mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance

 , , ,
, , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Models
2.2. Isolation of Hepatic Stellate Cells
2.3. Histology and Immunohistochemistry
2.4. Transcriptome Analysis on Hepatic Stellate Cells (Discovery)
2.5. Quantitative Real Time PCR (qRT-PCR) Validation of miRNAs in Total Livers
2.6. Bioinformatic Resources
2.7. In Vitro Treatments
2.8. Cell Proliferation Assay
2.9. Wound Healing Assay In Vitro
2.10. Statistical Analysis
3. Results
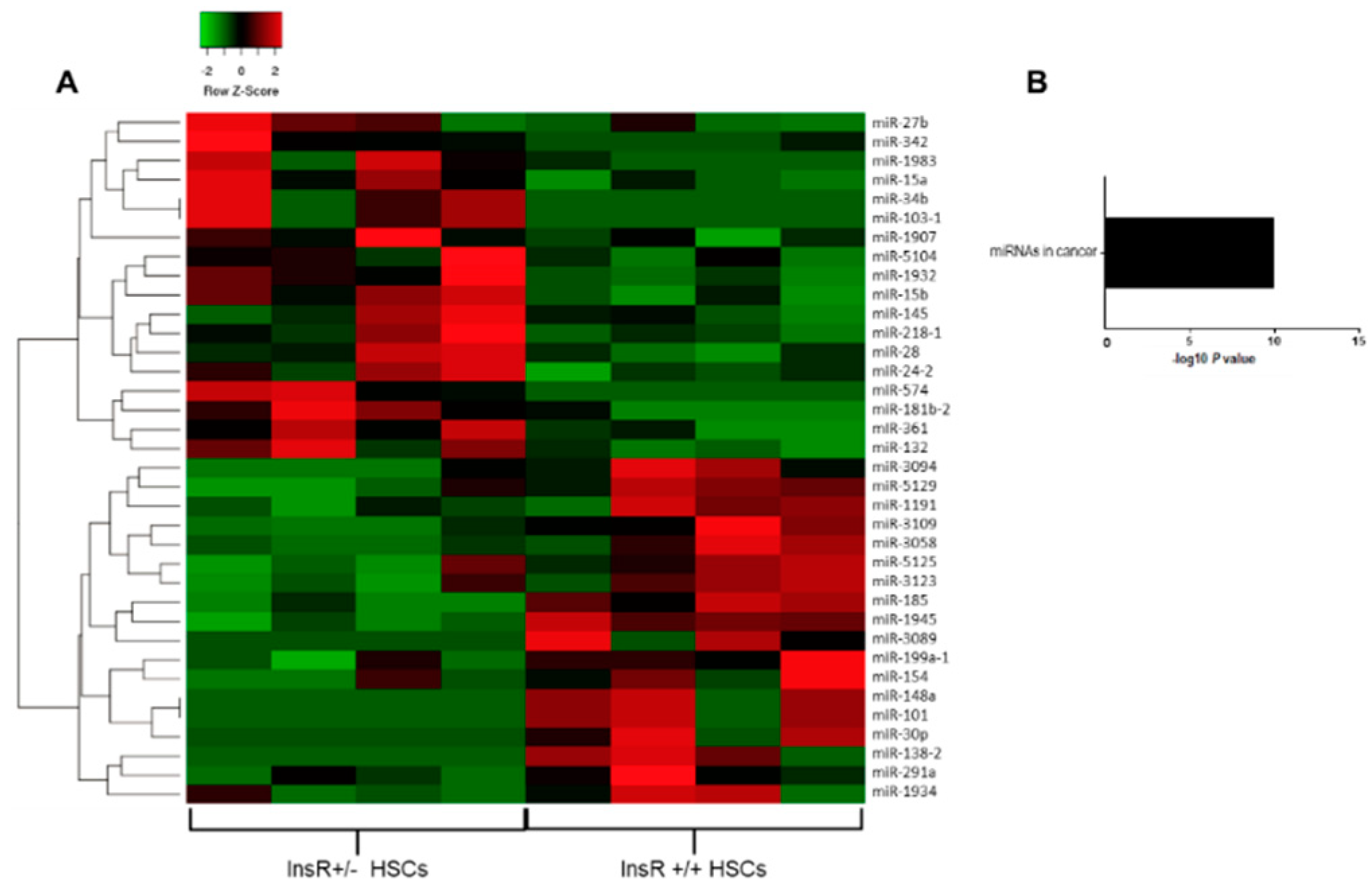
3.1. Hepatic Stellate Cells Isolated from InsR+/− Mice Show a Distinctive Pattern of miRNA Expression
3.2. Differentially Expressed Hepatic miRNAs in InsR+/− Mice
3.3. Pathway-Enriched Analysis of Differentially Expressed miRNAs in HSCs
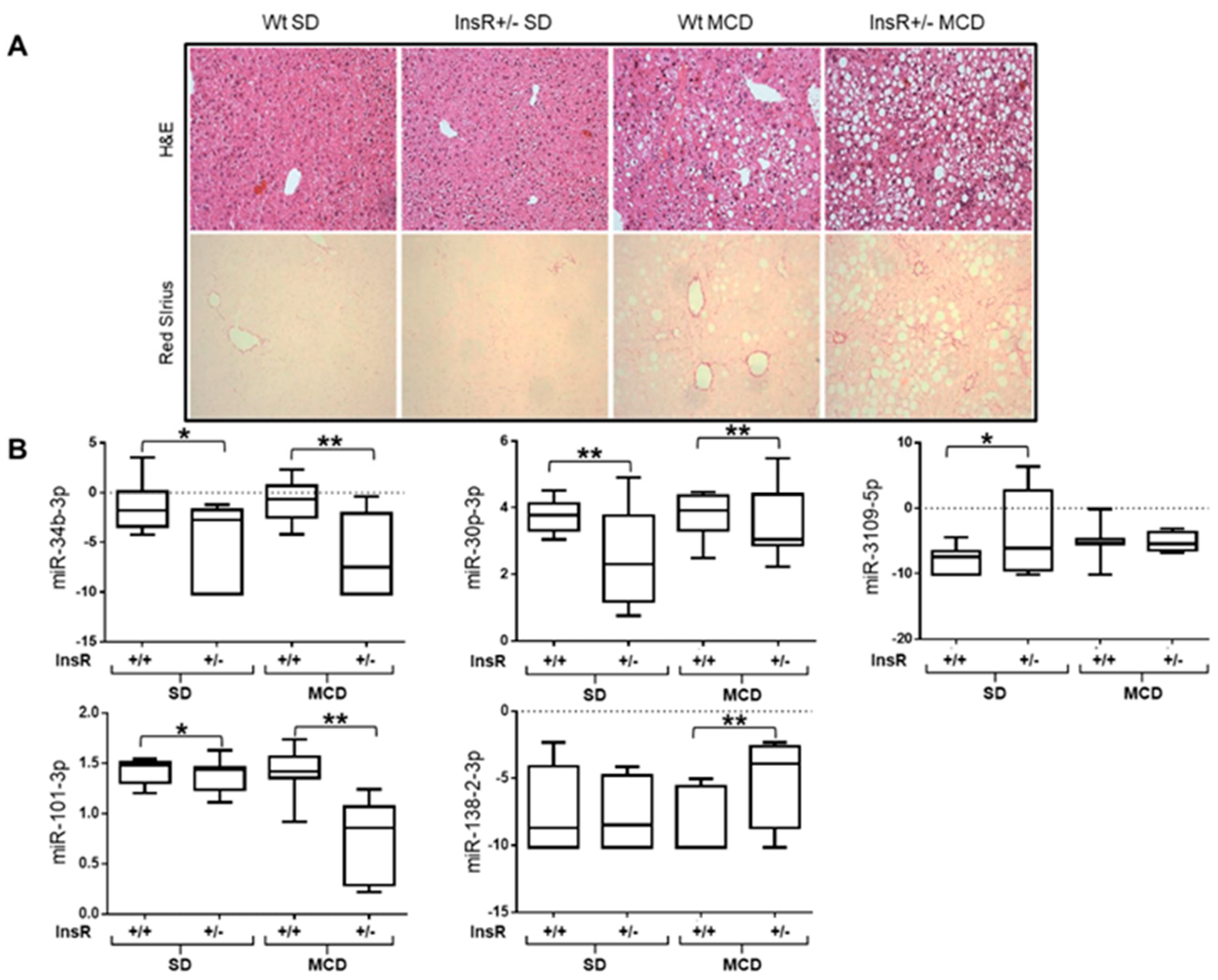
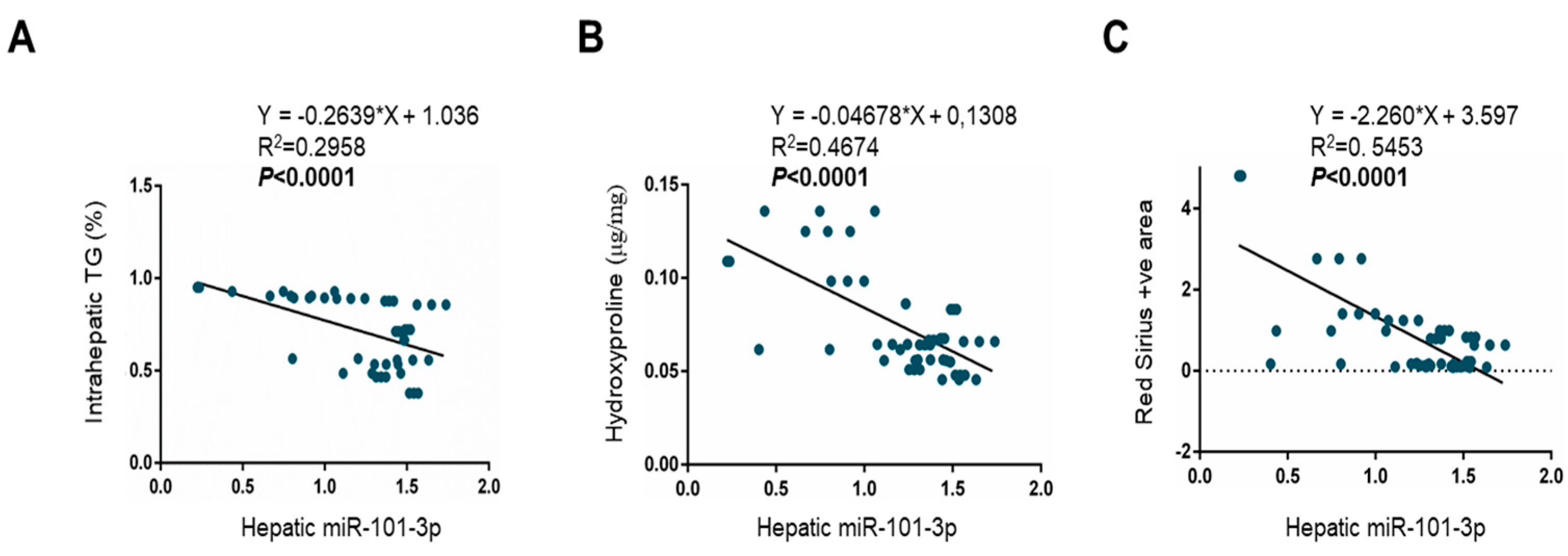
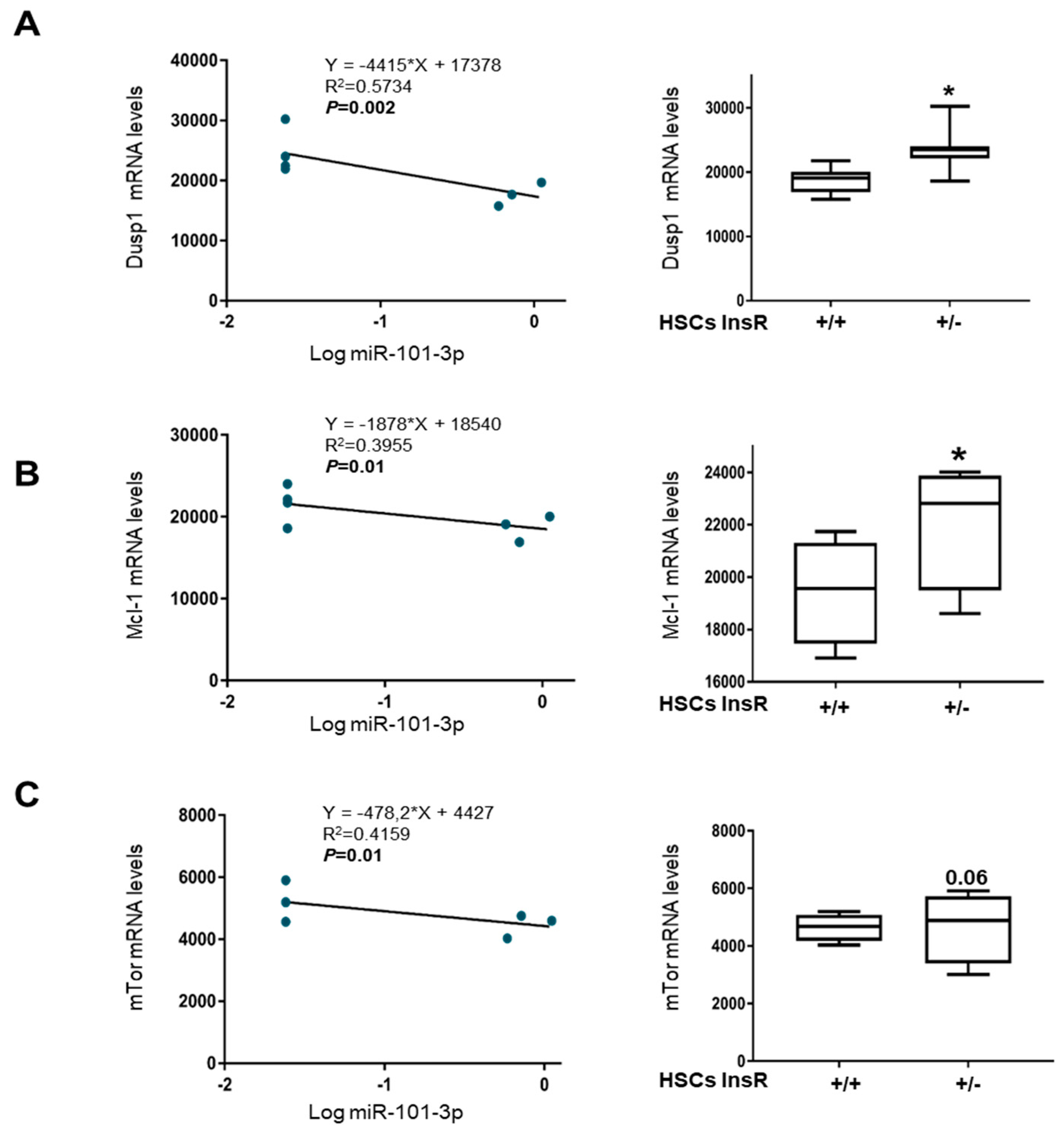
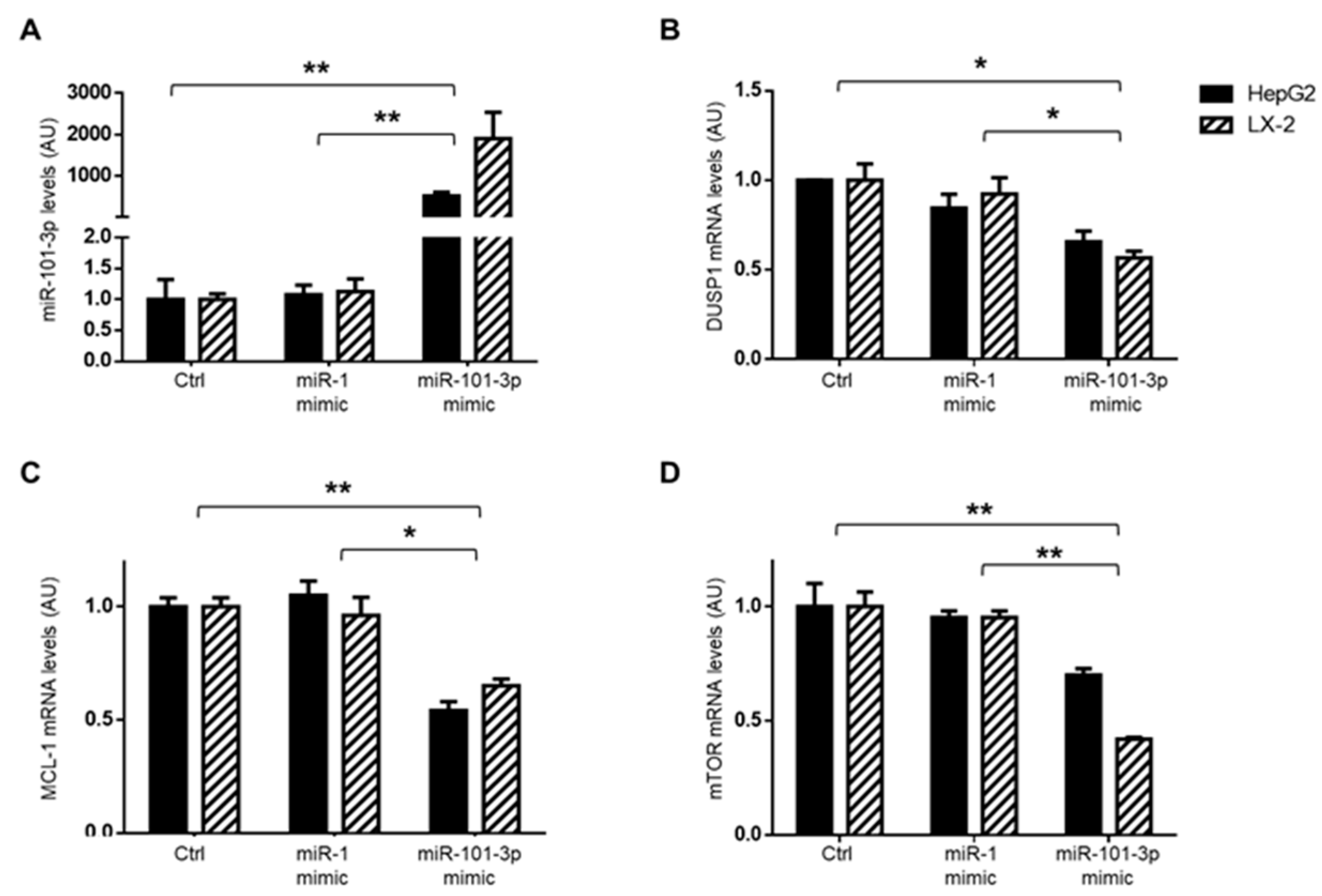
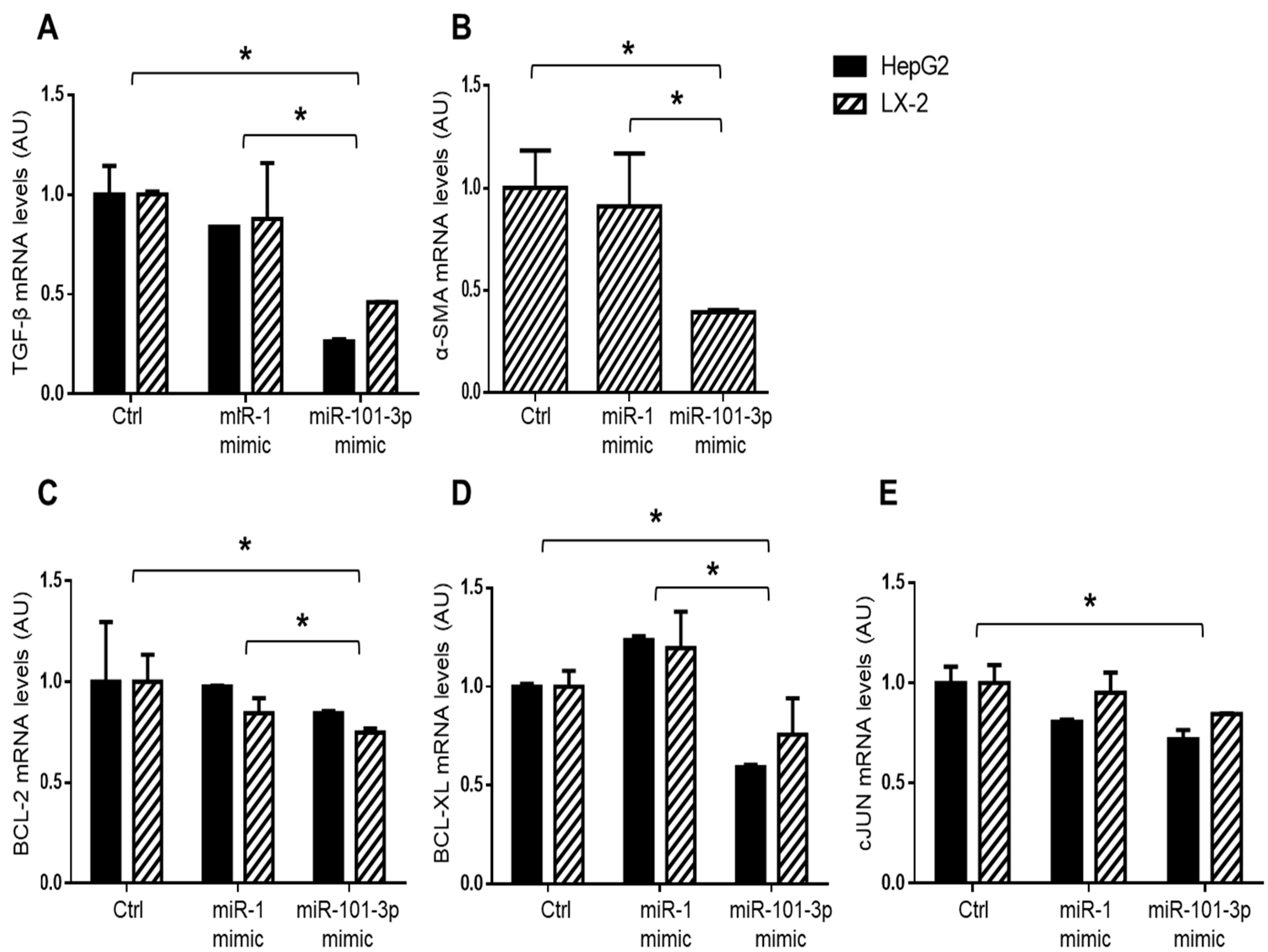
3.4. miR-101-3p and Pathway-Related Targets Involved in Fibrosis and Cell Proliferation in HSCs
3.5. miR-101-3p was Down-Regulated in HepG2 and LX-2 Cells
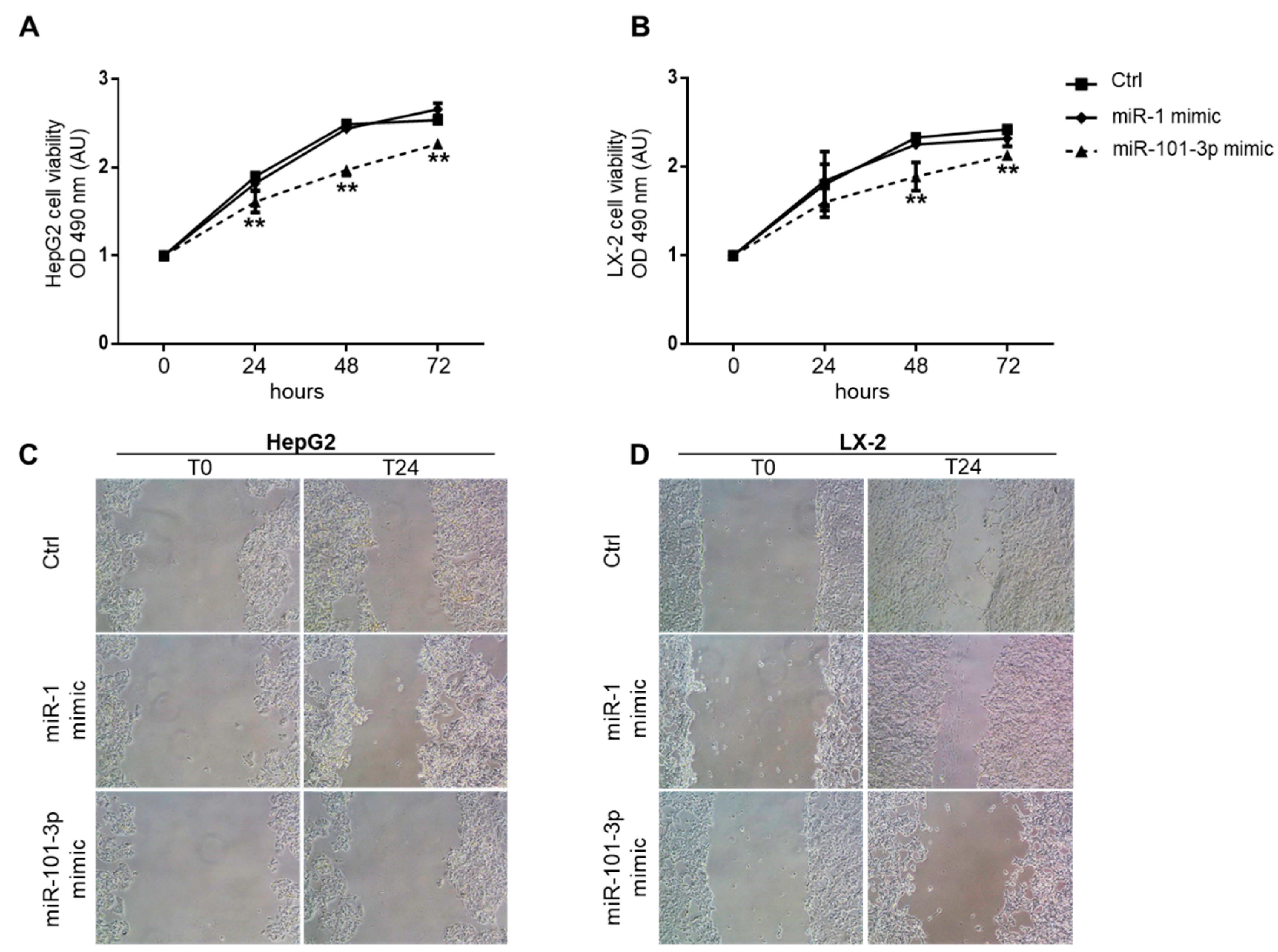
3.6. Overexpression of miR-101-3p Inhibited HepG2 and LX-2 Cell Proliferation and Migration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L. A Nutrigenomic Approach to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2017, 18, 1534. [Google Scholar] [Green Version]
