Incendiary Leptin

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Obesity Pandemic
2. Thermogenesis
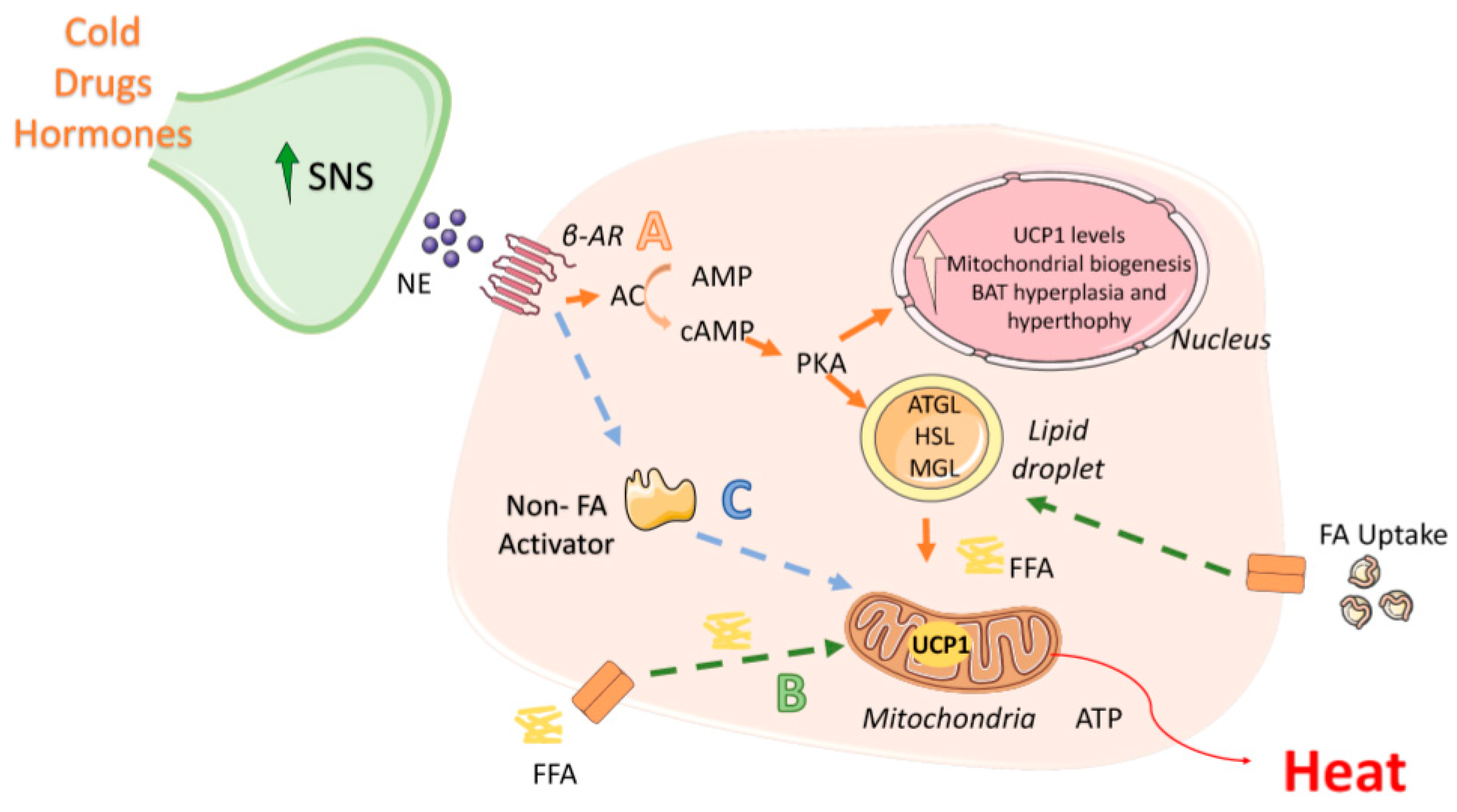
2.1. Activation of BAT Thermogenesis
2.2. Thermogenesis in Humans
2.3. Thermogenesis, a Therapeutic Treatment for Obesity?
3. Leptin History
4. Leptin Anorexigenic Effects
- (1)
- Pro-opiomelanocortin (POMC) neurons that express anorexigenic neuropeptides such as POMC and cocaine- and amphetamine-regulated transcript (CART) [137,138], and are stimulated by leptin through the release of α-melanocite-stimulating hormone (α-MSH) [139,140,141] that binds the melanocortin 3 receptors (MC3R) and MC4R. Several genetic variants of POMC and MC4R genes have been associated to human obesity, suggesting that the central melanocortin system is required for the anorexigenic effect of leptin.
- (2)
- Agouti-related protein (AgRP) neurons [142,143] which express orexigenic neuropeptides such as neuropeptide Y (NPY) and AgRP [144]. Leptin exerts