Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later?

 ,
,  , and
, and 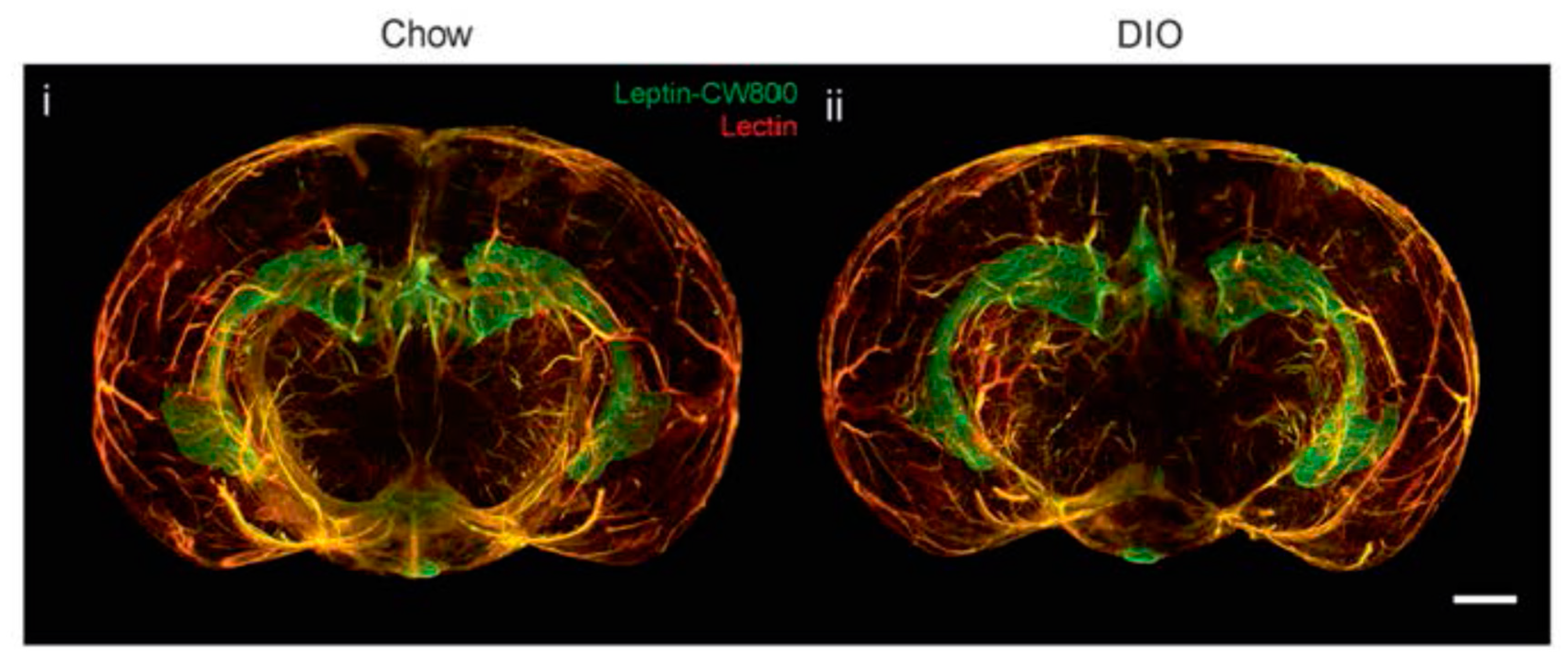{kind=link}
{kind=link}
Abstract
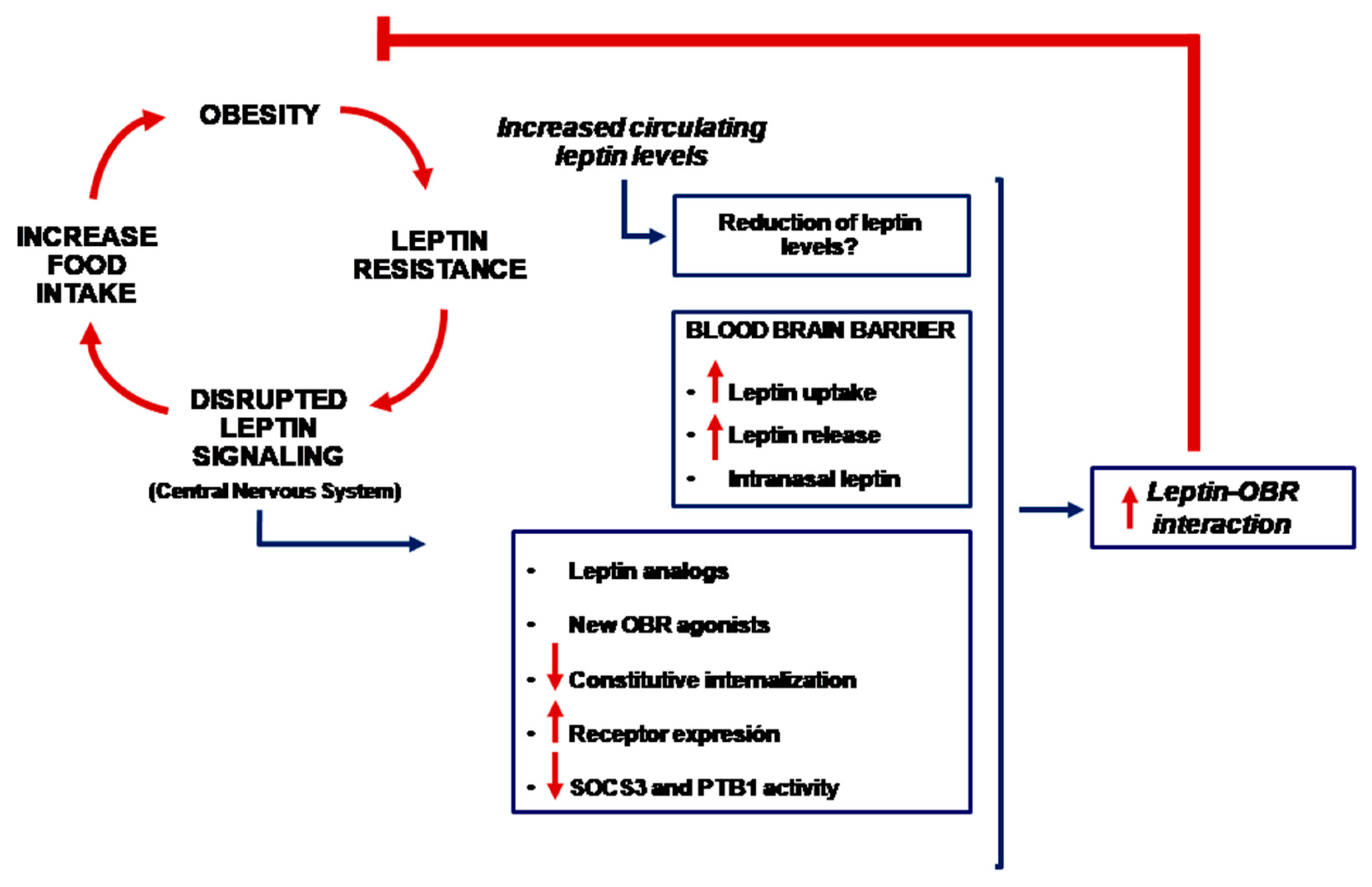
1. Introduction
2. Blood–Brain Barrier and Obesity
3. Leptin, Brain and Blood-Brain Barrier
4. Is It Possible to Use Leptin for the Treatment of Obesity?
5. Other Potential Therapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef]
- Woods, S.C.; Schwartz, M.W.; Baskin, D.G.; Seeley, R.J. Food intake and the regulation of body weight. Annu. Rev. Psychol. 2000, 51, 255–277. [Google Scholar] [CrossRef]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Bjorbaek, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Chen, K.; Li, F.; Li, J.; Cai, H.; Strom, S.; Bisello, A.; Kelley, D.E.; Friedman-Einat, M.; Skibinski, G.A.; McCrory, M.A.; et al. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat. Med. 2006, 12, 425–432. [Google Scholar] [CrossRef]
- Kabra, D.G.; Pfuhlmann, K.; Garcia-Caceres, C.; Schriever, S.C.; Casquero Garcia, V.; Kebede, A.F.; Fuente-Martin, E.; Trivedi, C.; Heppner, K.; Uhlenhaut, N.H.; et al. Hypothalamic leptin action is mediated by histone deacetylase 5. Nat. Commun. 2016, 7, 10782. [Google Scholar] [CrossRef]
- Rodriguez, E.M.; Blazquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776. [Google Scholar] [CrossRef]
- Banks, W.A.; Coon, A.B.; Robinson, S.M.; Moinuddin, A.; Shultz, J.M.; Nakaoke, R.; Morley, J.E. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes 2004, 53, 1253–1260. [Google Scholar] [CrossRef]
- Banks, W.A.; DiPalma, C.R.; Farrell, C.L. Impaired transport of leptin across the blood-brain barrier in obesity. Peptides 1999, 20, 1341–1345. [Google Scholar] [CrossRef]
- El-Haschimi, K.; Pierroz, D.D.; Hileman, S.M.; Bjorbaek, C.; Flier, J.S. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Investig. 2000, 105, 1827–1832. [Google Scholar] [CrossRef]
- Banks, W.A. Peptides and the blood-brain barrier. Peptides 2015, 72. [Google Scholar] [CrossRef]
- Moraes, J.C.; Coope, A.; Morari, J.; Cintra, D.E.; Roman, E.A.; Pauli, J.R.; Romanatto, T.; Carvalheira, J.B.; Oliveira, A.L.; Saad, M.J.; et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS ONE 2009, 4, e504. [Google Scholar] [CrossRef]
- Kim, D.W.; Glendining, K.A.; Grattan, D.R.; Jasoni, C.L. Maternal Obesity in the Mouse Compromises the Blood-Brain Barrier in the Arcuate Nucleus of Offspring. Endocrinology 2016, 157, 2229–2242. [Google Scholar] [CrossRef]
