Is an “Epigenetic Diet” for Migraines Justified? The Case of Folate and DNA Methylation



Abstract
:1. Introduction
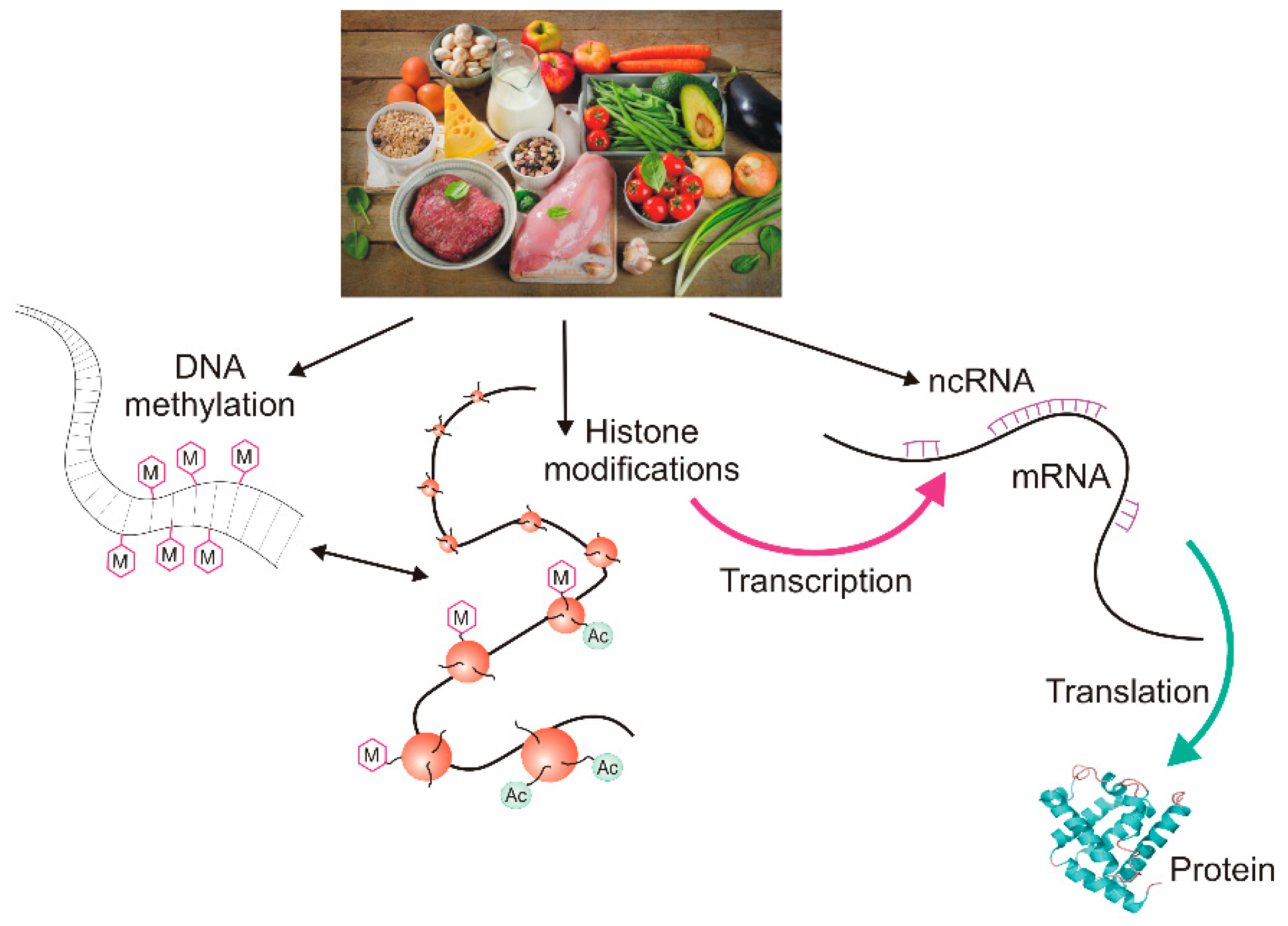
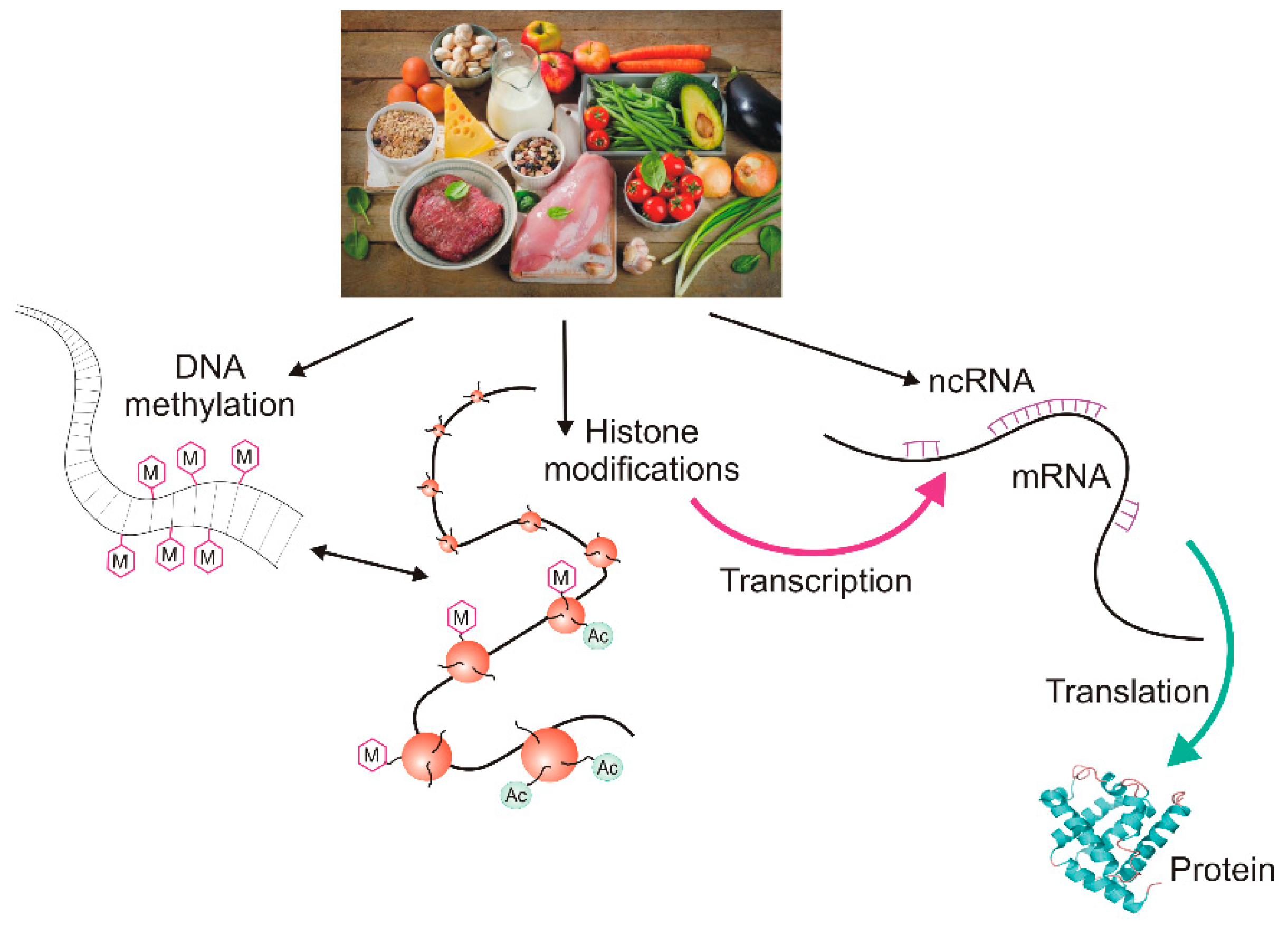
2. Epigenetic Regulation of Gene Expression
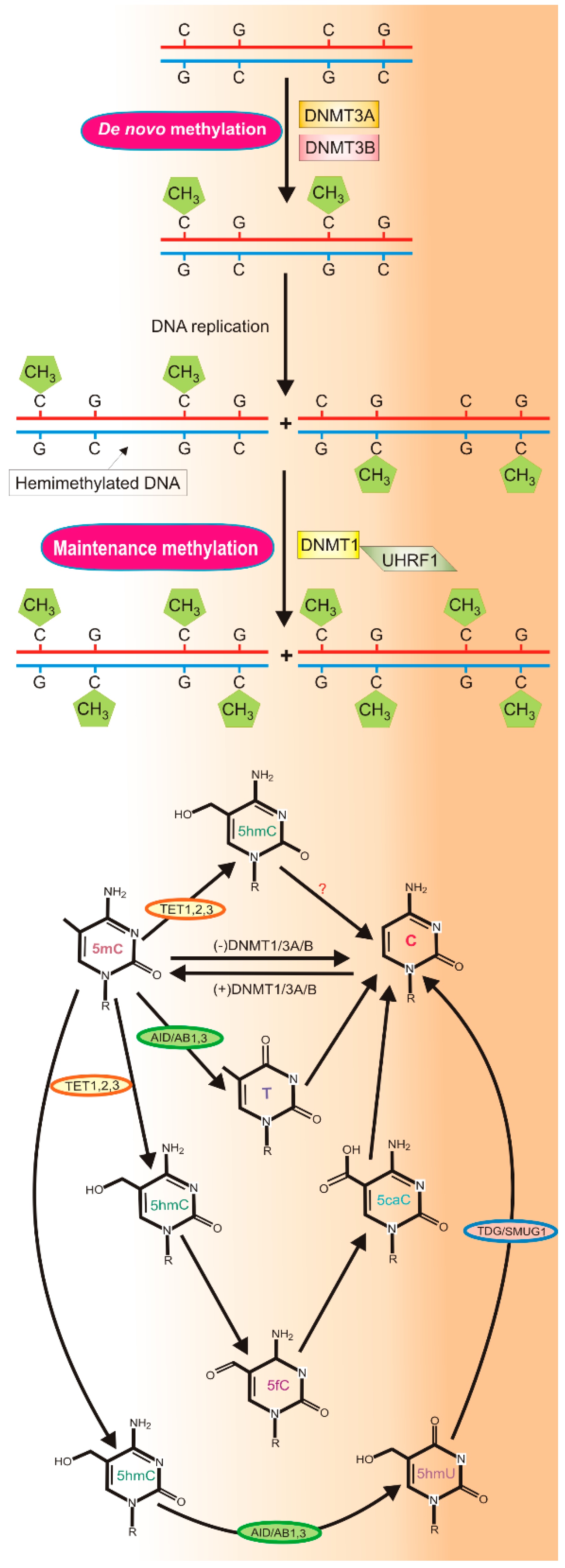
2.1. DNA Methylation and Demethylation
2.2. Histone Modifications and Non-Coding RNAs
3. The Epigenetic Diet—Does It Really Exist?
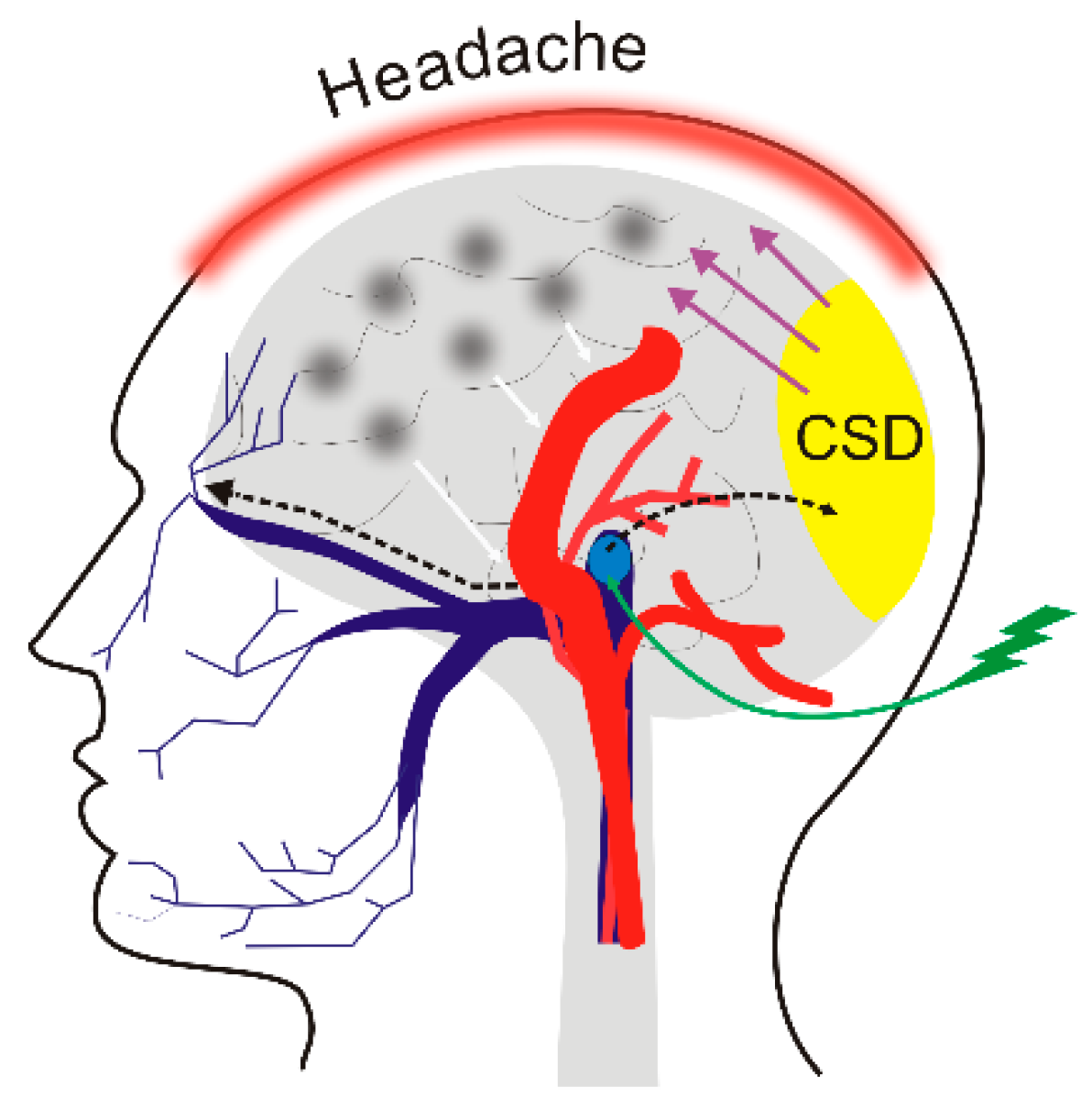
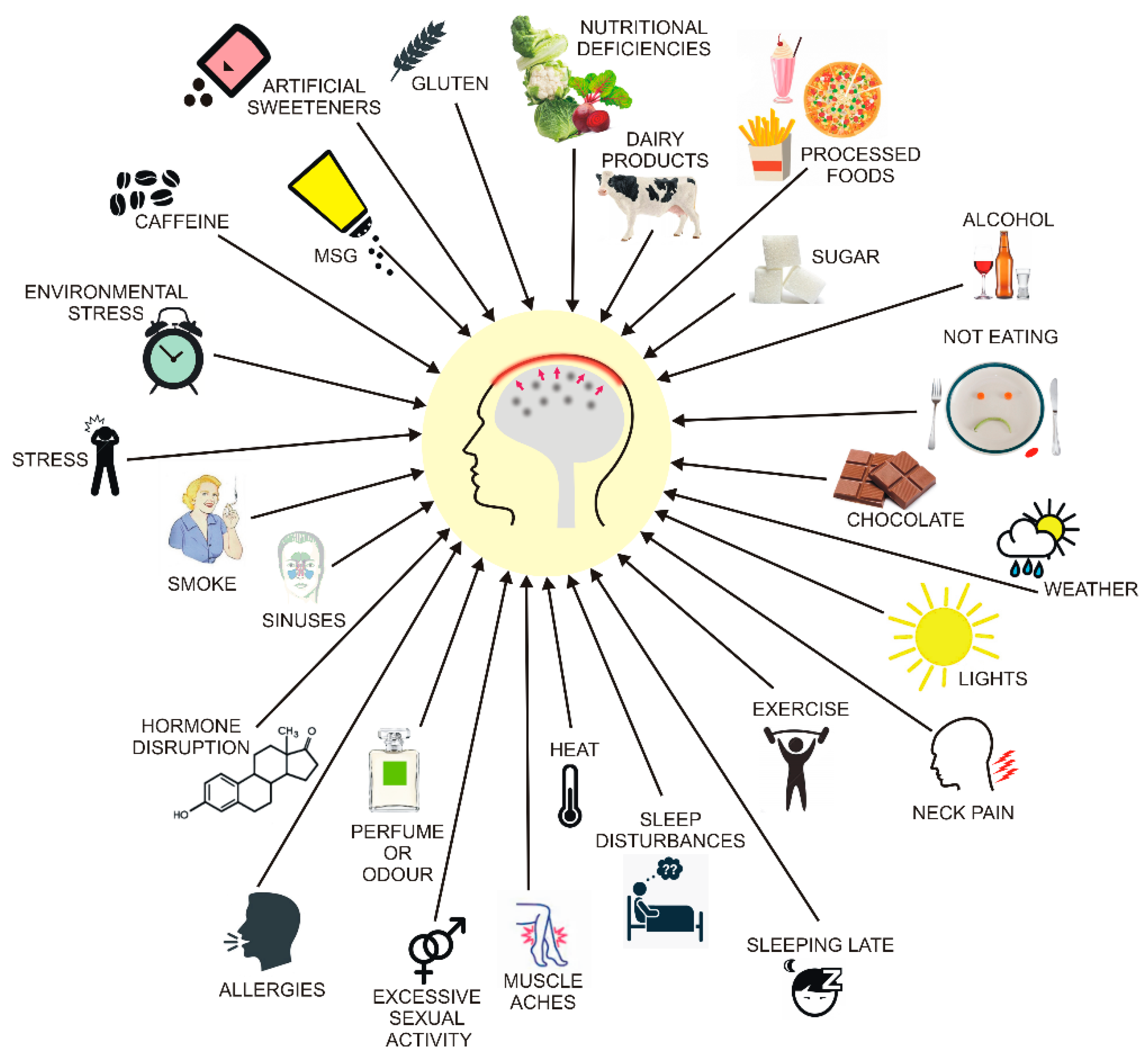
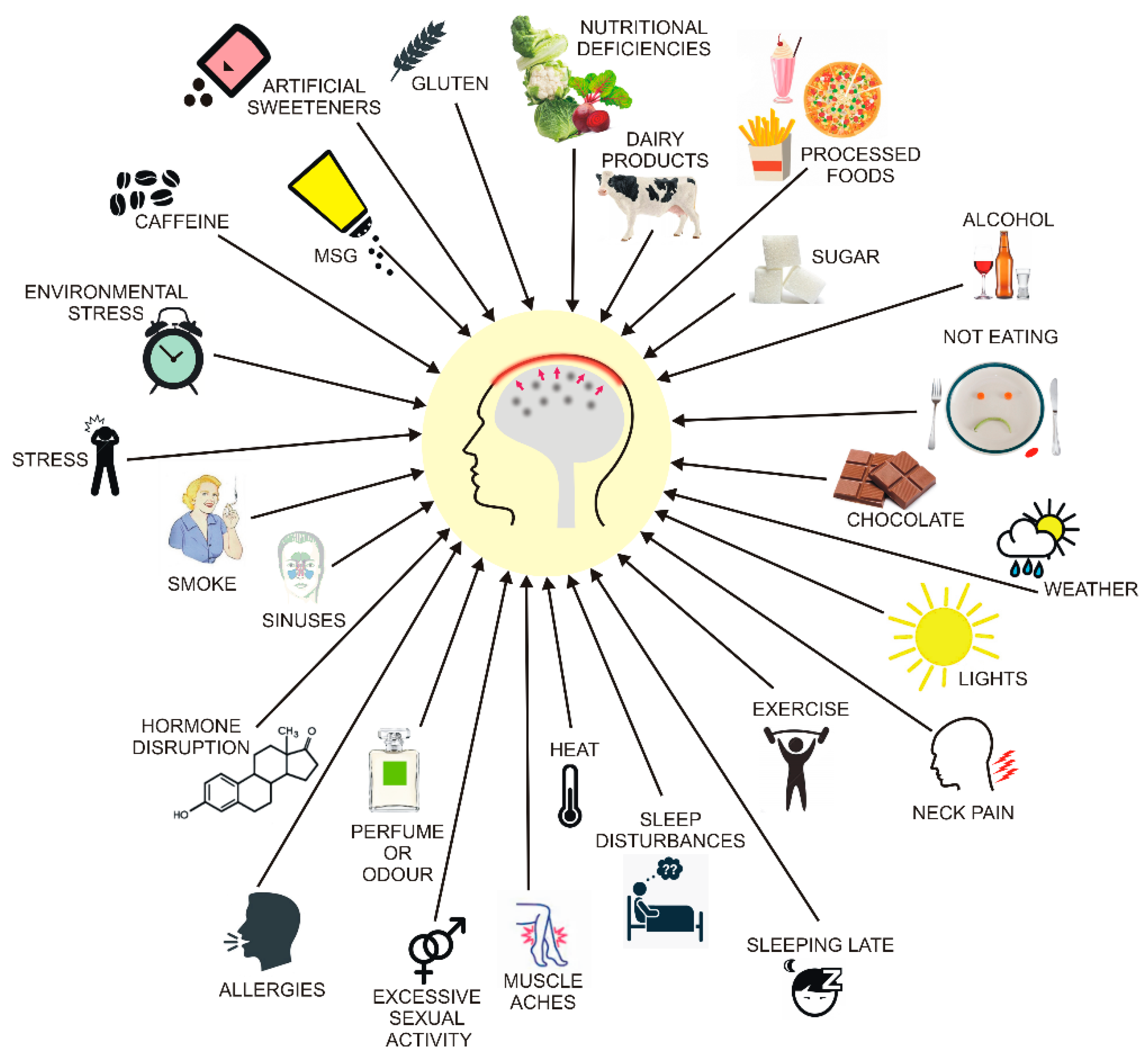
4. Migraine and Diet
5. DNA Methylation in Migraine
6. Folate and Its Role in DNA Methylation and Migraine Pathogenesis
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Remely, M.; Stefanska, B.; Lovrecic, L.; Magnet, U.; Haslberger, A.G. Nutriepigenomics: The role of nutrition in epigenetic control of human diseases. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Blot, W.J.; Tarone, R.E. Doll and Peto’s quantitative estimates of cancer risks: Holding generally true for 35 years. J. Natl. Cancer Inst. 2015, 107, 4. [Google Scholar] [CrossRef] [PubMed]
- Bektas, H.; Karabulut, H.; Doganay, B.; Acar, B. Allergens might trigger migraine attacks. Acta Neurol. Belg. 2017, 117, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Finocchi, C.; Sivori, G. Food as trigger and aggravating factor of migraine. Neurol. Sci. 2012, 33 (Suppl. 1), S77–S80. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.T.; Vij, B. Diet and Headache: Part 1. Headache 2016, 56, 1543–1552. [Google Scholar] [CrossRef]
- Martin, V.T.; Vij, B. Diet and Headache: Part 2. Headache 2016, 56, 1553–1562. [Google Scholar] [CrossRef]
- Owen, L.; Corfe, B. The role of diet and nutrition on mental health and wellbeing. Proc. Nutr. Soc. 2017, 76, 425–426. [Google Scholar] [CrossRef]