inhibitory effects on both AgRP and NPY neurons activity as well as on the release of the associated AgRP and NPY neuropeptides [142,145], resulting in a potent satiating effect.
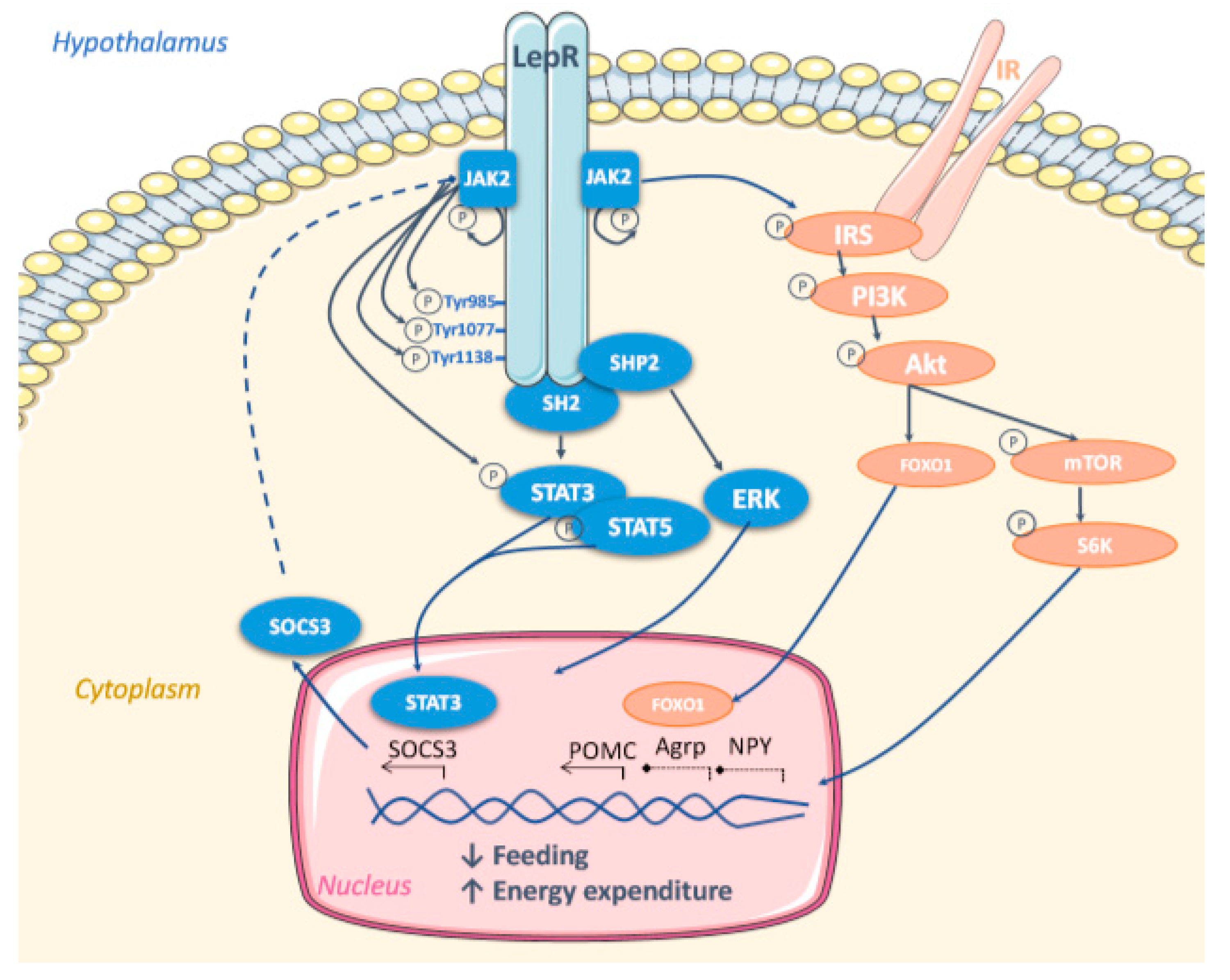
5. Molecular Mechanisms Mediating Leptin Effect
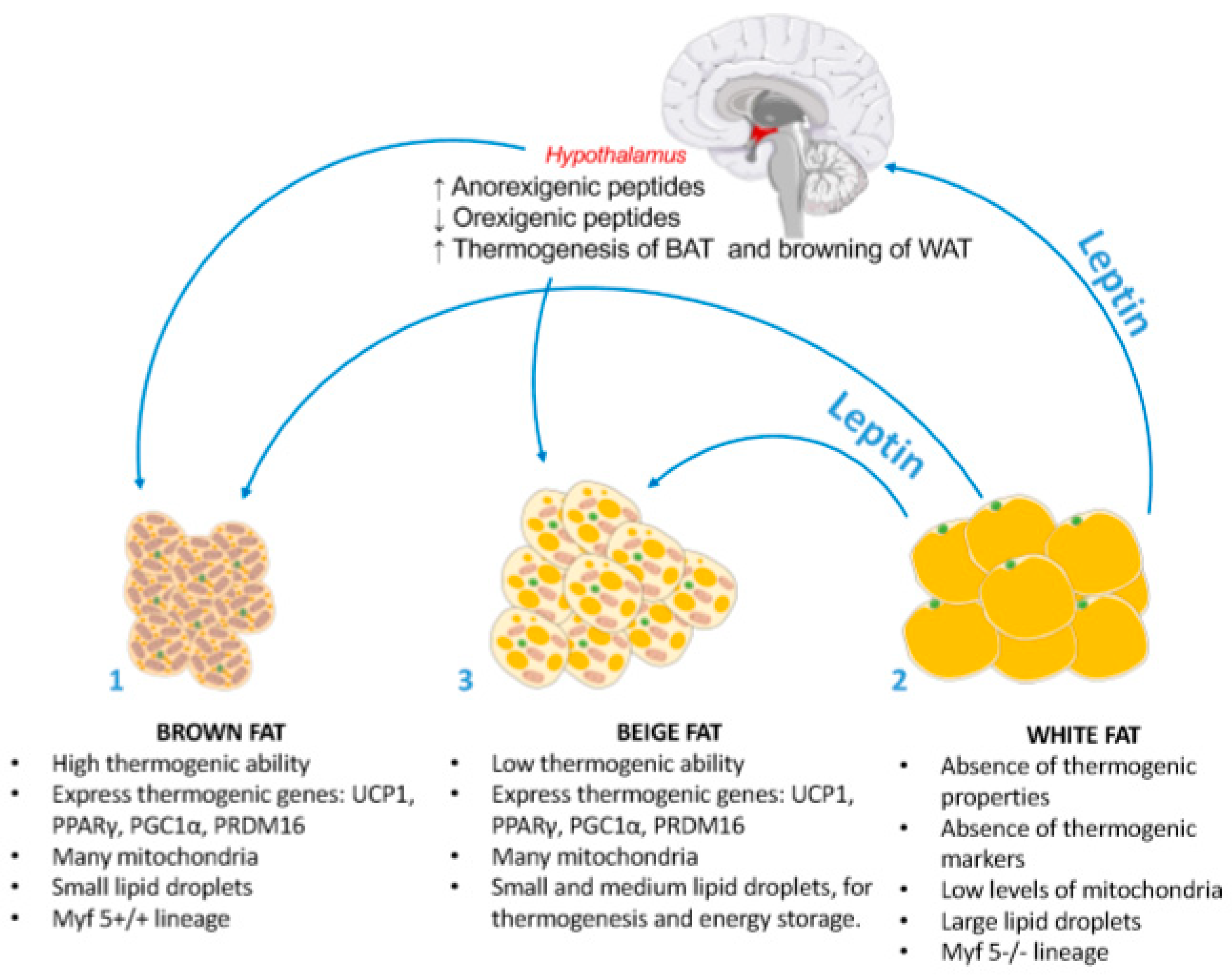
6. Leptin Effects on Thermogenesis and Browning
6.1. Thermogenic Effects of Leptin
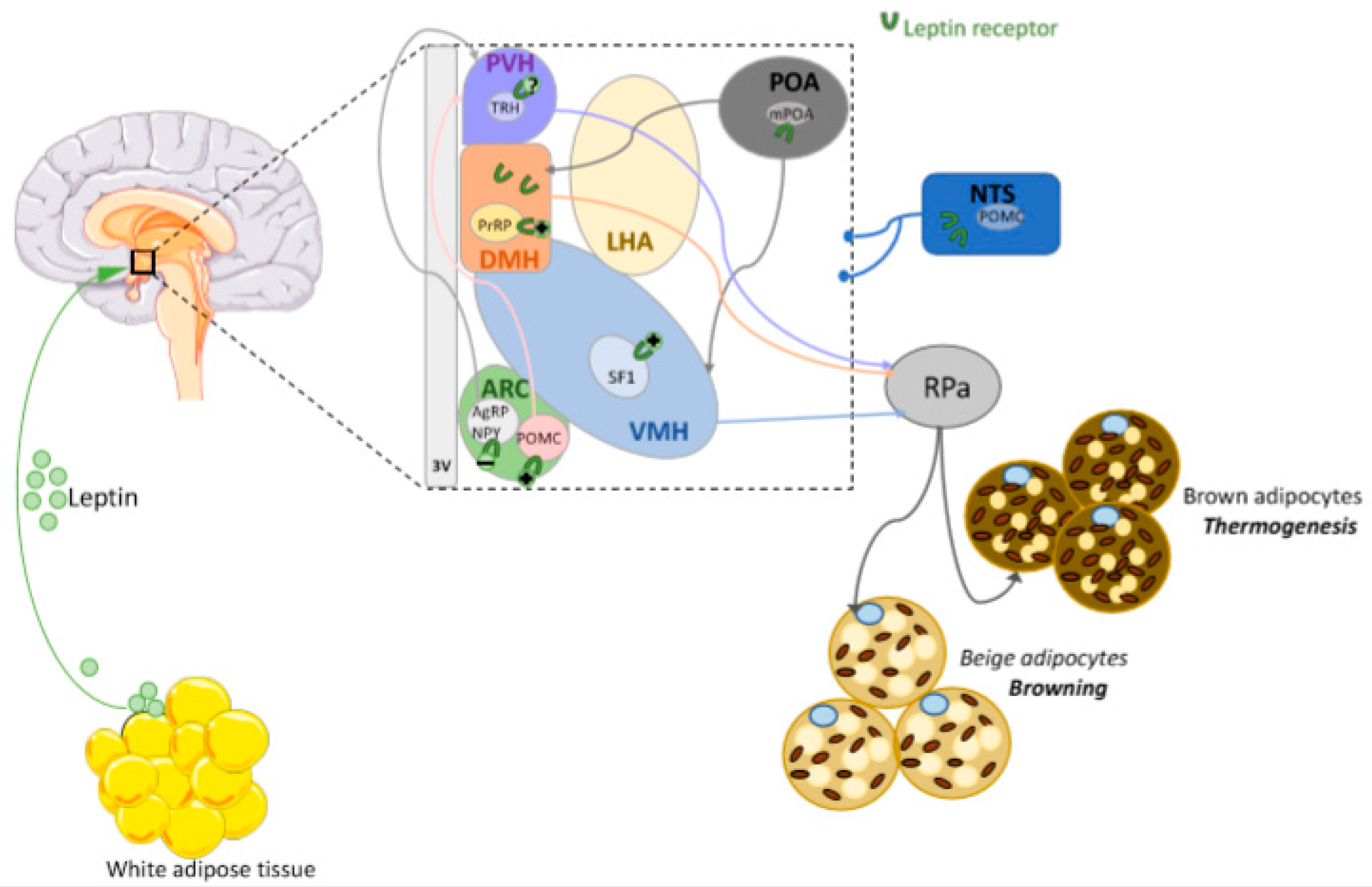
6.2. Leptin Actions in the DMH
6.3. Leptin Actions in the Preoptic Area (POA)
6.4. Leptin Actions in the PVH
6.5. Leptin Actions in the VMH
6.6. Leptin Actions in the ARC
6.7. Leptin Actions in Extra-Hypothalamic Areas
6.8. Browning and Leptin Central Action
6.9. Leptin Actions at Peripheral Level
7. Leptin Polymorphism and Metabolic Implications
8. Leptin as a Therapeutic Approach to Correct Obesity
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Obesity Data. Available online: https://www.who.int/topics/obesity/en/ (accessed on 12 February 2020).