- Van Heek, M.; Compton, D.S.; France, C.F.; Tedesco, R.P.; Fawzi, A.B.; Graziano, M.P.; Sybertz, E.J.; Strader, C.D.; Davis, H.R., Jr. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J. Clin. Investig. 1997, 99, 385–390. [Google Scholar] [CrossRef]
- Halaas, J.L.; Boozer, C.; Blair-West, J.; Fidahusein, N.; Denton, D.A.; Friedman, J.M. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8878–8883. [Google Scholar] [CrossRef]
- Caro, J.F.; Kolaczynski, J.W.; Nyce, M.R.; Ohannesian, J.P.; Opentanova, I.; Goldman, W.H.; Lynn, R.B.; Zhang, P.L.; Sinha, M.K.; Considine, R.V. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: A possible mechanism for leptin resistance. Lancet 1996, 348, 159–161. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Peskind, E.; Raskind, M.; Boyko, E.J.; Porte, D., Jr. Cerebrospinal fluid leptin levels: Relationship to plasma levels and to adiposity in humans. Nat. Med. 1996, 2, 589–593. [Google Scholar] [CrossRef]
- Faouzi, M.; Leshan, R.; Bjornholm, M.; Hennessey, T.; Jones, J.; Munzberg, H. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology 2007, 148, 5414–5423. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Huang, W.; Jaspan, J.B.; Maness, L.M. Leptin enters the brain by a saturable system independent of insulin. Peptides 1996, 17, 305–311. [Google Scholar] [CrossRef]
- Balland, E.; Dam, J.; Langlet, F.; Caron, E.; Steculorum, S.; Messina, A.; Rasika, S.; Falluel-Morel, A.; Anouar, Y.; Dehouck, B.; et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014, 19, 293–301. [Google Scholar] [CrossRef]
- Pan, W.; Hsuchou, H.; He, Y.; Sakharkar, A.; Cain, C.; Yu, C.; Kastin, A.J. Astrocyte leptin receptor (ObR) and leptin transport in adult-onset obese mice. Endocrinology 2008, 149, 2798–2806. [Google Scholar] [CrossRef]
- Hileman, S.M.; Tornoe, J.; Flier, J.S.; Bjorbaek, C. Transcellular transport of leptin by the short leptin receptor isoform ObRa in Madin-Darby Canine Kidney cells. Endocrinology 2000, 141, 1955–1961. [Google Scholar] [CrossRef]
- Boado, R.J.; Golden, P.L.; Levin, N.; Pardridge, W.M. Up-regulation of blood-brain barrier short-form leptin receptor gene products in rats fed a high fat diet. J. Neurochem. 1998, 71, 1761–1764. [Google Scholar] [CrossRef]
- Bjorbaek, C.; Elmquist, J.K.; Michl, P.; Ahima, R.S.; van Bueren, A.; McCall, A.L.; Flier, J.S. Expression of leptin receptor isoforms in rat brain microvessels. Endocrinology 1998, 139, 3485–3491. [Google Scholar] [CrossRef]
- Hileman, S.M.; Pierroz, D.D.; Masuzaki, H.; Bjorbaek, C.; El-Haschimi, K.; Banks, W.A.; Flier, J.S. Characterizaton of short isoforms of the leptin receptor in rat cerebral microvessels and of brain uptake of leptin in mouse models of obesity. Endocrinology 2002, 143, 775–783. [Google Scholar] [CrossRef]