- Paoli, A.; Moro, T.; Bosco, G.; Bianco, A.; Grimaldi, K.A.; Camporesi, E.; Mangar, D. Effects of n-3 polyunsaturated fatty acids (omega-3) supplementation on some cardiovascular risk factors with a ketogenic Mediterranean diet. Mar. Drugs 2015, 13, 996–1009. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Palomba, S.; Lagana, A.S.; Orio, F. Current Insights into Inositol Isoforms, Mediterranean and Ketogenic Diets for Polycystic Ovary Syndrome: From Bench to Bedside. Curr. Pharm. Des. 2016, 22, 5554–5557. [Google Scholar] [CrossRef]
- Castaldo, G.; Monaco, L.; Castaldo, L.; Galdo, G.; Cereda, E. An observational study of sequential protein-sparing, very low-calorie ketogenic diet (Oloproteic diet) and hypocaloric Mediterranean-like diet for the treatment of obesity. Int. J. Food Sci. Nutr. 2016, 67, 696–706. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys) function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.M.; Tollefsbol, T.O. Epigenetic diet: Impact on the epigenome and cancer. Epigenomics 2011, 3, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, Z.; Dincer, Y. Alzheimer’s disease and epigenetic diet. Neurochem. Int. 2014, 78, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Sapienza, C.; Issa, J.P. Diet, Nutrition, and Cancer Epigenetics. Annu. Rev. Nutr. 2016, 36, 665–681. [Google Scholar] [CrossRef]
- Ganesan, A. Multitarget Drugs: An Epigenetic Epiphany. ChemMedChem 2016, 11, 1227–1241. [Google Scholar] [CrossRef]
- Shaik, M.M.; Tan, H.L.; Kamal, M.A.; Gan, S.H. Do folate, vitamins B (6) and B (1)(2) play a role in the pathogenesis of migraine? The role of pharmacoepigenomics. CNS Neurol. Disord. Drug Targets 2014, 13, 828–835. [Google Scholar] [CrossRef]
