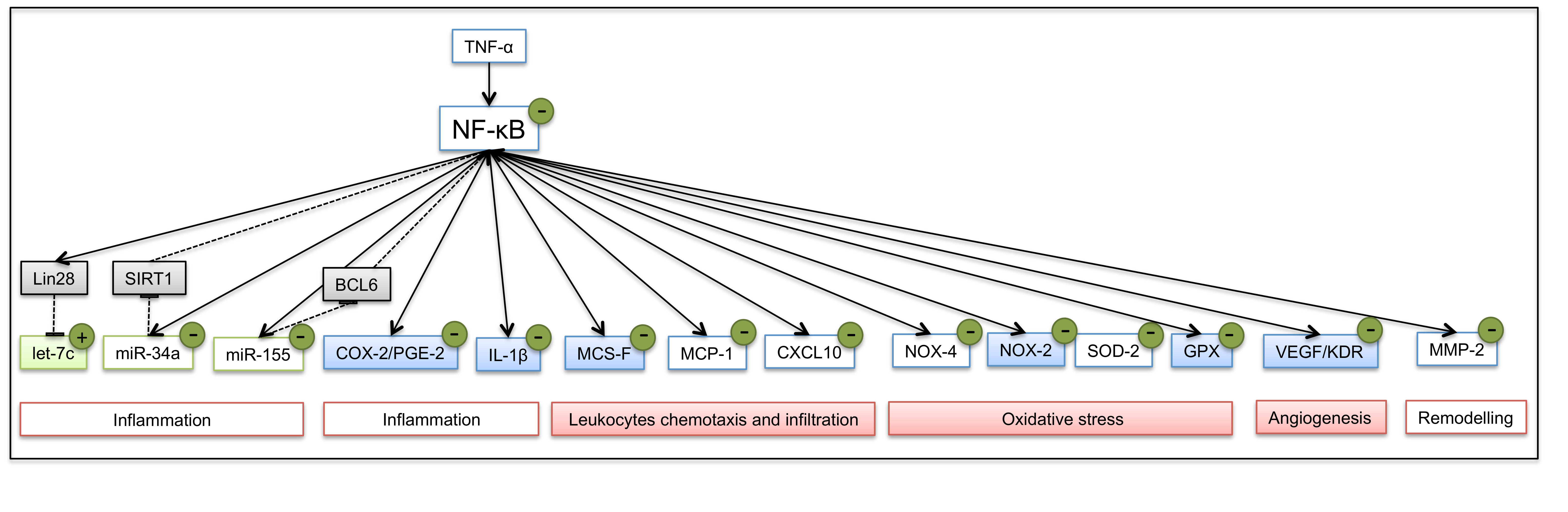
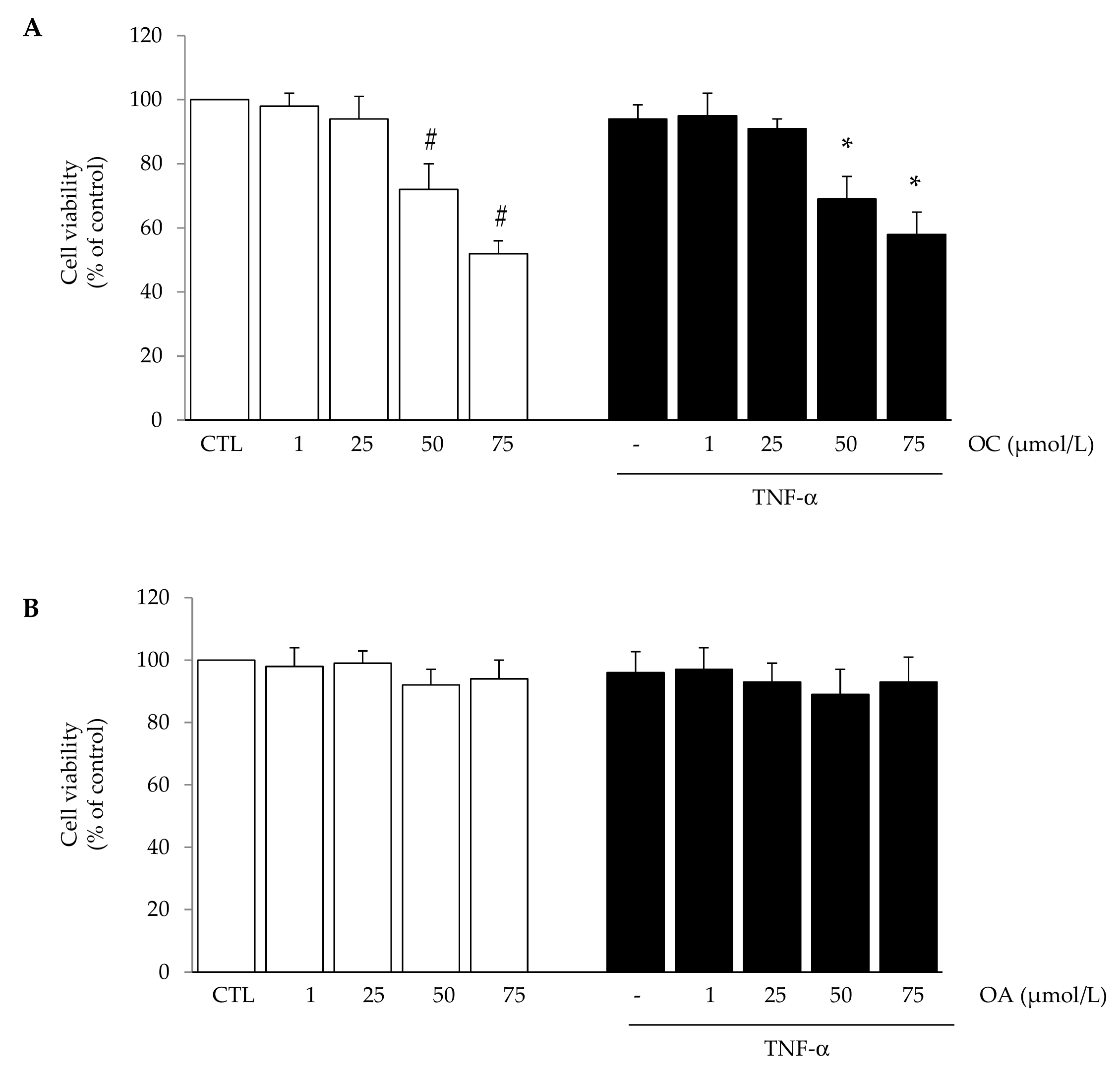
2.3. Cell Viability
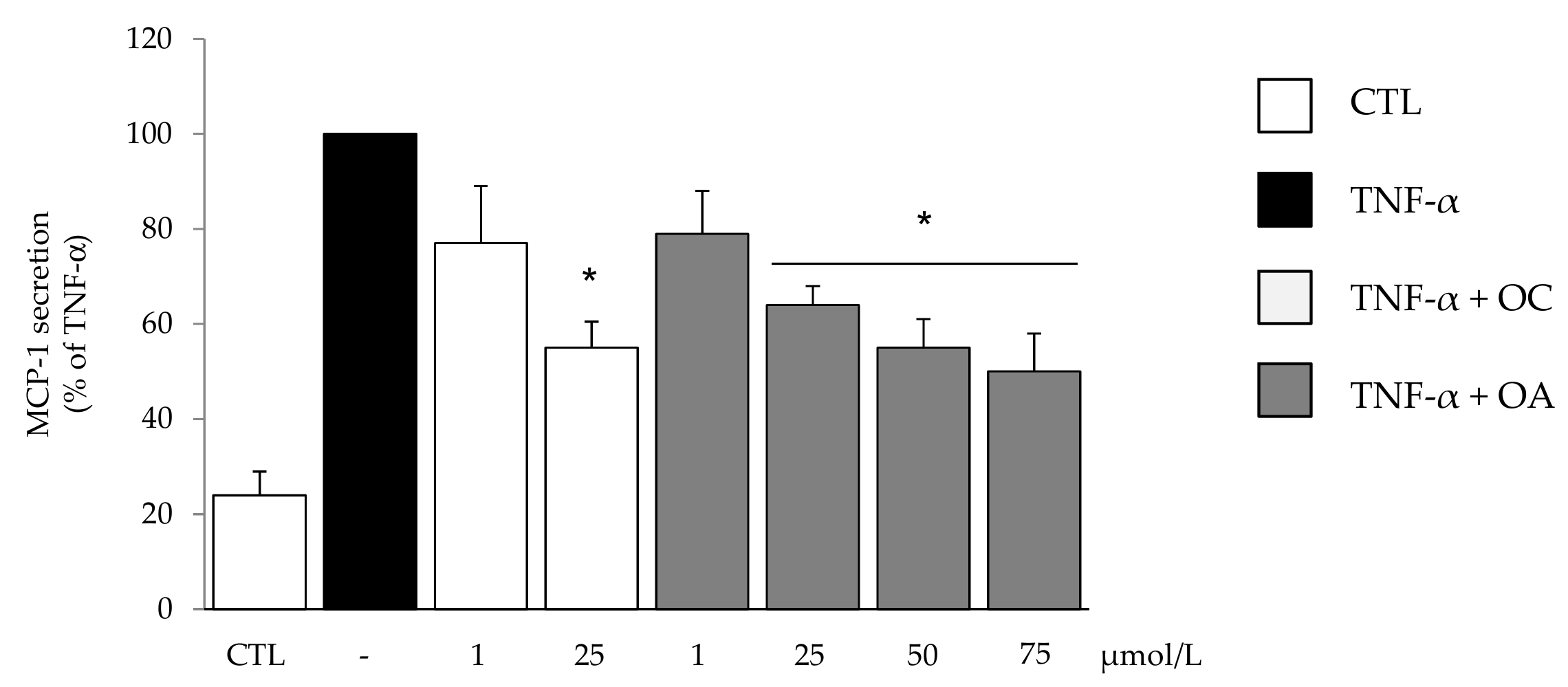
2.4. Measurement of MCP-1 in Culture Media
2.5. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction
2.6. Preparation of Nuclear Extracts and Measurement of NF-κB p65 DNA Binding Activity
2.7. Exosome Isolation from Cell Culture Supernatants
2.8. Evaluation of miRNA Expression
2.9. miRNA Target Prediction and Pathway Analysis
2.10. Molecular Docking
2.11. Molecular Dynamic Simulations
2.12. Binding Energy Evaluations
2.13. Statistical Analysis
3. Results
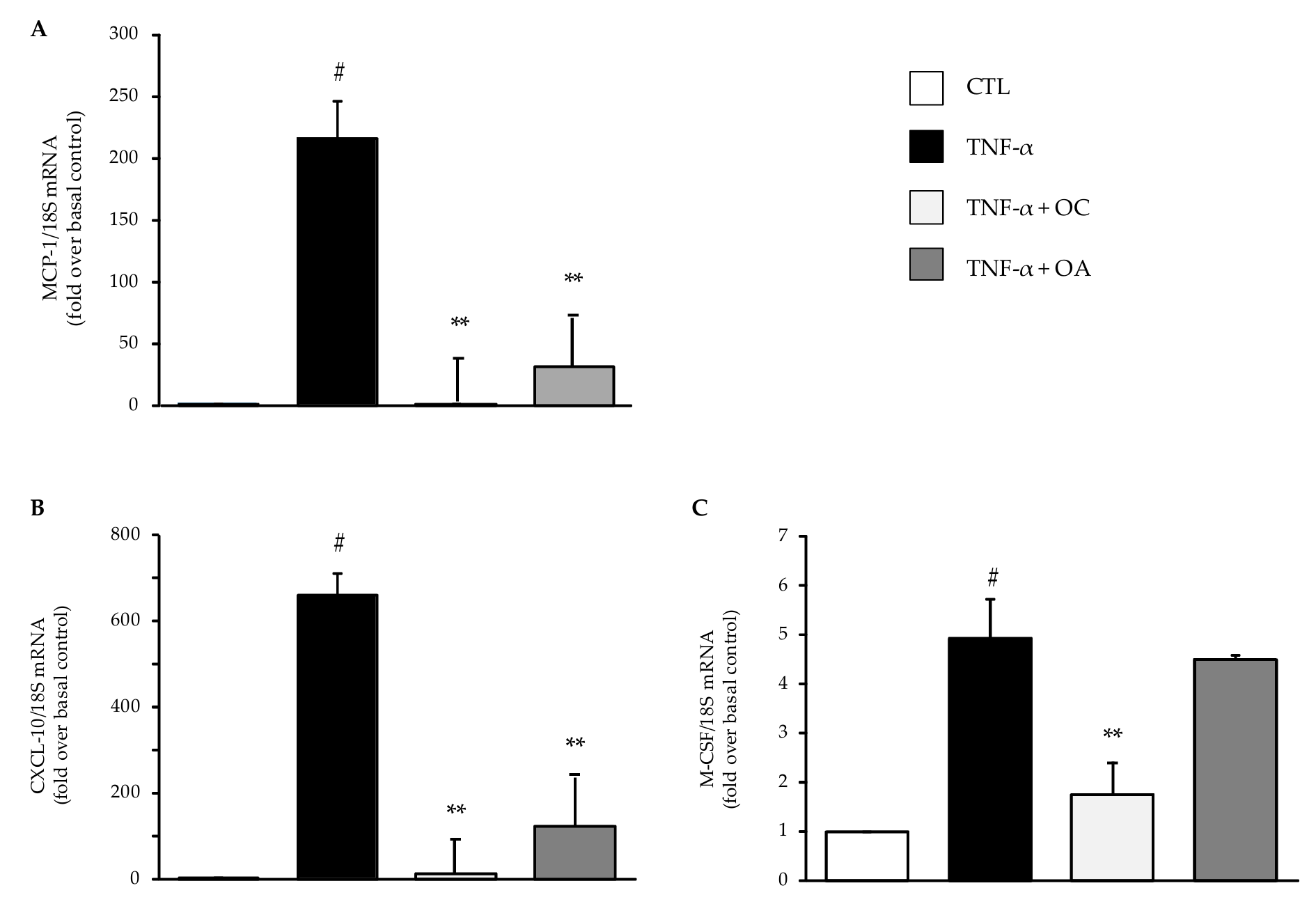
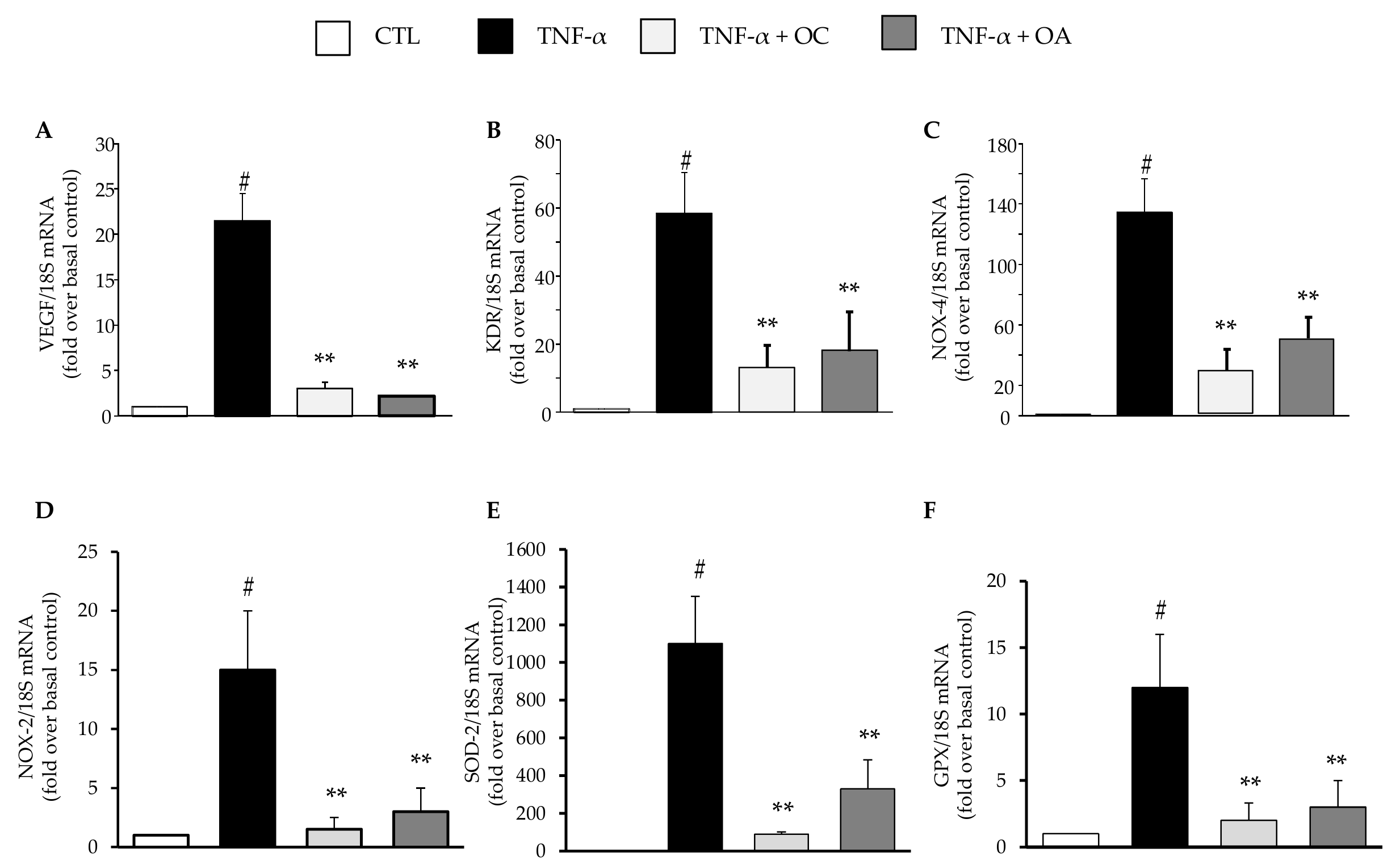
3.1. OC and OA Attenuate TNF-α–Mediated Inflammatory Gene Expression in Human Adipocytes
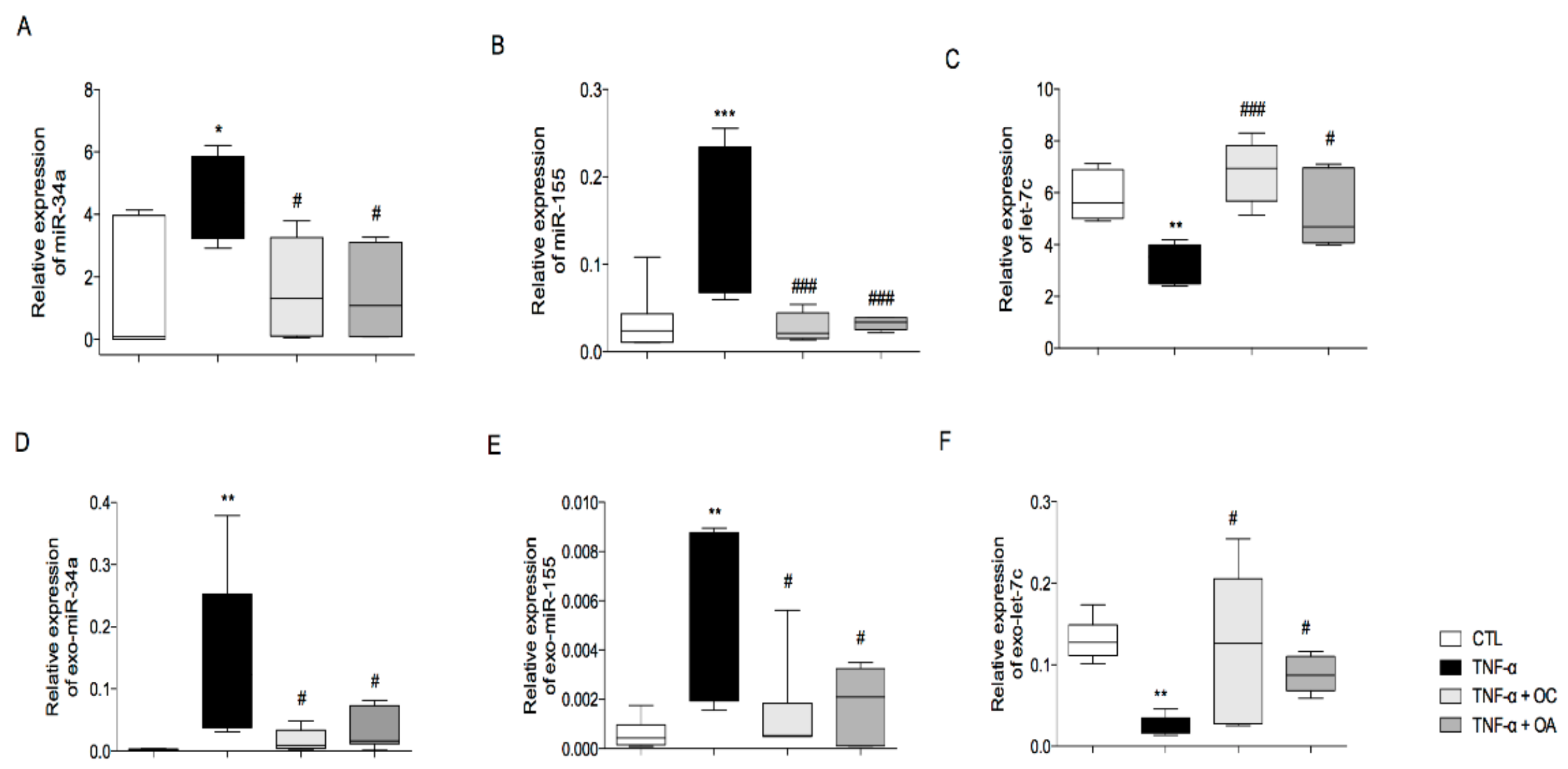
3.2. OC and OA Modulate TNF-α-Induced Inflammation-Linked miRNAs Expression in Adipocytes and Adipocyte-Derived Exosomes
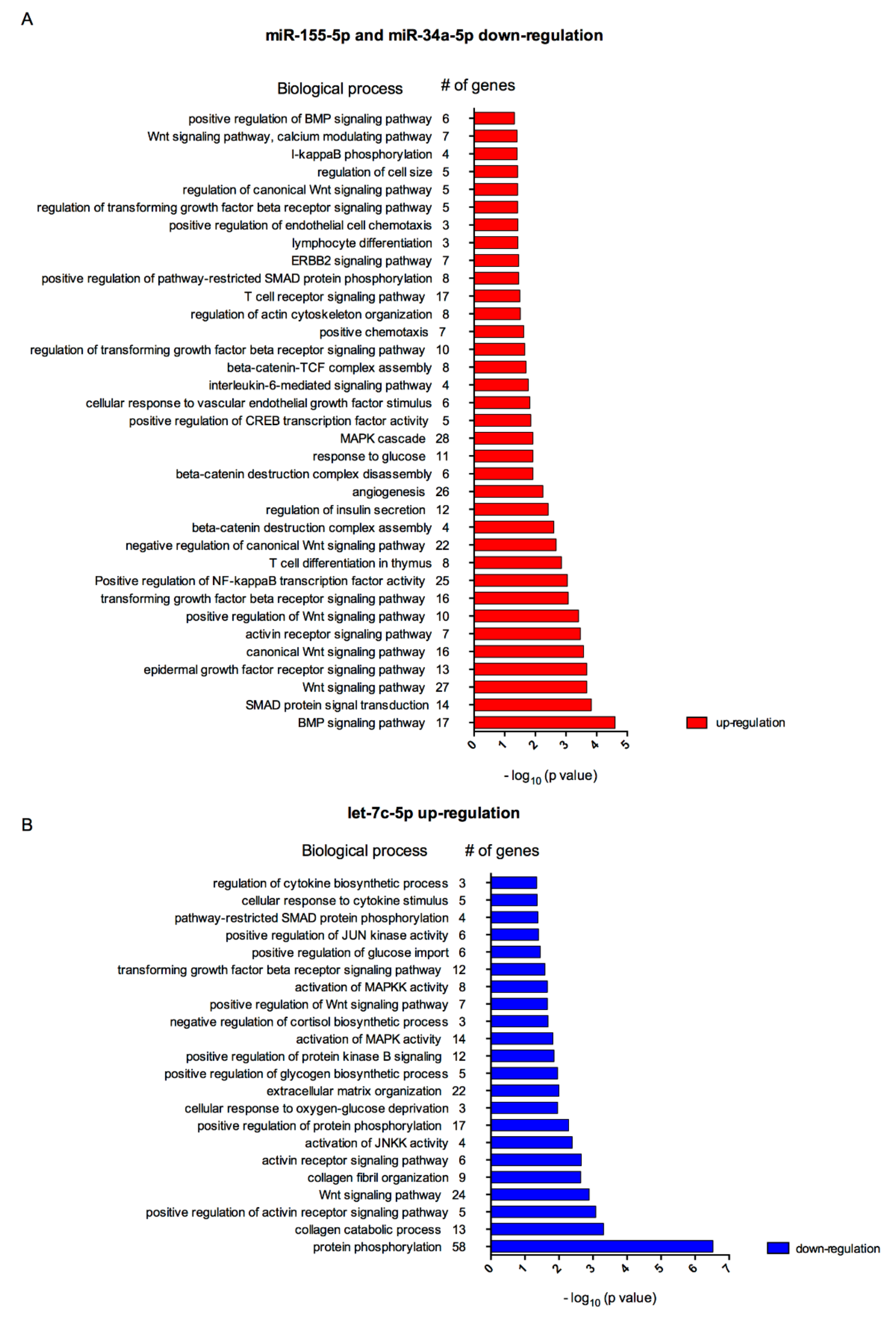
3.3. Biological Processes Associated to miRNAs Modulated by Polyphenols in Inflamed Adipocytes
3.4. NF-κB Inhibition by OC and OA in Human Adipocytes
3.4.1. OC and OC Attenuate TNF-α-Induced Activation of NF-κB
3.4.2. Molecular Modeling Studies of OC Interaction with NF-κB
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bluher, M. Adipose tissue inflammation: A cause or consequence of obesity-related insulin resistance? Clin. Sci. 2016, 130, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.B.; Lavie, C.J.; Blair, S.N. Obesity and cardiovascular disease. Circ. Res. 2016, 118, 1752–1770. [Google Scholar] [CrossRef] [PubMed]
- Scheja, L.; Heeren, J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat. Rev. Endocrinol. 2019, 15, 507–524. [Google Scholar] [CrossRef] [PubMed]
- Wensveen, F.M.; Valentić, S.; Šestan, M.; Turk Wensveen, T.; Polić, B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation: Highlights. Eur. J. Immunol. 2015, 45, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, X.; Ma, J.; Hassan, W.; Wu, H.; Ling, J.; Shang, J. β-Elemene attenuates atherosclerosis in apolipoprotein E-deficient mice via restoring NO levels and alleviating oxidative stress. Biomed. Pharmacother. 2017, 95, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, A.B. The effects of salsalate on glycemic control in patients with type 2 diabetes: A randomized trial. Ann. Intern. Med. 2010, 152, 346. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Ma, J.; Feng, B.; Zhang, H.; Alan Diehl, J.; Eugene Chin, Y.; Yan, W.; Xu, H. FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKβ pathways. Obesity 2011, 19, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.-W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Kulyté, A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat. Rev. Endocrinol. 2015, 11, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Moreno, M.; Mercader, J.M.; Moreno-Navarrete, J.M.; Fuentes-Batllevell, N.; Sabater, M.; Ricart, W.; Fernández-Real, J.M. Inflammation triggers specific microRNA profiles in human adipocytes and macrophages and in their supernatants. Clin. Epigenetics 2015, 7, 49. [Google Scholar] [CrossRef]
- Jayaseelan, V.P. Emerging role of exosomes as promising diagnostic tool for cancer. Cancer Gene Ther. 2019. [Online ahead of print]. [Google Scholar] [CrossRef] [PubMed]