- Byrne, D.C.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62 (Suppl. 1), S47–S64. [Google Scholar] [CrossRef] [Green Version]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Meroni, M.; Baselli, G.A.; Bassani, G.A.; Rametta, R.; Pietrelli, A.; Maggioni, M.; Facciotti, F.; Trunzo, V.; Badiali, S.; et al. Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin. Sci. 2017, 131, 1301–1315. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Panera, N.; Gnani, D.; Crudele, A.; Ceccarelli, S.; Nobili, V.; Alisi, A. MicroRNAs as controlled systems and controllers in non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15079–15086. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Fernández Gianotti, T.; Castaño, G.O.; Mallardi, P.; San Martino, J.; Mora Gonzalez Lopez Ledesma, M.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S.; et al. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Accili, D.; Drago, J.; Lee, E.J.; Johnson, M.D.; Cool, M.H.; Salvatore, P.; Asico, L.D.; José, P.A.; Taylor, S.; Westphal, H.; et al. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat. Genet. 1996, 12, 106–109. [Google Scholar] [CrossRef]
- Nakae, J.; Biggs, W.H.; Kitamura, T.; Cavenee, W.K.; Wright, C.V.; Arden, K.C.; Accili, D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat. Genet. 2002, 32, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Nakae, J.; Kitamura, T.; Park, B.C.; Dragatsis, I.; Accili, D. Transgenic rescue of insulin receptor-deficient mice. J. Clin. Invest. 2004, 114, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.Y.; Gonzalez-Martin, A.; Miletic, A.V.; Lai, M.; Knight, S.; Sabouri-Ghomi, M.; Head, S.R.; Macauley, M.S.; Rickert, R.C.; Xiao, C. Transfection of microRNA Mimics Should Be Used with Caution. Front Genet. 