- Schneeberger, M.; Gomis, R.; Claret, M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 2014, 220, T25–T46. [Google Scholar] [CrossRef]
- Tschop, M.H.; Finan, B.; Clemmensen, C.; Gelfanov, V.; Perez-Tilve, D.; Muller, T.D.; DiMarchi, R.D. Unimolecular Polypharmacy for Treatment of Diabetes and Obesity. Cell Metab. 2016, 24, 51–62. [Google Scholar] [CrossRef]
- Clemmensen, C.; Muller, T.D.; Woods, S.C.; Berthoud, H.R.; Seeley, R.J.; Tschop, M.H. Gut-Brain Cross-Talk in Metabolic Control. Cell 2017, 168, 758–774. [Google Scholar] [CrossRef]
- Lopez, M.; Tena-Sempere, M. Estradiol effects on hypothalamic AMPK and BAT thermogenesis: A gateway for obesity treatment? Pharmacol. Ther. 2017, 178, 109–122. [Google Scholar] [CrossRef]
- Cui, H.; Lopez, M.; Rahmouni, K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat. Rev. Endocrinol. 2017, 13, 338. [Google Scholar] [CrossRef]
- Hall, K.D.; Guo, J. Obesity Energetics: Body Weight Regulation and the Effects of Diet Composition. Gastroenterology 2017, 152, 1718–1727. [Google Scholar] [CrossRef]
- Sims, E.A.; Danforth, E., Jr.; Horton, E.S.; Bray, G.A.; Glennon, J.A.; Salans, L.B. Endocrine and metabolic effects of experimental obesity in man. Recent Prog. Horm. Res. 1973, 29, 457–496. [Google Scholar]
- Leibel, R.L.; Rosenbaum, M.; Hirsch, J. Changes in energy expenditure resulting from altered body weight. N. Engl. J. Med. 1995, 332, 621–628. [Google Scholar] [CrossRef]
- Ravussin, Y.; Leibel, R.L.; Ferrante, A.W., Jr. A missing link in body weight homeostasis: The catabolic signal of the overfed state. Cell Metab. 2014, 20, 565–572. [Google Scholar] [CrossRef]
- Fontaine, K.R.; Redden, D.T.; Wang, C.; Westfall, A.O.; Allison, D.B. Years of life lost due to obesity. JAMA 2003, 289, 187–193. [Google Scholar] [CrossRef]
- Bluher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Bellisari, A. Evolutionary origins of obesity. Obes. Rev. 2008, 9, 165–180. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Morrison, C. The brain, appetite, and obesity. Annu. Rev. Psychol. 2008, 59, 55–92. [Google Scholar] [CrossRef]
- Landsberg, L.; Young, J.B.; Leonard, W.R.; Linsenmeier, R.A.; Turek, F.W. Is obesity associated with lower body temperatures? Core temperature: A forgotten variable in energy balance. Metabolism 2009, 58, 871–876. [Google Scholar] [CrossRef]
- Doucet, E.; Imbeault, P.; St-Pierre, S.; Almeras, N.; Mauriege, P.; Despres, J.P.; Bouchard, C.; Tremblay, A. Greater than predicted decrease in energy expenditure during exercise after body weight loss in obese men. Clin. Sci. 2003, 105, 89–95. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Labbe, S.M.; Caron, A.; Lanfray, D.; Monge-Rofarello, B.; Bartness, T.J.; Richard, D. Hypothalamic control of brown adipose tissue thermogenesis. Front. Syst. Neurosci. 2015, 9, 150. [Google Scholar] [CrossRef]
- Lim, S.; Honek, J.; Xue, Y.; Seki, T.; Cao, Z.; Andersson, P.; Yang, X.; Hosaka, K.; Cao, Y. Cold-induced activation of brown adipose tissue and adipose angiogenesis in mice. Nat. Protoc. 2012, 7, 606–615. [Google Scholar] [CrossRef]
- Boyer, P.D. The ATP synthase—A splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef]
- Futai, M.; Noumi, T.; Maeda, M. ATP synthase (H+-ATPase): Results by combined biochemical and molecular biological approaches. Annu. Rev. Biochem. 1989, 58, 111–136. [Google Scholar] [CrossRef]
- von Ballmoos, C.; Wiedenmann, A.; Dimroth, P. Essentials for ATP synthesis by F1F0 ATP synthases. Annu. Rev. Biochem. 2009, 78, 649–672. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Locke, R.M. Thermogenic mechanisms in brown fat. Physiol. Rev. 1984, 64, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.E. Thermogenic mechanisms and their hormonal regulation. Physiol. Rev. 2006, 86, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Wennmalm, K.; Larsson, O.; Walden, T.B.; Lassmann, T.; Petrovic, N.; Hamilton, D.L.; Gimeno, R.E.; Wahlestedt, C.; Baar, K.; et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc. Natl. Acad. Sci. USA 2007, 104, 4401–4406. [Google Scholar] [CrossRef]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef]
- Shan, T.; Liang, X.; Bi, P.; Zhang, P.; Liu, W.; Kuang, S. Distinct populations of adipogenic and myogenic Myf5-lineage progenitors in white adipose tissues. J. Lipid Res. 2013, 54, 2214–2224. [Google Scholar] [CrossRef]
- Sanchez-Gurmaches, J.; Guertin, D.A. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat. Commun. 2014, 5, 4099. [Google Scholar] [CrossRef]
- Crisan, M.; Casteilla, L.; Lehr, L.; Carmona, M.; Paoloni-Giacobino, A.; Yap, S.; Sun, B.; Leger, B.; Logar, A.; Penicaud, L.; et al. A reservoir of brown adipocyte progenitors in human skeletal muscle. Stem Cells 2008, 26, 2425–2433. [Google Scholar] [CrossRef]
- Kajimura, S.; Saito, M. A new era in brown adipose tissue biology: Molecular control of brown fat development and energy homeostasis. Annu. Rev. Physiol. 2014, 76, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Werner, C.D.; Kebebew, E.; Celi, F.S. Functional thermogenic beige adipogenesis is inducible in human neck fat. Int. J. Obes. 2014, 38, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; Gonzalez, F.; Ferno, J.; Dieguez, C.; Rahmouni, K.; Nogueiras, R.; Lopez, M. The brain and brown fat. Ann. Med. 2015, 47, 150–168. [Google Scholar] [CrossRef]
- Walden, T.B.; Hansen, I.R.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Recruited versus nonrecruited molecular signatures of brown, “brite” and white adipose tissues. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, E19–E31. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Petrovic, N.; de Jong, J.M.; Kalinovich, A.V.; Cannon, B.; Nedergaard, J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep. 2013, 5, 1196–1203. [Google Scholar] [CrossRef]
- Nedergaard, J.; Cannon, B. UCP1 mRNA does not produce heat. Biochim. Biophys. Acta 2013, 1831, 943–949. [Google Scholar] [CrossRef]
- Keipert, S.; Jastroch, M. Brite/beige fat and UCP1—Is it thermogenesis? Biochim. Biophys. Acta 2014, 1837, 1075–1082. [Google Scholar] [CrossRef]
- Lidell, M.E.; Betz, M.J.; Enerback, S. Two types of brown adipose tissue in humans. Adipocyte 2014, 3, 63–66. [Google Scholar] [CrossRef]
- Zingaretti, M.C.; Crosta, F.; Vitali, A.; Guerrieri, M.; Frontini, A.; Cannon, B.; Nedergaard, J.; Cinti, S. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009, 23, 3113–3120. [Google Scholar] [CrossRef]
- Granneman, J.G. Norepinephrine infusions increase adenylate cyclase responsiveness in brown adipose tissue. J. Pharmacol. Exp. Ther. 1988, 245, 1075–1080. [Google Scholar] [PubMed]