- Thomou, T.; Mori, M.A.; Dreyfuss, J.M.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 2017, 542, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Pardo, G.; Sabater, M.; Hummel, M.; Ferrer, A.; Rodriguez-Hermosa, J.I.; Ruiz, B.; Ricart, W.; Peral, B.; et al. MiRNA Expression profile of human subcutaneous adipose and during adipocyte differentiation. PLoS ONE 2010, 5, e9022. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Mercader, J.M.; Catalan, V.; Moreno-Navarrete, J.M.; Pueyo, N.; Sabater, M.; Gomez-Ambrosi, J.; Anglada, R.; Fernandez-Formoso, J.A.; Ricart, W.; et al. Targeting the circulating microrna signature of obesity. Clin. Chem. 2013, 59, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Lorente-Cebrián, S.; Mejhert, N.; Kulyté, A.; Laurencikiene, J.; Åström, G.; Hedén, P.; Rydén, M.; Arner, P. MicroRNAs regulate human adipocyte lipolysis: Effects of miR-145 are linked to TNF-α. PLoS ONE 2014, 9, e86800. [Google Scholar] [CrossRef]
- Karkeni, E.; Astier, J.; Tourniaire, F.; El Abed, M.; Romier, B.; Gouranton, E.; Wan, L.; Borel, P.; Salles, J.; Walrand, S.; et al. Obesity-associated inflammation induces microRNA-155 expression in adipocytes and adipose tissue: Outcome on adipocyte function. J. Clin. Endocrinol. Metab. 2016, 101, 1615–1626. [Google Scholar] [CrossRef]
- Karkeni, E.; Bonnet, L.; Marcotorchino, J.; Tourniaire, F.; Astier, J.; Ye, J.; Landrier, J.-F. Vitamin D limits inflammation-linked microRNA expression in adipocytes In Vitro and In Vivo: A new mechanism for the regulation of inflammation by vitamin D. Epigenetics 2018, 13, 156–162. [Google Scholar] [CrossRef]
- Lavery, C.A.; Kurowska-Stolarska, M.; Holmes, W.M.; Donnelly, I.; Caslake, M.; Collier, A.; Baker, A.H.; Miller, A.M. miR-34a −/− mice are susceptible to diet-induced obesity: miR-34a −/− mice are susceptible to obesity. Obesity 2016, 24, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- McGregor, R.A.; Choi, M.S. microRNAs in the regulation of adipogenesis and obesity. Curr. Mol. Med. 2011, 11, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Fu, M.; Bookout, A.L.; Kliewer, S.A.; Mangelsdorf, D.J. MicroRNA let-7 regulates 3T3-L1 adipogenesis. Mol. Endocrinol. 2009, 23, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Croce, C.M.; Michaille, J.-J. miR-155: On the crosstalk between inflammation and cancer. Int. Rev. Immunol. 2009, 28, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, K.; Chen, X.; Meng, H.; Song, M.; Wang, Y.; Xu, X.; Bai, Y. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol. Biol. 2012, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-kappaB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef]
- Subedi, A.; Park, P.-H. Autocrine and paracrine modulation of microRNA-155 expression by globular adiponectin in RAW 264.7 macrophages: Involvement of MAPK/NF-κB pathway. Cytokine 2013, 64, 638–641. [Google Scholar] [CrossRef]
- Zhang, Y.; Tao, X.; Yin, L.; Xu, L.; Xu, Y.; Qi, Y.; Han, X.; Song, S.; Zhao, Y.; Lin, Y.; et al. Protective effects of dioscin against cisplatin-induced nephrotoxicity via the microRNA-34a/sirtuin 1 signalling pathway. Br. J. Pharmacol. 2017, 174, 2512–2527. [Google Scholar] [CrossRef]
- Martínez-González, M.A.; Salas-Salvadó, J.; Estruch, R.; Corella, D.; Fitó, M.; Ros, E. Benefits of the Mediterranean diet: Insights from the PREDIMED study. Prog. Cardiovasc. Dis. 2015, 58, 50–60. [Google Scholar] [CrossRef]
- Scoditti, E.; Nestola, A.; Massaro, M.; Calabriso, N.; Storelli, C.; De Caterina, R.; Carluccio, M.A. Hydroxytyrosol suppresses MMP-9 and COX-2 activity and expression in activated human monocytes via PKCα and PKCβ1 inhibition. Atherosclerosis 2014, 232, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, L.; Cicerale, S. The health benefiting mechanisms of virgin olive oil phenolic compounds. Molecules 2016, 21, 1734. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, G.K.; Keast, R.S.J.; Morel, D.; Lin, J.; Pika, J.; Han, Q.; Lee, C.-H.; Smith, A.B.; Breslin, P.A.S. Ibuprofen-like activity in extra-virgin olive oil. Nature 2005, 437, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, L.; Keast, R. Oleocanthal, a phenolic derived from virgin olive oil: A review of the beneficial effects on inflammatory disease. Int. J. Mol. Sci. 2014, 15, 12323–12334. [Google Scholar] [CrossRef] [PubMed]