2015, 6, 340. [Google Scholar] [CrossRef] [Green Version]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G.; et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatology 2008, 48, 792–798. [Google Scholar] [CrossRef]
- Mofrad, P.; Contos, M.J.; Haque, M.; Sargeant, C.; Fisher, R.A.; Luketic, V.A.; Sterling, R.K.; Shiffman, M.L.; Stravitz, R.T.; Sanyal, A.J. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003, 37, 1286–1292. [Google Scholar] [CrossRef]
- Maximos, M.; Bril, F.; Portillo Sanchez, P.; Lomonaco, R.; Orsak, B.; Biernacki, D.; Suman, A.; Weber, M.; Cusi, K. The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease. Hepatology 2015, 61, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.R.; Langlet, F.; Kido, Y.; Accili, D. Pathogenesis of selective insulin resistance in isolated hepatocytes. J. Biol. Chem. 2015, 290, 13972–13980. [Google Scholar] [CrossRef]
- Chakraborty, C.; Doss, C.G.; Bandyopadhyay, S.; Agoramoorthy, G. Influence of miRNA in insulin signaling pathway and insulin resistance: Micro-molecules with a major role in type-2 diabetes. Wiley Interdiscip. Rev. RNA 2014, 5, 697–712. [Google Scholar] [CrossRef]
- Li, C.Y.; Pang, Y.Y.; Yang, H.; Li, J.; Lu, H.X.; Wang, H.L.; Mo, W.J.; Huang, L.S.; Feng, Z.B.; Chen, G. Identification of miR-101-3p targets and functional features based on bioinformatics, meta-analysis and experimental verification in hepatocellular carcinoma. Am. J. Transl. Res. 2017, 9, 2088–2105. [Google Scholar]