- Sadeghi, O.; Maghsoudi, Z.; Khorvash, F.; Ghiasvand, R.; Askari, G. Assessment of pyridoxine and folate intake in migraine patients. Adv. Biomed. Res. 2016, 5, 47. [Google Scholar]
- Menon, S.; Lea, R.A.; Ingle, S.; Sutherland, M.; Wee, S.; Haupt, L.M.; Palmer, M.; Griffiths, L.R. Effects of dietary folate intake on migraine disability and frequency. Headache 2015, 55, 301–309. [Google Scholar] [CrossRef]
- Mandaviya, P.R.; Joehanes, R.; Brody, J.; Castillo-Fernandez, J.E.; Dekkers, K.F.; Do, A.N.; Graff, M.; Hanninen, I.K.; Tanaka, T.; de Jonge, E.A.L.; et al. Association of dietary folate and vitamin B-12 intake with genome-wide DNA methylation in blood: A large-scale epigenome-wide association analysis in 5841 individuals. Am. J. Clin. Nutr. 2019, 110, 437–450. [Google Scholar] [CrossRef]
- Epigenomics Fact Sheet. Available online: https://www.genome.gov/about-genomics/fact- sheets/Epigenomics-Fact-Sheet (accessed on 10 October 2019).
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Bronner, C.; Alhosin, M.; Hamiche, A.; Mousli, M. Coordinated Dialogue between UHRF1 and DNMT1 to Ensure Faithful Inheritance of Methylated DNA Patterns. Genes (Basel) 2019, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Veland, N.; Lu, Y.; Hardikar, S.; Gaddis, S.; Zeng, Y.; Liu, B.; Estecio, M.R.; Takata, Y.; Lin, K.; Tomida, M.W.; et al. DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 2019, 47, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, K.E.; Hobartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m5C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes (Basel) 2019, 10, 102. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Bird, A.P. CpG islands—‘a rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Fan, G.; Beard, C.; Chen, R.Z.; Csankovszki, G.; Sun, Y.; Siniaia, M.; Biniszkiewicz, D.; Bates, B.; Lee, P.P.; Kuhn, R.; et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J. Neurosci. 