- Fogli, S.; Arena, C.; Carpi, S.; Polini, B.; Bertini, S.; Digiacomo, M.; Gado, F.; Saba, A.; Saccomanni, G.; Breschi, M.C.; et al. Cytotoxic activity of oleocanthal isolated from virgin olive oil on human melanoma cells. Nutr. Cancer 2016, 68, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Sindona, G.; Caruso, A.; Cozza, A.; Fiorentini, S.; Lorusso, B.; Marini, E.; Nardi, M.; Procopio, A.; Zicari, S. Anti-inflammatory effect of 3,4-DHPEA-EDA [2-(3,4 -hydroxyphenyl) ethyl (3S, 4E)-4-formyl-3-(2-oxoethyl) hex-4-enoate] on primary human vascular endothelial cells. Curr. Med. Chem. 2012, 19, 4006–4013. [Google Scholar] [CrossRef]
- Lozano-Castellón, J.; López-Yerena, A.; de Alvarenga, J.F.R.; del Castillo-Alba, J.R.; Vallverdú-Queralt, A.; Escribano-Ferrer, E.; Lamuela-Raventós, R.M. Health-promoting properties of oleocanthal and oleacein: Two secoiridoids from extra-virgin olive oil. Crit. Rev. Food Sci. Nutr. 2019, 1–17. [Google Scholar] [CrossRef]
- Siriwardhana, N.; Kalupahana, N.S.; Cekanova, M.; LeMieux, M.; Greer, B.; Moustaid-Moussa, N. Modulation of adipose tissue inflammation by bioactive food compounds. J. Nutr. Biochem. 2013, 24, 613–623. [Google Scholar] [CrossRef]
- Milenkovic, D.; Deval, C.; Gouranton, E.; Landrier, J.-F.; Scalbert, A.; Morand, C.; Mazur, A. Modulation of miRNA expression by dietary polyphenols in apoE deficient mice: A new mechanism of the action of polyphenols. PLoS ONE 2012, 7, e29837. [Google Scholar] [CrossRef]
- Gentile, D.; Fornai, M.; Colucci, R.; Pellegrini, C.; Tirotta, E.; Benvenuti, L.; Segnani, C.; Ippolito, C.; Duranti, E.; Virdis, A.; et al. The flavonoid compound apigenin prevents colonic inflammation and motor dysfunctions associated with high fat diet-induced obesity. PLoS ONE 2018, 13, e0195502. [Google Scholar] [CrossRef]
- Gentile, D.; Fornai, M.; Pellegrini, C.; Colucci, R.; Benvenuti, L.; Duranti, E.; Masi, S.; Carpi, S.; Nieri, P.; Nericcio, A.; et al. Luteolin prevents cardiometabolic alterations and vascular dysfunction in mice with hfd-induced obesity. Front. Pharmacol. 2018, 9, 1094. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Shen, W.; Yu, G.; Jia, H.; Li, X.; Feng, Z.; Wang, Y.; Weber, P.; Wertz, K.; Sharman, E. Hydroxytyrosol promotes mitochondrial biogenesis and mitochondrial function in 3T3-L1 adipocytes. J. Nutr. Biochem. 2010, 21, 634–644. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Derraik, J.G.B.; Brennan, C.M.; Biggs, J.B.; Morgan, P.E.; Hodgkinson, S.C.; Hofman, P.L.; Cutfield, W.S. Olive (Olea europaea L.) leaf polyphenols improve insulin sensitivity in middle-aged overweight men: A randomized, placebo-controlled, crossover trial. PLoS ONE 2013, 8, e57622. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Song, S.J.; Keum, N.; Park, T. Olive leaf extract attenuates obesity in high-fat diet-fed mice by modulating the expression of molecules involved in Adipogenesis and thermogenesis. Evid. Based Complement. Altern. Med. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Scoditti, E.; Massaro, M.; Carluccio, M.A.; Pellegrino, M.; Wabitsch, M.; Calabriso, N.; Storelli, C.; De Caterina, R. Additive regulation of adiponectin expression by the mediterranean diet olive oil components oleic Acid and hydroxytyrosol in human adipocytes. PLoS ONE 2015, 10, e0128218. [Google Scholar] [CrossRef] [PubMed]