- Wroblewski, A.; Strycharz, J.; Świderska, E.; Drewniak, K.; Drzewoski, J.; Szemraj, J.; Kasznicki, J.; Śliwińska, A. Molecular Insight into the Interaction between Epigenetics and Leptin in Metabolic Disorders. Nutrients 2019, 11, 1872. [Google Scholar] [Green Version]
- Xu, X.; Tao, Y.; Shan, L.; Chen, R.; Jiang, H.Y.; Qian, Z.J.; Cai, F.; Ma, L.F.; Yu, Y.C. The Role of MicroRNAs in Hepatocellular Carcinoma. J. Cancer 2018, 9, 3557–3569. [Google Scholar] [CrossRef]
- Van Herck, A.M.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease—A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Rametta, R.; Meroni, M.; Valenti, L. The role of insulin resistance in nonalcoholic steatohepatitis and liver disease development—A potential therapeutic target? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 229–242. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, Z.; Tu, Y.; Shen, H.; Dai, Z.; Lin, J.; Zhou, Z. miR-101a and miR-30b contribute to inflammatory cytokine-mediated beta-cell dysfunction. Lab. Investig. 2015, 95, 1387–1397. [Google Scholar] [CrossRef]
- Higuchi, C.; Nakatsuka, A.; Eguchi, J.; Teshigawara, S.; Kanzaki, M.; Katayama, A.; Yamaguchi, S.; Takahashi, N.; Murakami, K.; Ogawa, D.; et al. Identification of circulating miR-101, miR-375 and miR-802 as biomarkers for type 2 diabetes. Metabolism 2015, 64, 489–497. [Google Scholar] [CrossRef]