2001, 21, 788–797. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Jin, Y.; Allen, E.G.; Jin, P. Diverse and Dynamic DNA Modifications in Brain and Diseases. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DesJarlais, R.; Tummino, P.J. Role of Histone-Modifying Enzymes and Their Complexes in Regulation of Chromatin Biology. Biochemistry 2016, 55, 1584–1599. [Google Scholar] [CrossRef] [PubMed]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Jiang, X.; Jiang, X.; Zhao, H. X-inactive-specific transcript: A long noncoding RNA with complex roles in human cancers. Gene 2018, 679, 28–35. [Google Scholar] [CrossRef]
- Sahakyan, A.; Yang, Y.; Plath, K. The Role of Xist in X-Chromosome Dosage Compensation. Trends Cell Biol. 2018, 28, 999–1013. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef]
- Zhao, L.; Duan, Y.T.; Lu, P.; Zhang, Z.J.; Zheng, X.K.; Wang, J.L.; Feng, W.S. Epigenetic Targets and their Inhibitors in Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 2395–2419. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Ross, S.A.; Wise, C.; Pogribna, M.; Jones, E.A.; Tryndyak, V.P.; James, S.J.; Dragan, Y.P.; Poirier, L.A. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat. Res. 2006, 593, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Corpe, C.P.; Eck, P.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J. Biol. Chem. 2013, 288, 9092–9101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpe, C.P.; Lee, J.H.; Kwon, O.; Eck, P.; Narayanan, J.; Kirk, K.L.; Levine, M. 6-Bromo-6-deoxy-L-ascorbic acid: An ascorbate analog specific for Na+-dependent vitamin C transporter but not glucose transporter pathways. J. Biol. Chem. 2005, 