- Stefanon, B.; Colitti, M. Original Research: Hydroxytyrosol, an ingredient of olive oil, reduces triglyceride accumulation and promotes lipolysis in human primary visceral adipocytes during differentiation. Exp. Biol. Med. 2016, 241, 1796–1802. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, G.E.; Lepore, S.M.; Morittu, V.M.; Arcidiacono, B.; Colica, C.; Procopio, A.; Maggisano, V.; Bulotta, S.; Costa, N.; Mignogna, C.; et al. Effects of Oleacein on high-fat diet-dependent steatosis, weight gain, and insulin resistance in mice. Front. Endocrinol. (Lausanne) 2018, 9, 116. [Google Scholar] [CrossRef]
- Polini, B.; Digiacomo, M.; Carpi, S.; Bertini, S.; Gado, F.; Saccomanni, G.; Macchia, M.; Nieri, P.; Manera, C.; Fogli, S. Oleocanthal and oleacein contribute to the in vitro therapeutic potential of extra virgin oil-derived extracts in non-melanoma skin cancer. Toxicol. In Vitro 2018, 52, 243–250. [Google Scholar] [CrossRef]
- Wabitsch, M.; Brenner, R.E.; Melzner, I.; Braun, M.; Möller, P.; Heinze, E.; Debatin, K.M.; Hauner, H. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 8–15. [Google Scholar] [CrossRef]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.F.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef]
- Adinolfi, B.; Carpi, S.; Romanini, A.; Da Pozzo, E.; Castagna, M.; Costa, B.; Martini, C.; Olesen, S.-P.; Schmitt, N.; Breschi, M.C.; et al. Analysis of the antitumor activity of clotrimazole on A375 human melanoma cells. Anticancer Res. 2015, 35, 3781–3786. [Google Scholar] [PubMed]
- Carpi, S.; Fogli, S.; Polini, B.; Montagnani, V.; Podestà, A.; Breschi, M.C.; Romanini, A.; Stecca, B.; Nieri, P. Tumor-promoting effects of cannabinoid receptor type 1 in human melanoma cells. Toxicol. In Vitro 2017, 40, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Maestro, version 9; Schrödinger Inc.: Portland, OR, USA, 2009.
- Macromodel, version 9.7; Schrödinger Inc.: Portland, OR, USA, 2009.
- Panne, D.; Maniatis, T.; Harrison, S.C. An atomic model of the interferon-beta enhanceosome. Cell 2007, 129, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Bononi, G.; Granchi, C.; Lapillo, M.; Giannotti, M.; Nieri, D.; Fortunato, S.; Boustani, M.E.; Caligiuri, I.; Poli, G.; Carlson, K.E.; et al. Discovery of long-chain salicylketoxime derivatives as monoacylglycerol lipase (MAGL) inhibitors. Eur. J. Med. Chem. 2018, 157, 817–836. [Google Scholar] [CrossRef] [PubMed]
- Luger, D.; Poli, G.; Wieder, M.; Stadler, M.; Ke, S.; Ernst, M.; Hohaus, A.; Linder, T.; Seidel, T.; Langer, T.; et al. Identification of the putative binding pocket of valerenic acid on GABAA receptors using docking studies and site-directed mutagenesis. Br. J. Pharmacol. 2015, 172, 5403–5413. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Liu, S.; Misquitta, Y.R.; Olland, A.; Johnson, M.A.; Kelleher, K.S.; Kriz, R.; Lin, L.L.; Stahl, M.; Mosyak, L. Crystal structure of a human IκB kinase β asymmetric dimer. J. Biol. Chem. 2013, 288, 22758–22767. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J. AMBER, version 14; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Dal Piaz, F.; Vera Saltos, M.B.; Franceschelli, S.; Forte, G.; Marzocco, S.; Tuccinardi, T.; Poli, G.; Nejad Ebrahimi, S.; Hamburger, M.; De Tommasi, N.; et al. Drug Affinity Responsive Target Stability (DARTS) identifies laurifolioside as a new clathrin heavy chain modulator. J. Nat. Prod. 2016, 79, 2681–2692. [Google Scholar] [CrossRef]