- Banks, W.A.; Niehoff, M.L.; Martin, D.; Farrell, C.L. Leptin transport across the blood-brain barrier of the Koletsky rat is not mediated by a product of the leptin receptor gene. Brain Res. 2002, 950, 130–136. [Google Scholar] [CrossRef]
- Maness, L.M.; Banks, W.A.; Kastin, A.J. Persistence of blood-to-brain transport of leptin in obese leptin-deficient and leptin receptor-deficient mice. Brain Res. 2000, 873, 165–167. [Google Scholar] [CrossRef]
- Banks, W.A.; Clever, C.M.; Farrell, C.L. Partial saturation and regional variation in the blood-to-brain transport of leptin in normal weight mice. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E1158–E1165. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Woods, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef]
- Ottaway, N.; Mahbod, P.; Rivero, B.; Norman, L.A.; Gertler, A.; D’Alessio, D.A.; Perez-Tilve, D. Diet-induced obese mice retain endogenous leptin action. Cell Metab. 2015, 21, 877–882. [Google Scholar] [CrossRef]
- Pan, W.W.; Myers, M.G., Jr. Leptin and the maintenance of elevated body weight. Nat. Rev. Neurosci. 2018, 19, 95–105. [Google Scholar] [CrossRef]
- Kleinert, M.; Kotzbeck, P.; Altendorfer-Kroath, T.; Birngruber, T.; Tschop, M.H.; Clemmensen, C. Time-resolved hypothalamic open flow micro-perfusion reveals normal leptin transport across the blood-brain barrier in leptin resistant mice. Mol. Metab. 2018, 13, 77–82. [Google Scholar] [CrossRef]
- Harrison, L.; Schriever, S.C.; Feuchtinger, A.; Kyriakou, E.; Baumann, P.; Pfuhlmann, K.; Messias, A.C.; Walch, A.; Tschop, M.H.; Pfluger, P.T. Fluorescent blood-brain barrier tracing shows intact leptin transport in obese mice. Int. J. Obes. 2019, 43, 1305–1318. [Google Scholar] [CrossRef]
- Muller, T.D.; Sullivan, L.M.; Habegger, K.; Yi, C.X.; Kabra, D.; Grant, E.; Ottaway, N.; Krishna, R.; Holland, J.; Hembree, J.; et al. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. J. Pept. Sci. 2012, 18, 383–393. [Google Scholar] [CrossRef]
- Elinav, E.; Niv-Spector, L.; Katz, M.; Price, T.O.; Ali, M.; Yacobovitz, M.; Solomon, G.; Reicher, S.; Lynch, J.L.; Halpern, Z.; et al. Pegylated leptin antagonist is a potent orexigenic agent: Preparation and mechanism of activity. Endocrinology 2009, 150, 3083–3091. [Google Scholar] [CrossRef]
- Hukshorn, C.J.; Westerterp-Plantenga, M.S.; Saris, W.H. Pegylated human recombinant leptin (PEG-OB) causes additional weight loss in severely energy-restricted, overweight men. Am. J. Clin. Nutr. 2003, 77, 771–776. [Google Scholar] [CrossRef]