- Whittle, A.J.; Lopez, M.; Vidal-Puig, A. Using brown adipose tissue to treat obesity - the central issue. Trends Mol. Med. 2011, 17, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Vidal-Puig, A. Beyond the sympathetic tone: The new brown fat activators. Cell Metab. 2013, 17, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.; Varela, L.; Vazquez, M.J.; Rodriguez-Cuenca, S.; Gonzalez, C.R.; Velagapudi, V.R.; Morgan, D.A.; Schoenmakers, E.; Agassandian, K.; Lage, R.; et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat. Med. 2010, 16, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; Gonzalez-Garcia, I.; Martinez-Sanchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central Ceramide-Induced Hypothalamic Lipotoxicity and ER Stress Regulate Energy Balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef]
- Martinez de Morentin, P.B.; Gonzalez-Garcia, I.; Martins, L.; Lage, R.; Fernandez-Mallo, D.; Martinez-Sanchez, N.; Ruiz-Pino, F.; Liu, J.; Morgan, D.A.; Pinilla, L.; et al. Estradiol regulates brown adipose tissue thermogenesis via hypothalamic AMPK. Cell Metab. 2014, 20, 41–53. [Google Scholar] [CrossRef]
- Martinez-Sanchez, N.; Seoane-Collazo, P.; Contreras, C.; Varela, L.; Villarroya, J.; Rial-Pensado, E.; Buque, X.; Aurrekoetxea, I.; Delgado, T.C.; Vazquez-Martinez, R.; et al. Hypothalamic AMPK-ER Stress-JNK1 Axis Mediates the Central Actions of Thyroid Hormones on Energy Balance. Cell Metab. 2017, 26, 212–229. [Google Scholar] [CrossRef]
- Seoane-Collazo, P.; Roa, J.; Rial-Pensado, E.; Linares-Pose, L.; Beiroa, D.; Ruiz-Pino, F.; Lopez-Gonzalez, T.; Morgan, D.A.; Pardavila, J.A.; Sanchez-Tapia, M.J.; et al. SF1-Specific AMPKalpha1 Deletion Protects Against Diet-Induced Obesity. Diabetes 2018, 67, 2213–2226. [Google Scholar] [CrossRef]
- Seoane-Collazo, P.; Linares-Pose, L.; Rial-Pensado, E.; Romero-Pico, A.; Moreno-Navarrete, J.M.; Martinez-Sanchez, N.; Garrido-Gil, P.; Iglesias-Rey, R.; Morgan, D.A.; Tomasini, N.; et al. Central nicotine induces browning through hypothalamic kappa opioid receptor. Nat. Commun. 2019, 10, 4037. [Google Scholar] [CrossRef]
- Cao, W.; Medvedev, A.V.; Daniel, K.W.; Collins, S. beta-Adrenergic activation of p38 MAP kinase in adipocytes: cAMP induction of the uncoupling protein 1 (UCP1) gene requires p38 MAP kinase. J. Biol. Chem. 2001, 276, 27077–27082. [Google Scholar] [CrossRef]
- Holm, C.; Fredrikson, G.; Cannon, B.; Belfrage, P. Hormone-sensitive lipase in brown adipose tissue: identification and effect of cold exposure. Biosci. Rep. 1987, 7, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Shih, M.F.; Taberner, P.V. Selective activation of brown adipocyte hormone-sensitive lipase and cAMP production in the mouse by beta 3-adrenoceptor agonists. Biochem. Pharmacol. 1995, 50, 601–608. [Google Scholar] [CrossRef]
- Clifford, G.M.; Londos, C.; Kraemer, F.B.; Vernon, R.G.; Yeaman, S.J. Translocation of hormone-sensitive lipase and perilipin upon lipolytic stimulation of rat adipocytes. J. Biol. Chem. 2000, 275, 5011–5015. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, C.; Kameda, K.; Tsujita, T.; Okuda, H. Relationships between lipolysis induced by various lipolytic agents and hormone-sensitive lipase in rat fat cells. J. Lipid Res. 2001, 42, 120–127. [Google Scholar] [PubMed]
- Cannon, B.; Nedergaard, J. What Ignites UCP1? Cell Metab. 2017, 26, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Diwoky, C.; Schoiswohl, G.; Feiler, U.; Wongsiriroj, N.; Abdellatif, M.; Kolb, D.; Hoeks, J.; Kershaw, E.E.; Sedej, S.; et al. Cold-Induced Thermogenesis Depends on ATGL-Mediated Lipolysis in Cardiac Muscle, but Not Brown Adipose Tissue. Cell Metab. 2017, 26, 753–763. [Google Scholar] [CrossRef]
- Shin, H.; Ma, Y.; Chanturiya, T.; Cao, Q.; Wang, Y.; Kadegowda, A.K.G.; Jackson, R.; Rumore, D.; Xue, B.; Shi, H.; et al. Lipolysis in Brown Adipocytes Is Not Essential for Cold-Induced Thermogenesis in Mice. Cell Metab. 2017, 26, 764–777. [Google Scholar] [CrossRef]
- Lowell, B.B.; Spiegelman, B.M. Towards a molecular understanding of adaptive thermogenesis. Nature 2000, 404, 652–660. [Google Scholar] [CrossRef]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, N.Z.; Larsen, T.J.; Peijs, L.; Daugaard, S.; Homoe, P.; Loft, A.; de Jong, J.; Mathur, N.; Cannon, B.; Nedergaard, J.; et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab. 2013, 17, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, P.; Hirvonen, J.; Kinnula, V. The occurrence of brown adipose tissue in outdoor workers. Eur. J. Appl. Physiol. Occup. Physiol. 1981, 46, 339–345. [Google Scholar] [CrossRef]
- Matsushita, M.; Yoneshiro, T.; Aita, S.; Kameya, T.; Sugie, H.; Saito, M. Impact of brown adipose tissue on body fatness and glucose metabolism in healthy humans. Int. J. Obes. 2014, 38, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Heaton, J.M. The distribution of brown adipose tissue in the human. J. Anat. 1972, 112, 35–39. [Google Scholar] [PubMed]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Okamatsu-Ogura, Y.; Kameya, T.; Kawai, Y.; Miyagawa, M.; Tsujisaki, M.; Saito, M. Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity 2011, 19, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Kayahara, T.; Kameya, T.; Kawai, Y.; Iwanaga, T.; Saito, M. Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Investig. 2013, 123, 3404–3408. [Google Scholar] [CrossRef]
- van der Lans, A.A.; Hoeks, J.; Brans, B.; Vijgen, G.H.; Visser, M.G.; Vosselman, M.J.; Hansen, J.; Jorgensen, J.A.; Wu, J.; Mottaghy, F.M.; et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J. Clin. Investig. 2013, 123, 3395–3403. [Google Scholar] [CrossRef]
- Lee, J.H.; Wei, L.; Gu, X.; Wei, Z.; Dix, T.A.; Yu, S.P. Therapeutic effects of pharmacologically induced hypothermia against traumatic brain injury in mice. J. Neurotrauma 2014, 31, 1417–1430. [Google Scholar] [CrossRef]
- Saito, M.; Yoneshiro, T.; Matsushita, M. Activation and recruitment of brown adipose tissue by cold exposure and food ingredients in humans. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 537–547. [Google Scholar] [CrossRef]
- van Marken Lichtenbelt, W.D.; Schrauwen, P. Implications of nonshivering thermogenesis for energy balance regulation in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R285–R296. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, G.H.; Bouvy, N.D.; Leenen, L.; Rijkers, K.; Cornips, E.; Majoie, M.; Brans, B.; van Marken Lichtenbelt, W.D. Vagus nerve stimulation increases energy expenditure: Relation to brown adipose tissue activity. PLoS ONE 2013, 8, e77221. [Google Scholar] [CrossRef]