- Poli, G.; Lapillo, M.; Jha, V.; Mouawad, N.; Caligiuri, I.; Macchia, M.; Minutolo, F.; Rizzolio, F.; Tuccinardi, T.; Granchi, C. Computationally driven discovery of phenyl (piperazin-1-yl) methanone derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Caligiuri, I.; Bertelli, E.; Poli, G.; Rizzolio, F.; Macchia, M.; Martinelli, A.; Minutolo, F.; Tuccinardi, T. Development of terphenyl-2-methyloxazol-5(4H)-one derivatives as selective reversible MAGL inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Lapillo, M.; Granchi, C.; Caciolla, J.; Mouawad, N.; Caligiuri, I.; Rizzolio, F.; Langer, T.; Minutolo, F.; Tuccinardi, T. Binding investigation and preliminary optimisation of the 3-amino-1,2,4-triazin-5(2H)-one core for the development of new Fyn inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- García-Piñeres, A.J.; Castro, V.; Mora, G.; Schmidt, T.J.; Strunck, E.; Pahl, H.L.; Merfort, I. Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 2001, 276, 39713–39720. [Google Scholar] [CrossRef] [PubMed]
- Ariga, A.; Namekawa, J.-I.; Matsumoto, N.; Inoue, J.-I.; Umezawa, K. Inhibition of tumor necrosis factor-alpha -induced nuclear translocation and activation of NF-kappa B by dehydroxymethylepoxyquinomicin. J. Biol. Chem. 2002, 277, 24625–24630. [Google Scholar] [CrossRef] [PubMed]
- Tzanavari, T.; Giannogonas, P.; Karalis, K.P. TNF-alpha and obesity. Curr. Dir. Autoimmun. 2010, 11, 145–156. [Google Scholar]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef]
- Kanda, H. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Tateya, S.; Tamori, Y.; Kawaguchi, T.; Kanda, H.; Kasuga, M. An increase in the circulating concentration of monocyte chemoattractant protein-1 elicits systemic insulin resistance irrespective of adipose tissue inflammation in mice. Endocrinology 2010, 151, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Öhman, M.K.; Wright, A.P.; Wickenheiser, K.J.; Luo, W.; Russo, H.M.; Eitzman, D.T. Monocyte chemoattractant protein-1 deficiency protects against visceral fat-induced atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1151–1158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Faber, D.R.; van der Graaf, Y.; Westerink, J.; Kanhai, D.A.; Monajemi, H.; Visseren, F.L.J.; SMART study Group. Hepatocyte growth factor and interferon-γ inducible protein-10 are related to visceral adiposity. Eur. J. Clin. Investig. 2013, 43, 369–378. [Google Scholar] [CrossRef]
- Scotece, M.; Gómez, R.; Conde, J.; Lopez, V.; Gómez-Reino, J.J.; Lago, F.; Smith, A.B.; Gualillo, O. Further evidence for the anti-inflammatory activity of oleocanthal: Inhibition of MIP-1α and IL-6 in J774 macrophages and in ATDC5 chondrocytes. Life Sci. 2012, 91, 1229–1235. [Google Scholar] [CrossRef]
- Scotece, M.; Conde, J.; Abella, V.; López, V.; Francisco, V.; Ruiz, C.; Campos, V.; Lago, F.; Gomez, R.; Pino, J.; et al. Oleocanthal inhibits catabolic and inflammatory mediators in LPS-activated human primary osteoarthritis (OA) chondrocytes through MAPKs/NF-κB pathways. Cell. Physiol. Biochem. 2018, 49, 2414–2426. [Google Scholar] [CrossRef]
- Czerwińska, M.E.; Kiss, A.K.; Naruszewicz, M. Inhibition of human neutrophils NEP activity, CD11b/CD18 expression and elastase release by 3,4-dihydroxyphenylethanol-elenolic acid dialdehyde, oleacein. Food Chem. 2014, 153, 1–8. [Google Scholar] [CrossRef]
- Filipek, A.; Czerwińska, M.E.; Kiss, A.K.; Wrzosek, M.; Naruszewicz, M. Oleacein enhances anti-inflammatory activity of human macrophages by increasing CD163 receptor expression. Phytomedicine 2015, 22, 1255–1261. [Google Scholar] [CrossRef]
- Filipek, A.; Czerwińska, M.E.; Kiss, A.K.; Polański, J.A.; Naruszewicz, M. Oleacein may inhibit destabilization of carotid plaques from hypertensive patients. Impact on high mobility group protein-1. Phytomedicine 2017, 32, 68–73. [Google Scholar] [CrossRef]