- Santos, A.S.; Cunha Neto, E.; Fukui, R.T.; Ferreira, L.R.P.; Silva, M.E.R. Increased Expression of Circulating microRNA 101-3p in Type 1 Diabetes Patients: New Insights Into miRNA-Regulated Pathophysiological Pathways for Type 1 Diabetes. Front. Immunol. 2019, 10, 1637. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.Y.; Liu, Q.; Chen, J.X.; Lan, K.; Ge, B.X. MicroRNA-101 targets MAPK phosphatase-1 to regulate the activation of MAPKs in macrophages. J. Immunol. 2010, 185, 7435–7442. [Google Scholar] [CrossRef]
- Wang, H.; Meng, Y.; Cui, Q.; Qin, F.; Yang, H.; Chen, Y.; Cheng, Y.; Shi, J.; Guo, Y. MiR-101 Targets the EZH2/Wnt/beta-Catenin the Pathway to Promote the Osteogenic Differentiation of Human Bone Marrow-Derived Mesenchymal Stem Cells. Sci. Rep. 2016, 6, 36988. [Google Scholar]
- Tu, X.; Zhang, H.; Zhang, J.; Zhao, S.; Zheng, X.; Zhang, Z.; Zhu, J.; Chen, J.; Dong, L.; Zang, Y.; et al. MicroRNA-101 suppresses liver fibrosis by targeting the TGFbeta signalling pathway. J. Pathol. 2014, 234, 46–59. [Google Scholar] [CrossRef]
- Huang, C.; Xiao, X.; Yang, Y.; Mishra, A.; Liang, Y.; Zeng, X.; Yang, X.; Xu, D.; Blackburn, M.R.; Henke, C.A.; et al. MicroRNA-101 attenuates pulmonary fibrosis by inhibiting fibroblast proliferation and activation. J. Biol. Chem. 2017, 292, 16420–16439. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Sun, X.; Shan, H.; Wang, N.; Wang, J.; Ren, J.; Feng, S.; Xie, L.; Lu, C.; Yuan, Y.; et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-beta1 pathway. Circulation 2012, 126, 840–850. [Google Scholar] [CrossRef]
- Su, H.; Yang, J.R.; Xu, T.; Huang, J.; Xu, L.; Yuan, Y.; Zhuang, S.M. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009, 69, 1135–1142. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Liu, Y.; Wu, W.; Li, X.; Wu, Y.; Chen, H.; Zhang, K.; Gu, L. miR-101 represses lung cancer by inhibiting interaction of fibroblasts and cancer cells by down-regulating CXCL12. Biomed. Pharmacother. 2015, 74, 215–221. [Google Scholar] [CrossRef]