- Orava, J.; Nuutila, P.; Noponen, T.; Parkkola, R.; Viljanen, T.; Enerback, S.; Rissanen, A.; Pietilainen, K.H.; Virtanen, K.A. Blunted metabolic responses to cold and insulin stimulation in brown adipose tissue of obese humans. Obesity 2013, 21, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
- Loh, R.K.C.; Kingwell, B.A.; Carey, A.L. Human brown adipose tissue as a target for obesity management; beyond cold-induced thermogenesis. Obes. Rev. 2017, 18, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, V.; Labbe, S.M.; Blondin, D.P.; Phoenix, S.; Guerin, B.; Haman, F.; Turcotte, E.E.; Richard, D.; Carpentier, A.C. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J. Clin. Investig. 2012, 122, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Orava, J.; Nuutila, P.; Lidell, M.E.; Oikonen, V.; Noponen, T.; Viljanen, T.; Scheinin, M.; Taittonen, M.; Niemi, T.; Enerback, S.; et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab. 2011, 14, 272–279. [Google Scholar] [CrossRef]
- Chondronikola, M.; Volpi, E.; Børsheim, E.; Porter, C.; Annamalai, P.; Enerbäck, S.; Lidell, M.E.; Saraf, M.K.; Labbe, S.M.; Hurren, N.M.; et al. Brown Adipose Tissue Improves Whole-Body Glucose Homeostasis and Insulin Sensitivity in Humans. Diabetes 2014, 63, 4089–4099. [Google Scholar] [CrossRef]
- Lee, P.; Smith, S.; Linderman, J.; Courville, A.B.; Brychta, R.J.; Dieckmann, W.; Werner, C.D.; Chen, K.Y.; Celi, F.S. Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes 2014, 63, 3686–3698. [Google Scholar] [CrossRef]
- Lee, P.; Bova, R.; Schofield, L.; Bryant, W.; Dieckmann, W.; Slattery, A.; Govendir, M.A.; Emmett, L.; Greenfield, J.R. Brown Adipose Tissue Exhibits a Glucose-Responsive Thermogenic Biorhythm in Humans. Cell Metab. 2016, 23, 602–609. [Google Scholar] [CrossRef]
- Hanssen, M.J.; van der Lans, A.A.; Brans, B.; Hoeks, J.; Jardon, K.M.; Schaart, G.; Mottaghy, F.M.; Schrauwen, P.; van Marken Lichtenbelt, W.D. Short-term Cold Acclimation Recruits Brown Adipose Tissue in Obese Humans. Diabetes 2016, 65, 1179–1189. [Google Scholar] [CrossRef]
- Hanssen, M.J.; Hoeks, J.; Brans, B.; van der Lans, A.A.; Schaart, G.; van den Driessche, J.J.; Jorgensen, J.A.; Boekschoten, M.V.; Hesselink, M.K.; Havekes, B.; et al. Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat. Med. 2015, 21, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Blondin, D.P.; Labbe, S.M.; Noll, C.; Kunach, M.; Phoenix, S.; Guerin, B.; Turcotte, E.E.; Haman, F.; Richard, D.; Carpentier, A.C. Selective Impairment of Glucose but Not Fatty Acid or Oxidative Metabolism in Brown Adipose Tissue of Subjects With Type 2 Diabetes. Diabetes 2015, 64, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Blondin, D.P.; Frisch, F.; Phoenix, S.; Guerin, B.; Turcotte, E.E.; Haman, F.; Richard, D.; Carpentier, A.C. Inhibition of Intracellular Triglyceride Lipolysis Suppresses Cold-Induced Brown Adipose Tissue Metabolism and Increases Shivering in Humans. Cell Metab. 2017, 25, 438–447. [Google Scholar] [CrossRef]
- Chondronikola, M.; Volpi, E.; Børsheim, E.; Porter, C.; Saraf, M.K.; Annamalai, P.; Yfanti, C.; Chao, T.; Wong, D.; Shinoda, K.; et al. Brown Adipose Tissue Activation Is Linked to Distinct Systemic Effects on Lipid Metabolism in Humans. Cell Metab. 2016, 23, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, F.; Mukherjee, M.; Kadziola, Z.; Sherwood, R.; Kakkar, V.V. Central cooling effects in patients with hypercholesterolaemia. Clin. Sci. 1998, 95, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Foster, M.T. Beiging of white adipose tissue as a therapeutic strategy for weight loss in humans. Horm. Mol. Biol. Clin. Investig. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Wijers, S.L.; Saris, W.H.; van Marken Lichtenbelt, W.D. Cold-induced adaptive thermogenesis in lean and obese. Obesity 2010, 18, 1092–1099. [Google Scholar] [CrossRef]
- Arch, J.R. The beta 3-adrenergic system and beta 3-adrenergic agonists. Rev. Endocr. Metab. Disord. 2001, 2, 385–393. [Google Scholar] [CrossRef]
- Cypess, A.M.; Weiner, L.S.; Roberts-Toler, C.; Franquet Elia, E.; Kessler, S.H.; Kahn, P.A.; English, J.; Chatman, K.; Trauger, S.A.; Doria, A.; et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. 2015, 21, 33–38. [Google Scholar] [CrossRef]
- Finlin, B.S.; Memetimin, H.; Confides, A.L.; Kasza, I.; Zhu, B.; Vekaria, H.J.; Harfmann, B.; Jones, K.A.; Johnson, Z.R.; Westgate, P.M.; et al. Human adipose beiging in response to cold and mirabegron. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Cypess, A.M.; Chen, Y.C.; Sze, C.; Wang, K.; English, J.; Chan, O.; Holman, A.R.; Tal, I.; Palmer, M.R.; Kolodny, G.M.; et al. Cold but not sympathomimetics activates human brown adipose tissue in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 10001–10005. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Aita, S.; Kawai, Y.; Iwanaga, T.; Saito, M. Nonpungent capsaicin analogs (capsinoids) increase energy expenditure through the activation of brown adipose tissue in humans. Am. J. Clin. Nutr. 2012, 95, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Galgani, J.E.; Ryan, D.H.; Ravussin, E. Effect of capsinoids on energy metabolism in human subjects. Br. J. Nutr. 2010, 103, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Camps, S.G.; Goh, H.J.; Govindharajulu, P.; Schaefferkoetter, J.D.; Townsend, D.W.; Verma, S.K.; Velan, S.S.; Sun, L.; Sze, S.K.; et al. Capsinoids activate brown adipose tissue (BAT) with increased energy expenditure associated with subthreshold 18-fluorine fluorodeoxyglucose uptake in BAT-positive humans confirmed by positron emission tomography scan. Am. J. Clin. Nutr. 2018, 107, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Nirengi, S.; Homma, T.; Inoue, N.; Sato, H.; Yoneshiro, T.; Matsushita, M.; Kameya, T.; Sugie, H.; Tsuzaki, K.; Saito, M.; et al. Assessment of human brown adipose tissue density during daily ingestion of thermogenic capsinoids using near-infrared time-resolved spectroscopy. J. Biomed. Opt. 2016, 21, 091305. [Google Scholar] [CrossRef]
- Broeders, E.P.; Nascimento, E.B.; Havekes, B.; Brans, B.; Roumans, K.H.; Tailleux, A.; Schaart, G.; Kouach, M.; Charton, J.; Deprez, B.; et al. The Bile Acid Chenodeoxycholic Acid Increases Human Brown Adipose Tissue Activity. Cell Metab. 2015, 22, 418–426. [Google Scholar] [CrossRef]
- Labruna, G.; Pasanisi, F.; Fortunato, G.; Nardelli, C.; Finelli, C.; Farinaro, E.; Contaldo, F.; Sacchetti, L. Sequence Analysis of the UCP1 Gene in a Severe Obese Population from Southern Italy. J. Obes. 2011, 2011, 269043. [Google Scholar] [CrossRef]
- Chathoth, S.; Ismail, M.H.; Vatte, C.; Cyrus, C.; Al Ali, Z.; Ahmed, K.A.; Acharya, S.; Al Barqi, A.M.; Al Ali, A. Association of Uncoupling Protein 1 (UCP1) gene polymorphism with obesity: A case-control study. BMC Med. Genet. 2018, 19, 203. [Google Scholar] [CrossRef]