- Vougogiannopoulou, K.; Lemus, C.; Halabalaki, M.; Pergola, C.; Werz, O.; Smith, A.B.; Michel, S.; Skaltsounis, L.; Deguin, B. One-step semisynthesis of oleacein and the determination as a 5-lipoxygenase inhibitor. J. Nat. Prod. 2014, 77, 441–445. [Google Scholar] [CrossRef]
- Rosignoli, P.; Fuccelli, R.; Fabiani, R.; Servili, M.; Morozzi, G. Effect of olive oil phenols on the production of inflammatory mediators in freshly isolated human monocytes. J. Nutr. Biochem. 2013, 24, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-S.; Jin, J.-S.; Chiang, C.-F.; Chan, P.-C.; Chen, C.-H.; Shih, K.-C. COX-2-mediated inflammation in fat is crucial for obesity-linked insulin resistance and fatty liver. Obesity 2009, 17, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.-C.; Hsiao, F.-C.; Chang, H.-M.; Wabitsch, M.; Hsieh, P.S. Importance of adipocyte cyclooxygenase-2 and prostaglandin E2–prostaglandin E receptor 3 signaling in the development of obesity-induced adipose tissue inflammation and insulin resistance. FASEB J. 2016, 30, 2282–2297. [Google Scholar] [CrossRef] [PubMed]
- Bouloumie, A.; Sengenes, C.; Portolan, G.; Galitzky, J.; Lafontan, M. Adipocyte produces matrix metalloproteinases 2 and 9: Involvement in adipose differentiation. Diabetes 2001, 50, 2080–2086. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, A.Y.; Ledoux, S.; Larger, E. Adipose tissue angiogenesis in obesity. Thromb. Haemost. 2013, 110, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Van Hul, M.; Lijnen, H.R. A functional role of gelatinase A in the development of nutritionally induced obesity in mice. J. Thromb. Haemost. 2008, 6, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Vincent, H.K.; Taylor, A.G. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int. J. Obes. 2006, 30, 400–418. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Den Hartigh, L.J.; Omer, M.; Goodspeed, L.; Wang, S.; Wietecha, T.; O’Brien, K.D.; Han, C.Y. Adipocyte-specific deficiency of NADPH oxidase 4 delays the onset of insulin resistance and attenuates adipose tissue inflammation in obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 466–475. [Google Scholar] [CrossRef]
- Han, C.Y.; Umemoto, T.; Omer, M.; Den Hartigh, L.J.; Chiba, T.; LeBoeuf, R.; Buller, C.L.; Sweet, I.R.; Pennathur, S.; Abel, E.D.; et al. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem. 2012, 287, 10379–10393. [Google Scholar] [CrossRef]
- Pepping, J.K.; Freeman, L.R.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. NOX2 deficiency attenuates markers of adiposopathy and brain injury induced by high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E392–E404. [Google Scholar] [CrossRef] [PubMed]
- Naruszewicz, M.; Czerwinska, M.; Kiss, A. Oleacein. Translation from Mediterranean Diet to Potential Antiatherosclerotic Drug. Curr. Pharm. Des. 2015, 21, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.-L.; Chin, K.-Y. The biological activities of oleocanthal from a molecular perspective. Nutrients 2018, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R.; Rosignoli, P.; De Bartolomeo, A.; Fuccelli, R.; Servili, M.; Montedoro, G.F.; Morozzi, G. Oxidative DNA damage is prevented by extracts of olive oil, hydroxytyrosol, and other olive phenolic compounds in human blood mononuclear cells and HL60 cells. J. Nutr. 2008, 138, 1411–1416. [Google Scholar] [CrossRef]
- Parzonko, A.; Czerwińska, M.E.; Kiss, A.K.; Naruszewicz, M. Oleuropein and oleacein may restore biological functions of endothelial progenitor cells impaired by angiotensin II via activation of Nrf2/heme oxygenase-1 pathway. Phytomedicine 2013, 20, 1088–1094. [Google Scholar] [CrossRef]