- He, H.; Tian, W.; Chen, H.; Deng, Y. MicroRNA-101 sensitizes hepatocellular carcinoma cells to doxorubicin-induced apoptosis via targeting Mcl-1. Mol. Med. Rep. 2016, 13, 1923–1929. [Google Scholar] [CrossRef]
- Konno, Y.; Dong, P.; Xiong, Y.; Suzuki, F.; Lu, J.B.; Cai, M.Y.; Watari, H.; Mitamura, Y.; Hosaka, M.; Hanley Sharon, J.B.; et al. MicroRNA-101 targets EZH2, MCL-1 and FOS to suppress proliferation, invasion and stem cell-like phenotype of aggressive endometrial cancer cells. Oncotarget 2014, 5, 6049–6062. [Google Scholar] [CrossRef]
- Xu, Y.; An, Y.; Wang, Y.; Zhang, C.; Zhang, H.; Huang, C.; Jiang, H.; Wang, X.; Li, X. miR-101 inhibits autophagy and enhances cisplatin-induced apoptosis in hepatocellular carcinoma cells. Oncol. Rep. 2013, 29, 2019–2024. [Google Scholar] [CrossRef]
- Xu, L.; Beckebaum, S.; Iacob, S.; Wu, G.; Kaiser, G.M.; Radtke, A.; Liu, C.; Kabar, I.; Schmidt, H.H.; Zhang, X.; et al. MicroRNA-101 inhibits human hepatocellular carcinoma progression through EZH2 downregulation and increased cytostatic drug sensitivity. J. Hepatol. 2014, 60, 590–598. [Google Scholar] [CrossRef]
- Hu, J.; Wu, C.; Zhao, X.; Liu, C. The prognostic value of decreased miR-101 in various cancers: A meta-analysis of 12 studies. Onco. Targets Ther. 2017, 10, 3709–3718. [Google Scholar] [CrossRef]
- Varambally, S.; Cao, Q.; Mani, R.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef]