- Samano, R.; Huesca-Gomez, C.; Lopez-Marure, R.; Hernandez-Cabrera, A.K.; Rodriguez-Ventura, A.; Tolentino, M.; Morales, R.M.; Gamboa, R. Association between UCP polymorphisms and adipokines with obesity in Mexican adolescents. J. Pediatr. Endocrinol. Metab. 2018, 31, 561–568. [Google Scholar] [CrossRef]
- Siiteri, P.K. Adipose tissue as a source of hormones. Am. J. Clin. Nutr. 1987, 45, 277–282. [Google Scholar] [CrossRef]
- Flier, J.S.; Cook, K.S.; Usher, P.; Spiegelman, B.M. Severely impaired adipsin expression in genetic and acquired obesity. Science 1987, 237, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.L. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 1973, 9, 294–298. [Google Scholar] [CrossRef]
- Chen, H.; Charlat, O.; Tartaglia, L.A.; Woolf, E.A.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; Moore, K.J.; Breitbart, R.E.; et al. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef]
- Lee, G.H.; Proenca, R.; Montez, J.M.; Carroll, K.M.; Darvishzadeh, J.G.; Lee, J.I.; Friedman, J.M. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996, 379, 632–635. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef]
- Maffei, M.; Fei, H.; Lee, G.H.; Dani, C.; Leroy, P.; Zhang, Y.; Proenca, R.; Negrel, R.; Ailhaud, G.; Friedman, J.M. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proc. Natl. Acad. Sci. USA 1995, 92, 6957–6960. [Google Scholar] [CrossRef]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef]
- Muruzabal, F.J.; Fruhbeck, G.; Gomez-Ambrosi, J.; Archanco, M.; Burrell, M.A. Immunocytochemical detection of leptin in non-mammalian vertebrate stomach. Gen. Comp. Endocrinol. 2002, 128, 149–152. [Google Scholar] [CrossRef]
- Allison, M.B.; Myers, M.G., Jr. 20 years of leptin: Connecting leptin signaling to biological function. J. Endocrinol. 2014, 223, T25–T35. [Google Scholar] [CrossRef]
- Chehab, F.F. 20 years of leptin: Leptin and reproduction: Past milestones, present undertakings, and future endeavors. J. Endocrinol. 2014, 223, T37–T48. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Mantzoros, C. 20 years of leptin: Role of leptin in human reproductive disorders. J. Endocrinol. 2014, 223, T49–T62. [Google Scholar] [CrossRef] [PubMed]
- DePaoli, A.M. 20 years of leptin: Leptin in common obesity and associated disorders of metabolism. J. Endocrinol. 2014, 223, T71–T81. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; O’Rahilly, S. 20 years of leptin: Human disorders of leptin action. J. Endocrinol. 2014, 223, T63–T70. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J. 20 years of leptin: Leptin at 20: An overview. J. Endocrinol. 2014, 223, T1–T8. [Google Scholar] [CrossRef]
- Gautron, L.; Elmquist, J.K. Sixteen years and counting: An update on leptin in energy balance. J. Clin. Investig. 2011, 121, 2087–2093. [Google Scholar] [CrossRef]
- Peelman, F.; Zabeau, L.; Moharana, K.; Savvides, S.N.; Tavernier, J. 20 years of leptin: Insights into signaling assemblies of the leptin receptor. J. Endocrinol. 2014, 223, T9–T23. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Leibel, R.L. 20 years of leptin: Role of leptin in energy homeostasis in humans. J. Endocrinol. 2014, 223, T83–T96. [Google Scholar] [CrossRef]
- Friedman, J.M.; Leibel, R.L.; Siegel, D.S.; Walsh, J.; Bahary, N. Molecular mapping of the mouse ob mutation. Genomics 1991, 11, 1054–1062. [Google Scholar] [CrossRef]
- Takaya, K.; Ogawa, Y.; Isse, N.; Okazaki, T.; Satoh, N.; Masuzaki, H.; Mori, K.; Tamura, N.; Hosoda, K.; Nakao, K. Molecular cloning of rat leptin receptor isoform complementary DNAs--identification of a missense mutation in Zucker fatty (fa/fa) rats. Biochem. Biophys. Res. Commun. 1996, 225, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Boozer, C.; Blair-West, J.; Fidahusein, N.; Denton, D.A.; Friedman, J.M. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8878–8883. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar] [CrossRef]
- Stephens, T.W.; Basinski, M.; Bristow, P.K.; Bue-Valleskey, J.M.; Burgett, S.G.; Craft, L.; Hale, J.; Hoffmann, J.; Hsiung, H.M.; Kriauciunas, A.; et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature 1995, 377, 530–532. [Google Scholar] [CrossRef]
- Cohen, S.L.; Halaas, J.L.; Friedman, J.M.; Chait, B.T.; Bennett, L.; Chang, D.; Hecht, R.; Collins, F. Human leptin characterization. Nature 1996, 382, 589. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Matarese, G.; Lord, G.M.; Keogh, J.M.; Lawrence, E.; Agwu, C.; Sanna, V.; Jebb, S.A.; Perna, F.; Fontana, S.; et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Investig. 2002, 110, 1093–1103. [Google Scholar] [CrossRef]
- Licinio, J.; Caglayan, S.; Ozata, M.; Yildiz, B.O.; de Miranda, P.B.; O’Kirwan, F.; Whitby, R.; Liang, L.; Cohen, P.; Bhasin, S.; et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc. Natl. Acad. Sci. USA 2004, 101, 4531–4536. [Google Scholar] [CrossRef]
- Elmquist, J.K.; Bjorbaek, C.; Ahima, R.S.; Flier, J.S.; Saper, C.B. Distributions of leptin receptor mRNA isoforms in the rat brain. J. Comp. Neurol. 1998, 395, 535–547. [Google Scholar] [CrossRef]
- Scott, M.M.; Lachey, J.L.; Sternson, S.M.; Lee, C.E.; Elias, C.F.; Friedman, J.M.; Elmquist, J.K. Leptin targets in the mouse brain. J. Comp. Neurol. 2009, 514, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Seeley, R.J.; Campfield, L.A.; Burn, P.; Baskin, D.G. Identification of targets of leptin action in rat hypothalamus. J. Clin. Investig. 1996, 98, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.G.; Hoggard, N.; Williams, L.M.; Lawrence, C.B.; Hannah, L.T.; Trayhurn, P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996, 387, 113–116. [Google Scholar] [CrossRef]
- Morton, G.J.; Niswender, K.D.; Rhodes, C.J.; Myers, M.G., Jr.; Blevins, J.E.; Baskin, D.G.; Schwartz, M.W. Arcuate nucleus-specific leptin receptor gene therapy attenuates the obesity phenotype of Koletsky (fa(k)/fa(k)) rats. Endocrinology 2003, 144, 2016–2024. [Google Scholar] [CrossRef]
- Elias, C.F.; Lee, C.; Kelly, J.; Aschkenasi, C.; Ahima, R.S.; Couceyro, P.R.; Kuhar, M.J.; Saper, C.B.; Elmquist, J.K. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron 1998, 21, 1375–1385. [Google Scholar] [CrossRef]
- Balthasar, N.; Coppari, R.; McMinn, J.; Liu, S.M.; Lee, C.E.; Tang, V.; Kenny, C.D.; McGovern, R.A.; Chua, S.C., Jr.; Elmquist, J.K.; et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 2004, 42, 983–991. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdan, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef]