- Calabriso, N.; Massaro, M.; Scoditti, E.; D’Amore, S.; Gnoni, A.; Pellegrino, M.; Storelli, C.; De Caterina, R.; Palasciano, G.; Carluccio, M.A. Extra virgin olive oil rich in polyphenols modulates VEGF-induced angiogenic responses by preventing NADPH oxidase activity and expression. J. Nutr. Biochem. 2016, 28, 19–29. [Google Scholar] [CrossRef]
- Carnevale, R.; Pignatelli, P.; Nocella, C.; Loffredo, L.; Pastori, D.; Vicario, T.; Petruccioli, A.; Bartimoccia, S.; Violi, F. Extra virgin olive oil blunt post-prandial oxidative stress via NOX2 down-regulation. Atherosclerosis 2014, 235, 649–658. [Google Scholar] [CrossRef]
- Sabir, J.S.M.; El Omri, A.; Shaik, N.A.; Banaganapalli, B.; Al-Shaeri, M.A.; Alkenani, N.A.; Hajrah, N.H.; Awan, Z.A.; Zrelli, H.; Elango, R.; et al. Identification of key regulatory genes connected to NF-κB family of proteins in visceral adipose tissues using gene expression and weighted protein interaction network. PLoS ONE 2019, 14, e0214337. [Google Scholar] [CrossRef]
- Madonna, R.; De Caterina, R. Relevance of new drug discovery to reduce NF-κB activation in cardiovascular disease. Vasc. Pharmacol. 2012, 57, 41–47. [Google Scholar] [CrossRef]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The immunomodulatory and anti-inflammatory role of polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef]
- Iacono, A.; Gómez, R.; Sperry, J.; Conde, J.; Bianco, G.; Meli, R.; Gómez-Reino, J.J.; Smith, A.B.; Gualillo, O. Effect of oleocanthal and its derivatives on inflammatory response induced by lipopolysaccharide in a murine chondrocyte cell line. Arthritis Rheum. 2010, 62, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Scoditti, E.; Carpi, S.; Massaro, M.; Pellegrino, M.; Polini, B.; Carluccio, M.A.; Wabitsch, M.; Verri, T.; Nieri, P.; De Caterina, R. Hydroxytyrosol modulates adipocyte gene and miRNA expression under inflammatory condition. Nutrients 2019, 11, 2493. [Google Scholar] [CrossRef] [PubMed]
- Scafuri, B.; Marabotti, A.; Carbone, V.; Minasi, P.; Dotolo, S.; Facchiano, A. A theoretical study on predicted protein targets of apple polyphenols and possible mechanisms of chemoprevention in colorectal cancer. Sci. Rep. 2016, 6, 32516. [Google Scholar] [CrossRef] [PubMed]
- Saibandith, B.; Spencer, J.P.E.; Rowland, I.R.; Commane, D.M. Olive Polyphenols and the Metabolic Syndrome. Molecules 2017, 22, 1082. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Glass, C.K. Nuclear receptors versus inflammation: Mechanisms of transrepression. Trends Endocrinol. Metab. 2006, 17, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Batarseh, Y.S.; Kaddoumi, A. Oleocanthal-rich extra-virgin olive oil enhances donepezil effect by reducing amyloid-β load and related toxicity in a mouse model of Alzheimer’s disease. J. Nutr. Biochem. 2018, 55, 113–123. [Google Scholar] [CrossRef]
- Lepore, S.M.; Maggisano, V.; Bulotta, S.; Mignogna, C.; Arcidiacono, B.; Procopio, A.; Brunetti, A.; Russo, D.; Celano, M. Oleacein prevents high fat diet-induced a diposity and ameliorates some biochemical parameters of insulin sensitivity in mice. Nutrients 2019, 11, 1829. [Google Scholar] [CrossRef]
- Lin, Q.; Ma, L.; Liu, Z.; Yang, Z.; Wang, J.; Liu, J.; Jiang, G. Targeting microRNAs: A new action mechanism of natural compounds. Oncotarget 2017, 8, 15961. [Google Scholar] [CrossRef]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef]