- Yi, X.; Yuan, D.; Farr, S.A.; Banks, W.A.; Poon, C.D.; Kabanov, A.V. Pluronic modified leptin with increased systemic circulation, brain uptake and efficacy for treatment of obesity. J. Control. Release 2014, 191, 34–46. [Google Scholar] [CrossRef]
- Kovalszky, I.; Surmacz, E.; Scolaro, L.; Cassone, M.; Ferla, R.; Sztodola, A.; Olah, J.; Hatfield, M.P.; Lovas, S.; Otvos, L., Jr. Leptin-based glycopeptide induces weight loss and simultaneously restores fertility in animal models. Diabetes Obes. Metab. 2010, 12, 393–402. [Google Scholar] [CrossRef]
- Zhang, C.; Su, Z.; Zhao, B.; Qu, Q.; Tan, Y.; Cai, L.; Li, X. Tat-modified leptin is more accessible to hypothalamus through brain-blood barrier with a significant inhibition of body-weight gain in high-fat-diet fed mice. Exp. Clin. Endocrinol. Diabetes 2010, 118, 31–37. [Google Scholar] [CrossRef]
- Price, T.O.; Farr, S.A.; Yi, X.; Vinogradov, S.; Batrakova, E.; Banks, W.A.; Kabanov, A.V. Transport across the blood-brain barrier of pluronic leptin. J. Pharmacol. Exp. Ther. 2010, 333, 253–263. [Google Scholar] [CrossRef]
- Morath, V.; Bolze, F.; Schlapschy, M.; Schneider, S.; Sedlmayer, F.; Seyfarth, K.; Klingenspor, M.; Skerra, A. PASylation of murine leptin leads to extended plasma half-life and enhanced in vivo efficacy. Mol. Pharm. 2015, 12, 1431–1442. [Google Scholar] [CrossRef]
- Castaigne, J.; Demeule, M.; Boivin, D.; Lawrence, B.; Che, C. Leptin and leptin analog conjugates and uses thereof. U.S. Patent 13/132,838, 24 November 2011. [Google Scholar]
- Castaigne, J.; Demeule, M.; Lawrence, B.; Boivin, D.; Che, C. Leptin and leptin analog conjugates and fusion proteins and uses thereof. WO 153642 A1, 2011. [Google Scholar]
- Grasso, P.; Lee, D.W.; Leinung, M.C. Leptin-related peptides. U.S. Patent No 7,790,683 B2, 2007. [Google Scholar]
- Grasso, P.; Rozhavskaya-Arena, M.; Leinung, M.C.; Lee, D.W. [D-LEU-4]-OB3, a synthetic leptin agonist, improves hyperglycemic control in C57BL/6Job/ob mice. Regul. Pept. 2001, 101, 123–129. [Google Scholar] [CrossRef]
- Leibel, R.L.; Rosenbaum, M.; Hirsch, J. Changes in energy expenditure resulting from altered body weight. New Engl. J. Med. 1995, 332, 621–628. [Google Scholar] [CrossRef]
- Lecoultre, V.; Ravussin, E.; Redman, L.M. The fall in leptin concentration is a major determinant of the metabolic adaptation induced by caloric restriction independently of the changes in leptin circadian rhythms. J. Clin. Endocrinol. Metab. 2011, 96, E1512–E1516. [Google Scholar] [CrossRef]