- Fan, W.; Boston, B.A.; Kesterson, R.A.; Hruby, V.J.; Cone, R.D. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 1997, 385, 165–168. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Woods, S.C.; Weigle, D.S.; Campfield, L.A.; Burn, P.; Baskin, D.G. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes 1997, 46, 2119–2123. [Google Scholar] [CrossRef]
- Morris, D.L.; Rui, L. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1247–E1259. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Munzberg, H.; Leinninger, G.M.; Leshan, R.L. The geometry of leptin action in the brain: More complicated than a simple ARC. Cell Metab. 2009, 9, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.M.; Breininger, J.F.; Baskin, D.G.; Schwartz, M.W. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1998, 1, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G.; Cowley, M.A.; Munzberg, H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef]
- Baumann, H.; Morella, K.K.; White, D.W.; Dembski, M.; Bailon, P.S.; Kim, H.; Lai, C.F.; Tartaglia, L.A. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 8374–8378. [Google Scholar] [CrossRef] [PubMed]
- Chua, S.C., Jr.; Chung, W.K.; Wu-Peng, X.S.; Zhang, Y.; Liu, S.M.; Tartaglia, L.; Leibel, R.L. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 1996, 271, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Bates, S.H.; Myers, M.G., Jr. The role of leptin receptor signaling in feeding and neuroendocrine function. Trends Endocrinol. Metab. 2003, 14, 447–452. [Google Scholar] [CrossRef]
- Uotani, S.; Bjorbaek, C.; Tornoe, J.; Flier, J.S. Functional properties of leptin receptor isoforms: Internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes 1999, 48, 279–286. [Google Scholar] [CrossRef]
- Schaab, M.; Kausch, H.; Klammt, J.; Nowicki, M.; Anderegg, U.; Gebhardt, R.; Rose-John, S.; Scheller, J.; Thiery, J.; Kratzsch, J. Novel regulatory mechanisms for generation of the soluble leptin receptor: Implications for leptin action. PLoS ONE 2012, 7, e34787. [Google Scholar] [CrossRef]
- Bjorbaek, C.; Uotani, S.; da Silva, B.; Flier, J.S. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, N.; Skoda, R.C. The leptin receptor activates janus kinase 2 and signals for proliferation in a factor-dependent cell line. Mol. Endocrinol. 1997, 11, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Friedman, J.M. Leptin receptor activation of SH2 domain containing protein tyrosine phosphatase 2 modulates Ob receptor signal transduction. Proc. Natl. Acad. Sci. USA 1999, 96, 9677–9682. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef]
- Bates, S.H.; Stearns, W.H.; Dundon, T.A.; Schubert, M.; Tso, A.W.; Wang, Y.; Banks, A.S.; Lavery, H.J.; Haq, A.K.; Maratos-Flier, E.; et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 2003, 421, 856–859. [Google Scholar] [CrossRef]
- Gao, Q.; Wolfgang, M.J.; Neschen, S.; Morino, K.; Horvath, T.L.; Shulman, G.I.; Fu, X.Y. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc. Natl. Acad. Sci. USA 2004, 101, 4661–4666. [Google Scholar] [CrossRef] [PubMed]
- Buettner, C.; Pocai, A.; Muse, E.D.; Etgen, A.M.; Myers, M.G., Jr.; Rossetti, L. Critical role of STAT3 in leptin’s metabolic actions. Cell Metab. 2006, 4, 49–60. [Google Scholar] [CrossRef]
- Zhang, J.; Scarpace, P.J. The soluble leptin receptor neutralizes leptin-mediated STAT3 signalling and anorexic responses in vivo. Br. J. Pharmacol. 2009, 158, 475–482. [Google Scholar] [CrossRef]
- Rahmouni, K.; Sigmund, C.D.; Haynes, W.G.; Mark, A.L. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes 2009, 58, 536–542. [Google Scholar] [CrossRef]
- Bjorbak, C.; Lavery, H.J.; Bates, S.H.; Olson, R.K.; Davis, S.M.; Flier, J.S.; Myers, M.G., Jr. SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J. Biol. Chem. 2000, 275, 40649–40657. [Google Scholar] [CrossRef]
- Bjornholm, M.; Munzberg, H.; Leshan, R.L.; Villanueva, E.C.; Bates, S.H.; Louis, G.W.; Jones, J.C.; Ishida-Takahashi, R.; Bjorbaek, C.; Myers, M.G., Jr. Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J. Clin. Investig. 2007, 117, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Munzberg, H.; Morrison, C.D. Structure, production and signaling of leptin. Metabolism 2015, 64, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Kellerer, M.; Koch, M.; Metzinger, E.; Mushack, J.; Capp, E.; Haring, H.U. Leptin activates PI-3 kinase in C2C12 myotubes via janus kinase-2 (JAK-2) and insulin receptor substrate-2 (IRS-2) dependent pathways. Diabetologia 1997, 40, 1358–1362. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, Y.B.; Uotani, S.; Pierroz, D.D.; Flier, J.S.; Kahn, B.B. In vivo administration of leptin activates signal transduction directly in insulin-sensitive tissues: Overlapping but distinct pathways from insulin. Endocrinology 2000, 141, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Anderwald, C.; Muller, G.; Koca, G.; Furnsinn, C.; Waldhausl, W.; Roden, M. Short-term leptin-dependent inhibition of hepatic gluconeogenesis is mediated by insulin receptor substrate-2. Mol. Endocrinol. 2002, 16, 1612–1628. [Google Scholar] [CrossRef]
- Carvalheira, J.B.; Ribeiro, E.B.; Folli, F.; Velloso, L.A.; Saad, M.J. Interaction between leptin and insulin signaling pathways differentially affects JAK-STAT and PI 3-kinase-mediated signaling in rat liver. Biol. Chem. 2003, 384, 151–159. [Google Scholar] [CrossRef]
- Duan, C.; Li, M.; Rui, L. SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J. Biol. Chem. 2004, 279, 43684–43691. [Google Scholar] [CrossRef]
- Wauman, J.; Zabeau, L.; Tavernier, J. The Leptin Receptor Complex: Heavier Than Expected? Front. Endocrinol. 2017, 8, 30. [Google Scholar] [CrossRef]
- Kim, M.S.; Pak, Y.K.; Jang, P.G.; Namkoong, C.; Choi, Y.S.; Won, J.C.; Kim, K.S.; Kim, S.W.; Kim, H.S.; Park, J.Y.; et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat. Neurosci. 2006, 9, 901–906. [Google Scholar] [CrossRef]
- Plum, L.; Lin, H.V.; Dutia, R.; Tanaka, J.; Aizawa, K.S.; Matsumoto, M.; Kim, A.J.; Cawley, N.X.; Paik, J.H.; Loh, Y.P.; et al. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat. Med. 2009, 15, 1195–1201. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Feng, Y.; Kitamura, Y.I.; Chua, S.C., Jr.; Xu, A.W.; Barsh, G.S.; Rossetti, L.; Accili, D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat. Med. 2006, 12, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Blouet, C.; Ono, H.; Schwartz, G.J. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab. 2008, 8, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferre, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Dagon, Y.; Hur, E.; Zheng, B.; Wellenstein, K.; Cantley, L.C.; Kahn, B.B. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 2012, 16, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M. Hypothalamic AMPK and energy balance. Eur. J. Clin. Investig. 2018, 48, e12996. [Google Scholar] [CrossRef]