280, 5211–5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Kwon, O.; Chen, S.; Daruwala, R.; Eck, P.; Park, J.B.; Levine, M. Flavonoid inhibition of sodium-dependent vitamin C transporter 1 (SVCT1) and glucose transporter isoform 2 (GLUT2), intestinal transporters for vitamin C and Glucose. J. Biol. Chem. 2002, 277, 15252–15260. [Google Scholar] [CrossRef] [Green Version]
- Yung, S.; Mayersohn, M.; Robinson, J.B. Ascorbic acid absorption in humans: A comparison among several dosage forms. J. Pharm. Sci. 1982, 71, 282–285. [Google Scholar] [CrossRef]
- Beker, B.Y.; Sonmezoglu, I.; Imer, F.; Apak, R. Protection of ascorbic acid from copper(II)-catalyzed oxidative degradation in the presence of flavonoids: Quercetin, catechin and morin. Int. J. Food Sci. Nutr. 2011, 62, 504–512. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- Steiner, T.J.; Stovner, L.J.; Birbeck, G.L. Migraine: The seventh disabler. J. Headache Pain 2013, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Kissoon, N.R.; Cutrer, F.M. Aura and Other Neurologic Dysfunction in or with Migraine. Headache 2017, 57, 1179–1194. [Google Scholar] [CrossRef]
- Seyed Forootan, N.S.; Lee, M.; Guyuron, B. Migraine headache trigger site prevalence analysis of 2590 sites in 1010 patients. J. Plast. Reconstr. Aesthet. Surg. 2017, 70, 152–158. [Google Scholar] [CrossRef]
- Levy, D.; Strassman, A.M.; Burstein, R. A critical view on the role of migraine triggers in the genesis of migraine pain. Headache 2009, 49, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Lipton, R.B.; Ferrari, M.D. Migraine—current understanding and treatment. N. Engl. J. Med. 2002, 346, 257–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugs@FDA: FDA Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 10 October 2019).