- Doucet, E.; St Pierre, S.; Almeras, N.; Mauriege, P.; Richard, D.; Tremblay, A. Changes in energy expenditure and substrate oxidation resulting from weight loss in obese men and women: Is there an important contribution of leptin? J. Clin. Endocrinol. Metab. 2000, 85, 1550–1556. [Google Scholar]
- Leibel, R.L. The role of leptin in the control of body weight. Nutr. Rev. 2002, 60, S15–S19. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Sy, M.; Pavlovich, K.; Leibel, R.L.; Hirsch, J. Leptin reverses weight loss-induced changes in regional neural activity responses to visual food stimuli. J. Clin. Investig. 2008, 118, 2583–2591. [Google Scholar] [CrossRef]
- Ahima, R.S.; Prabakaran, D.; Mantzoros, C.; Qu, D.; Lowell, B.; Maratos-Flier, E.; Flier, J.S. Role of leptin in the neuroendocrine response to fasting. Nature 1996, 382, 250–252. [Google Scholar] [CrossRef]
- Ravussin, Y.; Gutman, R.; Diano, S.; Shanabrough, M.; Borok, E.; Sarman, B.; Lehmann, A.; LeDuc, C.A.; Rosenbaum, M.; Horvath, T.L.; et al. Effects of chronic weight perturbation on energy homeostasis and brain structure in mice. Am. J. Physiol. 2011, 300, R1352–R1362. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Murphy, E.M.; Heymsfield, S.B.; Matthews, D.E.; Leibel, R.L. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J. Clin. Endocrinol. Metab. 2002, 87, 2391–2394. [Google Scholar] [CrossRef]
- Aronne, L.; Fujioka, K.; Aroda, V.; Chen, K.; Halseth, A.; Kesty, N.C.; Burns, C.; Lush, C.W.; Weyer, C. Progressive reduction in body weight after treatment with the amylin analog pramlintide in obese subjects: A phase 2, randomized, placebo-controlled, dose-escalation study. J. Clin. Endocrinol. Metab. 2007, 92, 2977–2983. [Google Scholar] [CrossRef]
- Ravussin, E.; Smith, S.R.; Mitchell, J.A.; Shringarpure, R.; Shan, K.; Maier, H.; Koda, J.E.; Weyer, C. Enhanced weight loss with pramlintide/metreleptin: An integrated neurohormonal approach to obesity pharmacotherapy. Obesity 2009, 17, 1736–1743. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Turek, V.F.; Griffin, P.S.; Wittmer, C.; Parkes, D.G.; Roth, J.D. Multi-hormonal weight loss combinations in diet-induced obese rats: Therapeutic potential of cholecystokinin? Physiol. Behav. 2010, 100, 187–195. [Google Scholar] [CrossRef]
- Clemmensen, C.; Chabenne, J.; Finan, B.; Sullivan, L.; Fischer, K.; Kuchler, D.; Sehrer, L.; Ograjsek, T.; Hofmann, S.M.; Schriever, S.C.; et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes 2014, 63, 1422–1427. [Google Scholar] [CrossRef]