- Wang, R.; Wang, H.B.; Hao, C.J.; Cui, Y.; Han, X.C.; Hu, Y.; Li, F.F.; Xia, H.F.; Ma, X. MiR-101 is involved in human breast carcinogenesis by targeting Stathmin1. PLoS ONE 2012, 7, e46173. [Google Scholar] [CrossRef]
- Zheng, F.; Liao, Y.J.; Cai, M.Y.; Liu, T.H.; Chen, S.P.; Wu, P.H.; Wu, L.; Bian, X.W.; Guan, X.Y.; Zeng, Y.X.; et al. Systemic delivery of microRNA-101 potently inhibits hepatocellular carcinoma in vivo by repressing multiple targets. PLoS Genet. 2015, 11, e1004873. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAs | InsR+/− Genotype | MCD Diet | Interaction (InsR+/− * MCD) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| β | 95% CI | p * | β | 95% CI | p * | β | 95% CI | p * | |
| miR-34b-3p | −1.71 | −2.54 to −0.89 | 0.0001 | −0.23 | −1.06 to 0.59 | 0.57 | −0.41 | −0.24 to 0.41 | 0.32 |
| miR-101b-3p | 0.13 | −0.21 to −0.05 | 0.0009 | −0.15 | −0.23 to −0.07 | 0.0002 | −0.15 | −0.23 to −0.08 | 0.0001 |
| miR-30p-3p | −0.28 | −0.55 to −0.02 | 0.03 | 0.17 | −0.09 to 0.44 | 0.19 | 0.33 | 0.06 to 0.59 | 0.01 |
| miR-3109-5p | 0.92 | 0.02 to 1.84 | 0.04 | 0.29 | −0.61 to 1.20 | 0.51 | −0.81 | −1.71 to 0.09 | 0.07 |
| miR-138-2-3p | 0.68 | −0.07 to 1.43 | 0.07 | 0.47 | −0.28 to 1.22 | 0.22 | 0.91 | 0.15 to 1.66 | 0.02 |
| Steatosis | Lobular Inflammation | Fibrosis | NAS | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | 95% CI | p * | β | 95% CI | p * | β | 95% CI | p * | β | 95% CI | p * | |
| miR-34b-3p | −2.20 | −3.71 to −0.66 | 0.006 | −2.50 | −4.30 to −0.66 | 0.009 | −0.54 | −2.13 to 1.03 | 0.49 | −1.41 | −2.30 to −0.51 | 0.003 |