- Are the New Migraine Medications Working? Available online: https://www.health.harvard.edu/diseases-and-conditions/are-the-new-migraine-medications-working (accessed on 10 October 2019).
- Kelman, L. The triggers or precipitants of the acute migraine attack. Cephalalgia Int. J. Headache 2007, 27, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Vetvik, K.G.; MacGregor, E.A. Sex differences in the epidemiology, clinical features, and pathophysiology of migraine. Lancet Neurol. 2017, 16, 76–87. [Google Scholar] [CrossRef]
- Fila, M.; Pawlowska, E.; Blasiak, J. Mitochondria in migraine pathophysiology - does epigenetics play a role? Arch. Med. Sci. AMS 2019, 15, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.G.; Albury, C.L.; Griffiths, L.R. Advances in genetics of migraine. J. Headache Pain 2019, 20, 72. [Google Scholar] [CrossRef]
- Gormley, P.; Anttila, V.; Winsvold, B.S.; Palta, P.; Esko, T.; Pers, T.H.; Farh, K.H.; Cuenca-Leon, E.; Muona, M.; Furlotte, N.A.; et al. Corrigendum: Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 2016, 48, 856–866. [Google Scholar] [CrossRef] [Green Version]
- de Vries, B.; Frants, R.R.; Ferrari, M.D.; van den Maagdenberg, A.M. Molecular genetics of migraine. Hum. Genet. 2009, 126, 115–132. [Google Scholar] [CrossRef]
- Kocerha, J.; Aggarwal, N. Chapter 8—Epigenetics in Neurobehavioral Disease. In Epigenetics in Human Disease, 2nd ed.; Tollefsbol, T.O., Ed.; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Kidlington, UK, 2018; Volume 6, pp. 251–267. [Google Scholar]
- Majchrzak-Celinska, A.; Baer-Dubowska, W. Pharmacoepigenetics: An element of personalized therapy? Expert Opin. Drug Metab. Toxicol. 2017, 13, 387–398. [Google Scholar] [CrossRef]
- Tomson, T.; Battino, D.; Perucca, E. Valproic acid after five decades of use in epilepsy: Time to reconsider the indications of a time-honoured drug. Lancet Neurol. 2016, 15, 210–218. [Google Scholar] [CrossRef]
- Liu, F.; Ma, T.; Che, X.; Wang, Q.; Yu, S. The Efficacy of Venlafaxine, Flunarizine, and Valproic Acid in the Prophylaxis of Vestibular Migraine. Front. Neurol. 2017, 8, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eising, E.; Datson, N.A.; van den Maagdenberg, A.M.J.M.; Ferrari, M.D. Epigenetic mechanisms in migraine: A promising avenue? BMC Med. 2013, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.S.; Ferguson, L.R. The interaction between epigenetics, nutrition and the development of cancer. Nutrients 2015, 7, 922–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nattagh-Eshtivani, E.; Sani, M.A.; Dahri, M.; Ghalichi, F.; Ghavami, A.; Arjang, P.; Tarighat-Esfanjani, A. The role of nutrients in the pathogenesis and treatment of migraine headaches: Review. Biomed. Pharmacother. 2018, 102, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Barbanti, P.; Fofi, L.; Aurilia, C.; Egeo, G.; Caprio, M. Ketogenic diet in migraine: Rationale, findings and perspectives. Neurol. Sci. 2017, 38 (Suppl. 1), 111–115. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.B.; Crawford, P.A. Ketone bodies as epigenetic modifiers. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 260–266. [Google Scholar] [CrossRef]
- Andreeva, V.A.; Szabo de Edelenyi, F.; Druesne-Pecollo, N.; Touvier, M.; Hercberg, S.; Galan, P. Macronutrient Intake in Relation to Migraine and Non-Migraine Headaches. Nutrients 2018, 10, 1309. [Google Scholar] [CrossRef] [Green Version]
- Winsvold, B.S.; Palta, P.; Eising, E.; Page, C.M.; van den Maagdenberg, A.M.; Palotie, A.; Zwart, J.A. Epigenetic DNA methylation changes associated with headache chronification: A retrospective case-control study. Cephalalgia Int. J. Headache 2018, 38, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Terlizzi, R.; Bacalini, M.G.; Pirazzini, C.; Giannini, G.; Pierangeli, G.; Garagnani, P.; Franceschi, C.; Cevoli, S.; Cortelli, P. Epigenetic DNA methylation changes in episodic and chronic migraine. Neurol. Sci. 2018, 39 (Suppl. 1), 67–68. [Google Scholar] [CrossRef]
- Liao, Y.J.; Jiang, J.R.; Jin, S.Q. The association between COMT Val158Met polymorphism and migraine risk: A meta-analysis. Cephalalgia 2017, 37, 592–598. [Google Scholar] [CrossRef]
- Chen, H.; Ji, C.X.; Zhao, L.L.; Kong, X.J.; Zeng, X.T. Association Between Polymorphisms of DRD2, COMT, DBH, and MAO-A Genes and Migraine Susceptibility: A Meta-Analysis. Medicine (Baltimore) 2015, 94, e2012. [Google Scholar] [CrossRef] [PubMed]
- Gerring, Z.F.; McRae, A.F.; Montgomery, G.W.; Nyholt, D.R. Genome-wide DNA methylation profiling in whole blood reveals epigenetic signatures associated with migraine. BMC Genom. 2018, 19, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, D.; Hou, L.; Zhang, X.; Han, X.; Chen, M.; Tang, W.; Liu, R.; Dong, Z.; Yu, S. DNA methylation of RAMP1 gene in migraine: An exploratory analysis. J. Headache Pain 2015, 16, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Lasaosa, S.; Belvis, R.; Cuadrado, M.L.; Diaz-Insa, S.; Gago-Veiga, A.; Guerrero-Peral, A.L.; Huerta, M.; Irimia, P.; Lainez, J.M.; Latorre, G.; et al. Calcitonin gene-related peptide in migraine: From pathophysiology to treatment. Neurologia. 2019. [Google Scholar] [CrossRef]
- Park, K.Y.; Fletcher, J.R.; Raddant, A.C.; Russo, A.F. Epigenetic regulation of the calcitonin gene-related peptide gene in trigeminal glia. Cephalalgia 2011, 31, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Labruijere, S.; Stolk, L.; Verbiest, M.; de Vries, R.; Garrelds, I.M.; Eilers, P.H.; Danser, A.H.; Uitterlinden, A.G.; MaassenVanDenBrink, A. Methylation of migraine-related genes in different tissues of the rat. PLoS ONE 2014, 9, e87616. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Field, M.S.; Stover, P.J. Cell cycle regulation of folate-mediated one-carbon metabolism. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1426. [Google Scholar] [CrossRef]