- Byun, K.; Gil, S.Y.; Namkoong, C.; Youn, B.S.; Huang, H.; Shin, M.S.; Kang, G.M.; Kim, H.K.; Lee, B.; Kim, Y.B.; et al. Clusterin/ApoJ enhances central leptin signaling through Lrp2-mediated endocytosis. EMBO Rep. 2014, 15, 801–808. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Munzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef]
- Yan, C.; Yang, Y.; Saito, K.; Xu, P.; Wang, C.; Hinton, A.O., Jr.; Yan, X.; Wu, Q.; Tong, Q.; Elmquist, J.K.; et al. Meta-chlorophenylpiperazine enhances leptin sensitivity in diet-induced obese mice. Br. J. Pharmacol. 2015, 172, 3510–3521. [Google Scholar] [CrossRef]
- Lundin, A.; Rondahl, H.; Walum, E.; Wilcke, M. Expression and intracellular localization of leptin receptor long isoform-GFP chimera. Biochim. Biophys. Acta 2000, 1499, 130–138. [Google Scholar] [CrossRef]
- Belouzard, S.; Delcroix, D.; Rouille, Y. Low levels of expression of leptin receptor at the cell surface result from constitutive endocytosis and intracellular retention in the biosynthetic pathway. J. Biol. Chem. 2004, 279, 28499–28508. [Google Scholar] [CrossRef]
- Belouzard, S.; Rouille, Y. Ubiquitylation of leptin receptor OB-Ra regulates its clathrin-mediated endocytosis. EMBO J. 2006, 25, 932–942. [Google Scholar] [CrossRef]
- Couturier, C.; Sarkis, C.; Seron, K.; Belouzard, S.; Chen, P.; Lenain, A.; Corset, L.; Dam, J.; Vauthier, V.; Dubart, A.; et al. Silencing of OB-RGRP in mouse hypothalamic arcuate nucleus increases leptin receptor signaling and prevents diet-induced obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 19476–19481. [Google Scholar] [CrossRef]
- Seo, S.; Guo, D.F.; Bugge, K.; Morgan, D.A.; Rahmouni, K.; Sheffield, V.C. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum. Mol. Genet. 2009, 18, 1323–1331. [Google Scholar] [CrossRef]
- Haft, C.R.; de la Luz Sierra, M.; Barr, V.A.; Haft, D.H.; Taylor, S.I. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol. Cell. Biol. 1998, 18, 7278–7287. [Google Scholar] [CrossRef]
- Parks, W.T.; Frank, D.B.; Huff, C.; Renfrew Haft, C.; Martin, J.; Meng, X.; de Caestecker, M.P.; McNally, J.G.; Reddi, A.; Taylor, S.I.; et al. Sorting nexin 6, a novel SNX, interacts with the transforming growth factor-beta family of receptor serine-threonine kinases. J. Biol. Chem. 2001, 276, 19332–19339. [Google Scholar] [CrossRef]
- Barr, V.A.; Lane, K.; Taylor, S.I. Subcellular localization and internalization of the four human leptin receptor isoforms. J. Biol. Chem. 1999, 274, 21416–21424. [Google Scholar] [CrossRef]
- De Ceuninck, L.; Wauman, J.; Masschaele, D.; Peelman, F.; Tavernier, J. Reciprocal cross-regulation between RNF41 and USP8 controls cytokine receptor sorting and processing. J. Cell Sci. 2013, 126, 3770–3781. [Google Scholar] [CrossRef]