| miR-101-3p | −0.16 | −0.33 to 0.001 | 0.05 | −0.21 | −0.41 to −0.02 | 0.03 | −0.30 | −0.44 to −0.15 | 0.0001 | −0.12 | −0.21 to −0.01 | 0.02 |
| miR-30p-3p | −0.33 | −0.86 to 0.19 | 0.22 | 0.26 | −0.37 to 0.90 | 0.41 | −0.19 | −0.71 to 0.32 | 0.45 | −0.05 | −0.37 to 0.27 | 0.74 |
| miR-3109-5p | 0.23 | −1.47 to 1.95 | 0.78 | −0.52 | −3.52 to 0.47 | 0.13 | −0.16 | −1.83 to 1.5 | 0.84 | −0.30 | −1.32 to 0.72 | 0.56 |
| miR-138-2-3p | 0.19 | 1.30 to 1.69 | 0.79 | 0.78 | −0.98 to 2.54 | 0.34 | 1.37 | −0.03 to 2.77 | 0.05 | 0.27 | −0.62 to 1.16 | 0.55 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meroni, M.; Longo, M.; Erconi, V.; Valenti, L.; Gatti, S.; Fracanzani, A.L.; Dongiovanni, P. mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance. Nutrients 2019, 11, 2597. https://doi.org/10.3390/nu11112597
Meroni M, Longo M, Erconi V, Valenti L, Gatti S, Fracanzani AL, Dongiovanni P. mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance. Nutrients. 2019; 11(11):2597. https://doi.org/10.3390/nu11112597
Chicago/Turabian StyleMeroni, Marica, Miriam Longo, Veronica Erconi, Luca Valenti, Stefano Gatti, Anna Ludovica Fracanzani, and Paola Dongiovanni. 2019. "mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance" Nutrients 11, no. 11: 2597. https://doi.org/10.3390/nu11112597
APA StyleMeroni, M., Longo, M., Erconi, V., Valenti, L., Gatti, S., Fracanzani, A. L., & Dongiovanni, P. (2019). mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance. Nutrients, 11(11), 2597. https://doi.org/10.3390/nu11112597