- Field, M.S.; Kamynina, E.; Chon, J.; Stover, P.J. Nuclear Folate Metabolism. Annu. Rev. Nutr. 2018, 38, 219–243. [Google Scholar] [CrossRef]
- Scaglione, F.; Panzavolta, G. Folate, folic acid and 5-methyltetrahydrofolate are not the same thing. Xenobiotica 2014, 44, 480–488. [Google Scholar] [CrossRef]
- Soda, K. Polyamine Metabolism and Gene Methylation in Conjunction with One-Carbon Metabolism. Int. J. Mol. Sci. 2018, 19, 3106. [Google Scholar] [CrossRef] [Green Version]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieroth, R.; Paver, S.; Day, S.; Lammersfeld, C. Folate and Its Impact on Cancer Risk. Curr. Nutr. Rep. 2018, 7, 70–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capelli, I.; Cianciolo, G.; Gasperoni, L.; Zappulo, F.; Tondolo, F.; Cappuccilli, M.; La Manna, G. Folic Acid and Vitamin B12 Administration in CKD, Why Not? Nutrients 2019, 11, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, J.; Mason, J.B.; Choi, S.-W. Too much folate: A risk factor for cancer and cardiovascular disease? Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.; Hughes, C.F.; Ward, M.; Hoey, L.; McNulty, H. Diet, nutrition and the ageing brain: Current evidence and new directions. Proc. Nutr. Soc. 2018, 77, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Balashova, O.A.; Visina, O.; Borodinsky, L.N. Folate action in nervous system development and disease. Dev. Neurobiol. 2018, 78, 391–402. [Google Scholar] [CrossRef]
- Stover, P.J.; Durga, J.; Field, M.S. Folate nutrition and blood-brain barrier dysfunction. Curr. Opin. Biotechnol. 2017, 44, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Joachim, E.; Goldenberg, N.A.; Bernard, T.J.; Armstrong-Wells, J.; Stabler, S.; Manco-Johnson, M.J. The Methylenetetrahydrofolate Reductase Polymorphism (MTHFR c.677C>T) and Elevated Plasma Homocysteine Levels in a U.S. Pediatric Population with Incident Thromboembolism. Thromb. Res. 2013, 132, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Esse, R.; Barroso, M.; Tavares de Almeida, I.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef] [Green Version]
- Siennicka, A.; Zuchowski, M.; Chelstowski, K.; Cnotliwy, M.; Clark, J.S.; Jastrzebska, M. Homocysteine-Enhanced Proteolytic and Fibrinolytic Processes in Thin Intraluminal Thrombus and Adjacent Wall of Abdominal Aortic Aneurysm: Study In Vitro. Biomed. Res. Int. 2018, 2018, 3205324. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Gruenewald-Schneider, U.; De Sousa, D.; Webb, S.; Merusi, C.; Kerr, A.R.; James, K.D.; Smith, C.; Walker, R.; Andrews, R.; et al. Inter-individual variability contrasts with regional homogeneity in the human brain DNA methylome. Nucleic Acids Res. 2015, 43, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainero, I.; Vacca, A.; Roveta, F.; Govone, F.; Gai, A.; Rubino, E. Targeting MTHFR for the treatment of migraines. Expert Opin. Ther. Targets 2019, 23, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Geng, P.; Ma, M.; Yu, S.; Yang, M.; He, M.; Dong, Z.; Zhang, W. MTHFR C677T polymorphism and migraine risk: A meta-analysis. J. Neurol. Sci. 2014, 336, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, G.; Attina, S.; Spano, M.; Ingegneri, G.; Sgro, D.L.; Pustorino, G.; Bonsignore, M.; Trapani-Lombardo, V.; Tortorella, G. Efficacy of folic acid in children with migraine, hyperhomocysteinemia and MTHFR polymorphisms. Headache 2007, 47, 1342–1344. [Google Scholar] [CrossRef] [PubMed]
- Lea, R.; Colson, N.; Quinlan, S.; Macmillan, J.; Griffiths, L. The effects of vitamin supplementation and MTHFR (C677T) genotype on homocysteine-lowering and migraine disability. Pharmacogenet. Genom. 2009, 19, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Askari, G.; Nasiri, M.; Mozaffari-Khosravi, H.; Rezaie, M.; Bagheri-Bidakhavidi, M.; Sadeghi, O. The effects of folic acid and pyridoxine supplementation on characteristics of migraine attacks in migraine patients with aura: A double-blind, randomized placebo-controlled, clinical trial. Nutrition 2017, 38, 74–79. [Google Scholar] [CrossRef]
- Sutherland, H.G.; Hermile, H.; Sanche, R.; Menon, S.; Lea, R.A.; Haupt, L.M.; Griffiths, L.R. Association study of MTHFD1 coding polymorphisms R134K and R653Q with migraine susceptibility. Headache 2014, 54, 1506–1514. [Google Scholar] [CrossRef] [Green Version]
- Liew, S.C.; Gupta, E.D. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: Epidemiology, metabolism and the associated diseases. Eur. J. Med. Genet. 2015, 58, 1–10. [Google Scholar] [CrossRef]
- McNulty, H.; Dowey le, R.C.; Strain, J.J.; Dunne, A.; Ward, M.; Molloy, A.M.; McAnena, L.B.; Hughes, J.P.; Hannon-Fletcher, M.; Scott, J.M. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C->T polymorphism. Circulation 2006, 113, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Isobe, C.; Terayama, Y. A remarkable increase in total homocysteine concentrations in the CSF of migraine patients with aura. Headache 2010, 50, 1561–1569. [Google Scholar] [CrossRef]
- Sadeghi, O.; Maghsoudi, Z.; Askari, G.; Khorvash, F.; Feizi, A. Association between serum levels of homocysteine with characteristics of migraine attacks in migraine with aura. J. Res. Med. Sci. 2014, 19, 1041–1045. [Google Scholar] [PubMed]