- Seron, K.; Couturier, C.; Belouzard, S.; Bacart, J.; Monte, D.; Corset, L.; Bocquet, O.; Dam, J.; Vauthier, V.; Lecoeur, C.; et al. Endospanins regulate a postinternalization step of the leptin receptor endocytic pathway. J. Biol. Chem. 2011, 286, 17968–17981. [Google Scholar] [CrossRef]
- Vauthier, V.; Swartz, T.D.; Chen, P.; Roujeau, C.; Pagnon, M.; Mallet, J.; Sarkis, C.; Jockers, R.; Dam, J. Endospanin 1 silencing in the hypothalamic arcuate nucleus contributes to sustained weight loss of high fat diet obese mice. Gene Ther. 2014, 21, 638–644. [Google Scholar] [CrossRef]
- Kim, T.H.; Choi, D.H.; Vauthier, V.; Dam, J.; Li, X.; Nam, Y.J.; Ko, Y.; Kwon, H.J.; Shin, S.H.; Cechetto, J.; et al. Anti-obesity phenotypic screening looking to increase OBR cell surface expression. J. Biomol. Screen. 2014, 19, 88–99. [Google Scholar] [CrossRef]
- Bjornholm, M.; Munzberg, H.; Leshan, R.L.; Villanueva, E.C.; Bates, S.H.; Louis, G.W.; Jones, J.C.; Ishida-Takahashi, R.; Bjorbaek, C.; Myers, M.G., Jr. Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J. Clin. Investig. 2007, 117, 1354–1360. [Google Scholar] [CrossRef]
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L.; Myers, M.G., Jr.; Xu, A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 2010, 59, 894–906. [Google Scholar] [CrossRef]
- Mori, H.; Hanada, R.; Hanada, T.; Aki, D.; Mashima, R.; Nishinakamura, H.; Torisu, T.; Chien, K.R.; Yasukawa, H.; Yoshimura, A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 2004, 10, 739–743. [Google Scholar] [CrossRef]
- Howard, J.K.; Cave, B.J.; Oksanen, L.J.; Tzameli, I.; Bjorbaek, C.; Flier, J.S. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat. Med. 2004, 10, 734–738. [Google Scholar] [CrossRef]
- Marine, J.C.; McKay, C.; Wang, D.; Topham, D.J.; Parganas, E.; Nakajima, H.; Pendeville, H.; Yasukawa, H.; Sasaki, A.; Yoshimura, A.; et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell 1999, 98, 617–627. [Google Scholar] [CrossRef]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C.; et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.B.; Sharpe, A.H.; et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924. [Google Scholar] [CrossRef]
- Banno, R.; Zimmer, D.; De Jonghe, B.C.; Atienza, M.; Rak, K.; Yang, W.; Bence, K.K. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J. Clin. Investig. 2010, 120, 720–734. [Google Scholar] [CrossRef]
- De Jonghe, B.C.; Hayes, M.R.; Banno, R.; Skibicka, K.P.; Zimmer, D.J.; Bowen, K.A.; Leichner, T.M.; Alhadeff, A.L.; Kanoski, S.E.; Cyr, N.E.; et al. Deficiency of PTP1B in POMC neurons leads to alterations in energy balance and homeostatic response to cold exposure. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E1002–E1011. [Google Scholar] [CrossRef]