- La, X.; Zhang, L.; Li, Z.; Li, H.; Yang, Y. (−)-Epigallocatechin Gallate (EGCG) enhances the sensitivity of colorectal cancer cells to 5-FU by inhibiting GRP78/NF-κB/miR-155-5p/MDR1 pathway. J. Agric. Food Chem. 2019, 67, 2510–2518. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.; Wu, S.; He, J.; Lou, L.; Ye, W.; Wang, J. microRNA-34a and microRNA-34c promote the activation of human hepatic stellate cells by targeting peroxisome proliferator-activated receptor γ. Mol. Med. Rep. 2015, 11, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective blockade of microRNA processing by Lin28. Science 2008, 320, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.-J. Promiscuous effects of some phenolic natural products on inflammation at least in part arise from their ability to modulate the expression of global regulators, namely microRNAs. Molecules 2016, 21, 1263. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Carrasco-Pancorbo, A.; Nevedomskaya, E.; Mayboroda, O.A.; Deelder, A.M.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Exploratory analysis of human urine by LC–ESI-TOF MS after high intake of olive oil: Understanding the metabolism of polyphenols. Anal. Bioanal. Chem. 2010, 398, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.; Paiva-Martins, F.; Corona, G.; Debnam, E.S.; Jose Oruna-Concha, M.; Vauzour, D.; Gordon, M.H.; Spencer, J.P.E. Absorption and metabolism of olive oil secoiridoids in the small intestine. Br. J. Nutr. 2011, 105, 1607–1618. [Google Scholar] [CrossRef]
- Bartholomé, R.; Haenen, G.; Hollman, C.H.; Bast, A.; Dagnelie, P.C.; Roos, D.; Keijer, J.; Kroon, P.A.; Needs, P.W.; Arts, I.C.W. Deconjugation kinetics of glucuronidated phase II flavonoid metabolites by beta-glucuronidase from neutrophils. Drug Metab. Pharmacokinet. 2010, 25, 379–387. [Google Scholar] [CrossRef]
- Kano, S.; Komada, H.; Yonekura, L.; Sato, A.; Nishiwaki, H.; Tamura, H. Absorption, metabolism, and excretion by freely moving rats of 3,4-DHPEA-EDA and related polyphenols from olive fruits (Olea europaea). J. Nutr. Metab. 2016, 2016, 1–10. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpi, S.; Scoditti, E.; Massaro, M.; Polini, B.; Manera, C.; Digiacomo, M.; Esposito Salsano, J.; Poli, G.; Tuccinardi, T.; Doccini, S.; et al. The Extra-Virgin Olive Oil Polyphenols Oleocanthal and Oleacein Counteract Inflammation-Related Gene and miRNA Expression in Adipocytes by Attenuating NF-κB Activation. Nutrients 2019, 11, 2855. https://doi.org/10.3390/nu11122855
Carpi S, Scoditti E, Massaro M, Polini B, Manera C, Digiacomo M, Esposito Salsano J, Poli G, Tuccinardi T, Doccini S, et al. The Extra-Virgin Olive Oil Polyphenols Oleocanthal and Oleacein Counteract Inflammation-Related Gene and miRNA Expression in Adipocytes by Attenuating NF-κB Activation. Nutrients. 2019; 11(12):2855. https://doi.org/10.3390/nu11122855
Chicago/Turabian StyleCarpi, Sara, Egeria Scoditti, Marika Massaro, Beatrice Polini, Clementina Manera, Maria Digiacomo, Jasmine Esposito Salsano, Giulio Poli, Tiziano Tuccinardi, Stefano Doccini, and et al. 2019. "The Extra-Virgin Olive Oil Polyphenols Oleocanthal and Oleacein Counteract Inflammation-Related Gene and miRNA Expression in Adipocytes by Attenuating NF-κB Activation" Nutrients 11, no. 12: 2855. https://doi.org/10.3390/nu11122855
APA StyleCarpi, S., Scoditti, E., Massaro, M., Polini, B., Manera, C., Digiacomo, M., Esposito Salsano, J., Poli, G., Tuccinardi, T., Doccini, S., Santorelli, F. M., Carluccio, M. A., Macchia, M., Wabitsch, M., De Caterina, R., & Nieri, P. (2019). The Extra-Virgin Olive Oil Polyphenols Oleocanthal and Oleacein Counteract Inflammation-Related Gene and miRNA Expression in Adipocytes by Attenuating NF-κB Activation. Nutrients, 11(12), 2855. https://doi.org/10.3390/nu11122855