- Ho, P.I.; Ortiz, D.; Rogers, E.; Shea, T.B. Multiple aspects of homocysteine neurotoxicity: Glutamate excitotoxicity, kinase hyperactivation and DNA damage. J. Neurosci. Res. 2002, 70, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.; Penix, L.; Mattson, M.P. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Res. Med. Sci. 2000, 20, 6920–6926. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Mattiuzzi, C.; Meschi, T.; Cervellin, G.; Borghi, L. Homocysteine and migraine. A narrative review. Clin. Chim. Acta 2014, 433, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Meijers, J.M.; van Bokhorst-de van der Schueren, M.A.; Schols, J.M.; Soeters, P.B.; Halfens, R.J. Defining malnutrition: Mission or mission impossible? Nutrition 2010, 26, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Stratton, R.J.; Hackston, A.; Longmore, D.; Dixon, R.; Price, S.; Stroud, M.; King, C.; Elia, M. Malnutrition in hospital outpatients and inpatients: Prevalence, concurrent validity and ease of use of the ‘malnutrition universal screening tool’ (‘MUST’) for adults. Br. J. Nutr. 2004, 92, 799–808. [Google Scholar] [CrossRef]
- Trinh, K.V.; Diep, D.; Chen, K.J.Q. Systematic Review of Episodic Migraine Prophylaxis: Efficacy of Conventional Treatments Used in Comparisons with Acupuncture. Med. Acupunct. 2019, 31, 85–97. [Google Scholar] [CrossRef]
- Silberstein, S.D. Preventive Migraine Treatment. Continuum (Minneap Minn) 2015, 21, 973–989. [Google Scholar] [CrossRef]
- Ramachandran, R. Neurogenic inflammation and its role in migraine. Semin. Immunopathol. 2018, 40, 301–314. [Google Scholar] [CrossRef]
- Johannessen Landmark, C. Antiepileptic drugs in non-epilepsy disorders: Relations between mechanisms of action and clinical efficacy. CNS Drugs 2008, 22, 27–47. [Google Scholar] [CrossRef]
- Detich, N.; Bovenzi, V.; Szyf, M. Valproate induces replication-independent active DNA demethylation. J. Biol. Chem. 2003, 278, 27586–27592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milutinovic, S.; D’Alessio, A.C.; Detich, N.; Szyf, M. Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis 2007, 28, 560–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Tian, Y.; Chlenski, A.; Salwen, H.R.; Lu, Z.; Raj, J.U.; Yang, Q. Valproic acid shows a potent antitumor effect with alteration of DNA methylation in neuroblastoma. Anticancer Drugs 2012, 23, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, G.; Kim, S.H.; Chang, M.J. Effect of ketogenic diet and other dietary therapies on anti-epileptic drug concentrations in patients with epilepsy. J. Clin. Pharm. Ther. 2017, 42, 758–764. [Google Scholar] [CrossRef]
- Ki, S.; Kwon, S.H.; Eum, J.; Raslan, A.A.; Kim, K.N.; Hwang, B.J.; Kee, Y. 3D light-sheet assay assessing novel valproate-associated cardiotoxicity and folic acid relief in zebrafish embryogenesis. Chemosphere 2019, 227, 551–560. [Google Scholar] [CrossRef]
- Ornoy, A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod. Toxicol. (Elmsford, N. Y.) 2009, 28, 1–10. [Google Scholar] [CrossRef]
- Shona, S.I.; Rizk, A.A.; El Sadik, A.O.; Emam, H.Y.; Ali, E.N. Effect of valproic acid administration during pregnancy on postnatal development of cerebellar cortex and the possible protective role of folic acid. Folia Morphol. 2018, 77, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, K.; Stuber, T.; Rehn, M.; Frieauff, E. Fetal Valproate Syndrome - Still a Problem Today! Zeitschrift fur Geburtshilfe und Neonatologie 2017, 221, 243–246. [Google Scholar] [CrossRef]
- Krushkal, J.; Zhao, Y.; Hose, C.; Monks, A.; Doroshow, J.H.; Simon, R. Concerted changes in transcriptional regulation of genes involved in DNA methylation, demethylation, and folate-mediated one-carbon metabolism pathways in the NCI-60 cancer cell line panel in response to cancer drug treatment. Clin. Epigenetics 2016, 8, 73. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Full Name | Reference | |
|---|---|---|
| SH2D5 | SH2 domain containing 5 | [72] |
| COMT * | catechol-O-methyltransferase | [73] |
| ZNF234 | zinc finger protein 234 | [73] |
| SOCS1 | suppressor of cytokine signaling 1 | [73] |
| SLC2A9, SLC38A4, SLC6A5 | solute carrier family 2,38A,6A member 9,4,5 | [76] |
| DGKG | diacylglycerol kinase gamma | [76] |
| KIF26A | kinesin family member 26A | [76] |
| DOCK6 | dedicator of cytokinesis 6 | [76] |
| CFD | complement factor D | [76] |
| RAMP1 * | receptor activity modifying protein 1 | [77] |
| CGRP * | calcitonin gene related peptide | [80] |
| CRCP *1) | CGRP receptor component | [80] |
| CALCRL *1) | calcitonin receptor like receptor | [80] |
| ESR1 *1) | estrogen receptor 1 | [80] |
| NOS3 *1) | nitric oxide synthase 3 | [80] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fila, M.; Chojnacki, C.; Chojnacki, J.; Blasiak, J. Is an “Epigenetic Diet” for Migraines Justified? The Case of Folate and DNA Methylation. Nutrients 2019, 11, 2763. https://doi.org/10.3390/nu11112763
Fila M, Chojnacki C, Chojnacki J, Blasiak J. Is an “Epigenetic Diet” for Migraines Justified? The Case of Folate and DNA Methylation. Nutrients. 2019; 11(11):2763. https://doi.org/10.3390/nu11112763
Chicago/Turabian StyleFila, Michal, Cezary Chojnacki, Jan Chojnacki, and Janusz Blasiak. 2019. "Is an “Epigenetic Diet” for Migraines Justified? The Case of Folate and DNA Methylation" Nutrients 11, no. 11: 2763. https://doi.org/10.3390/nu11112763
APA StyleFila, M., Chojnacki, C., Chojnacki, J., & Blasiak, J. (2019). Is an “Epigenetic Diet” for Migraines Justified? The Case of Folate and DNA Methylation. Nutrients, 11(11), 2763. https://doi.org/10.3390/nu11112763