- Cho, H. Protein tyrosine phosphatase 1B (PTP1B) and obesity. Vitam. Horm. 2013, 91, 405–424. [Google Scholar] [PubMed]
- Lantz, K.A.; Hart, S.G.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 2010, 18, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, D.H. Chemical chaperones: A pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Bailey, E.C.; McCune, S.L.; Dong, J.Y.; Townes, T.M. Reactivation of silenced, virally transduced genes by inhibitors of histone deacetylase. Proc. Natl. Acad. Sci. USA 1997, 94, 5798–5803. [Google Scholar] [CrossRef] [PubMed]
- Maestri, N.E.; Brusilow, S.W.; Clissold, D.B.; Bassett, S.S. Long-term treatment of girls with ornithine transcarbamylase deficiency. N. Engl. J. Med. 1996, 335, 855–859. [Google Scholar] [CrossRef]
- Hosoi, T.; Yamaguchi, R.; Noji, K.; Matsuo, S.; Baba, S.; Toyoda, K.; Suezawa, T.; Kayano, T.; Tanaka, S.; Ozawa, K. Flurbiprofen ameliorated obesity by attenuating leptin resistance induced by endoplasmic reticulum stress. EMBO Mol. Med. 2014, 6, 335–346. [Google Scholar] [CrossRef]
- Hosoi, T.; Baba, S.; Ozawa, K. Therapeutic potential of flurbiprofen against obesity in mice. Biochem. Biophys. Res. Commun. 2014, 449, 132–134. [Google Scholar] [CrossRef]
- Schulz, C.; Paulus, K.; Johren, O.; Lehnert, H. Intranasal leptin reduces appetite and induces weight loss in rats with diet-induced obesity (DIO). Endocrinology 2012, 153, 143–153. [Google Scholar] [CrossRef]
- Hackl, M.T.; Furnsinn, C.; Schuh, C.M.; Krssak, M.; Carli, F.; Guerra, S.; Freudenthaler, A.; Baumgartner-Parzer, S.; Helbich, T.H.; Luger, A.; et al. Brain leptin reduces liver lipids by increasing hepatic triglyceride secretion and lowering lipogenesis. Nat. Commun. 2019, 10, 2717. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. https://doi.org/10.3390/nu11112704
Izquierdo AG, Crujeiras AB, Casanueva FF, Carreira MC. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients. 2019; 11(11):2704. https://doi.org/10.3390/nu11112704
Chicago/Turabian StyleIzquierdo, Andrea G., Ana B. Crujeiras, Felipe F. Casanueva, and Marcos C. Carreira. 2019. "Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later?" Nutrients 11, no. 11: 2704. https://doi.org/10.3390/nu11112704
APA StyleIzquierdo, A. G., Crujeiras, A. B., Casanueva, F. F., & Carreira, M. C. (2019). Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients, 11(11), 2704. https://doi.org/10.3390/nu11112704