- Su, H.; Jiang, L.; Carter-Su, C.; Rui, L. Glucose enhances leptin signaling through modulation of AMPK activity. PLoS ONE 2012, 7, e31636. [Google Scholar] [CrossRef]
- Thurlby, P.L.; Trayhurn, P. The role of thermoregulatory thermogenesis in the development of obesity in genetically-obese (ob/ob) mice pair-fed with lean siblings. Br. J. Nutr. 1979, 42, 377–385. [Google Scholar] [CrossRef]
- Breslow, M.J.; Min-Lee, K.; Brown, D.R.; Chacko, V.P.; Palmer, D.; Berkowitz, D.E. Effect of leptin deficiency on metabolic rate in ob/ob mice. Am. J. Physiol. 1999, 276, E443–E449. [Google Scholar] [CrossRef]
- Breslow, M.J.; An, Y.; Berkowitz, D.E. Beta-3 adrenoceptor (beta-3AR) expression in leptin treated OB/OB mice. Life Sci. 1997, 61, 59–64. [Google Scholar] [CrossRef]
- Collins, S.; Daniel, K.W.; Rohlfs, E.M.; Ramkumar, V.; Taylor, I.L.; Gettys, T.W. Impaired expression and functional activity of the beta 3- and beta 1-adrenergic receptors in adipose tissue of congenitally obese (C57BL/6J ob/ob) mice. Mol. Endocrinol. 1994, 8, 518–527. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trayhurn, P.; Thurlby, P.L.; James, W.P. Thermogenic defect in pre-obese ob/ob mice. Nature 1977, 266, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.; Zhou, J.; Redmann, S.M., Jr.; Smagin, G.N.; Smith, S.R.; Rodgers, E.; Zachwieja, J.J. A leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology 1998, 139, 8–19. [Google Scholar] [CrossRef]
- Contreras, C.; Nogueiras, R.; Dieguez, C.; Rahmouni, K.; Lopez, M. Traveling from the hypothalamus to the adipose tissue: The thermogenic pathway. Redox Biol. 2017, 12, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, N.; Moreno-Navarrete, J.M.; Contreras, C.; Rial-Pensado, E.; Ferno, J.; Nogueiras, R.; Dieguez, C.; Fernandez-Real, J.M.; Lopez, M. Thyroid hormones induce browning of white fat. J. Endocrinol. 2017, 232, 351–362. [Google Scholar] [CrossRef]
- Kaisanlahti, A.; Glumoff, T. Browning of white fat: Agents and implications for beige adipose tissue to type 2 diabetes. J. Physiol. Biochem. 2019, 75, 1–10. [Google Scholar] [CrossRef]
- Haynes, W.G.; Morgan, D.A.; Walsh, S.A.; Mark, A.L.; Sivitz, W.I. Receptor-mediated regional sympathetic nerve activation by leptin. J. Clin. Investig. 1997, 100, 270–278. [Google Scholar] [CrossRef]
- Dunbar, J.C.; Hu, Y.; Lu, H. Intracerebroventricular leptin increases lumbar and renal sympathetic nerve activity and blood pressure in normal rats. Diabetes 1997, 46, 2040–2043. [Google Scholar] [CrossRef]
- Richards, R.J.; Blalock, A.; Liao, J.; Reisin, E. Leptin: Sympathetic and cardiovascular effects. Curr. Cardiol. Rep. 2003, 5, 453–458. [Google Scholar] [CrossRef]
- Commins, S.P.; Watson, P.M.; Levin, N.; Beiler, R.J.; Gettys, T.W. Central leptin regulates the UCP1 and ob genes in brown and white adipose tissue via different beta-adrenoceptor subtypes. J. Biol. Chem. 2000, 275, 33059–33067. [Google Scholar] [CrossRef]
- Enriori, P.J.; Sinnayah, P.; Simonds, S.E.; Garcia Rudaz, C.; Cowley, M.A. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J. Neurosci. 2011, 31, 12189–12197. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.W.; Hoefig, C.S.; Abreu-Vieira, G.; de Jong, J.M.A.; Petrovic, N.; Mittag, J.; Cannon, B.; Nedergaard, J. Leptin Raises Defended Body Temperature without Activating Thermogenesis. Cell Rep. 2016, 14, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Doring, H.; Schwarzer, K.; Nuesslein-Hildesheim, B.; Schmidt, I. Leptin selectively increases energy expenditure of food-restricted lean mice. Int. J. Obes. 1998, 22, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Mistry, A.M.; Swick, A.G.; Romsos, D.R. Leptin rapidly lowers food intake and elevates metabolic rates in lean and ob/ob mice. J. Nutr. 1997, 127, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Rafael, J.; Herling, A.W. Leptin effect in ob/ob mice under thermoneutral conditions depends not necessarily on central satiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R790–R795. [Google Scholar] [CrossRef]
- Hogberg, H.; Engblom, L.; Ekdahl, A.; Lidell, V.; Walum, E.; Alberts, P. Temperature dependence of O2 consumption; opposite effects of leptin and etomoxir on respiratory quotient in mice. Obesity 2006, 14, 673–682. [Google Scholar] [CrossRef]
- Ukropec, J.; Anunciado, R.V.; Ravussin, Y.; Kozak, L.P. Leptin is required for uncoupling protein-1-independent thermogenesis during cold stress. Endocrinology 2006, 147, 2468–2480. [Google Scholar] [CrossRef]
- Kaiyala, K.J.; Ogimoto, K.; Nelson, J.T.; Muta, K.; Morton, G.J. Physiological role for leptin in the control of thermal conductance. Mol. Metab. 2016, 5, 892–902. [Google Scholar] [CrossRef]
- Dimicco, J.A.; Zaretsky, D.V. The dorsomedial hypothalamus: A new player in thermoregulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R47–R63. [Google Scholar] [CrossRef]
- Morrison, S.F.; Madden, C.J.; Tupone, D. Central control of brown adipose tissue thermogenesis. Front. Endocrinol. 2012, 3, 5. [Google Scholar] [CrossRef]
- Morrison, S.F.; Nakamura, K.; Madden, C.J. Central control of thermogenesis in mammals. Exp. Physiol. 2008, 93, 773–797. [Google Scholar] [CrossRef] [PubMed]
- Gautron, L.; Lazarus, M.; Scott, M.M.; Saper, C.B.; Elmquist, J.K. Identifying the efferent projections of leptin-responsive neurons in the dorsomedial hypothalamus using a novel conditional tracing approach. J. Comp. Neurol. 2010, 518, 2090–2108. [Google Scholar] [CrossRef] [PubMed]
- Rezai-Zadeh, K.; Yu, S.; Jiang, Y.; Laque, A.; Schwartzenburg, C.; Morrison, C.D.; Derbenev, A.V.; Zsombok, A.; Munzberg, H. Leptin receptor neurons in the dorsomedial hypothalamus are key regulators of energy expenditure and body weight, but not food intake. Mol. Metab. 2014, 3, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Cakir, I.; Diaz Martinez, M.; Lining Pan, P.; Welch, E.B.; Patel, S.; Ghamari-Langroudi, M. Leptin Receptor Signaling in Sim1-expressing Neurons Regulates Body Temperature and Adaptive Thermogenesis. Endocrinology 2019, 160, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Munzberg, H.; Flier, J.S.; Bjorbaek, C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 2004, 145, 4880–4889. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Munzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef]
- Elmquist, J.K.; Ahima, R.S.; Elias, C.F.; Flier, J.S.; Saper, C.B. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc. Natl. Acad. Sci. USA 1998, 95, 741–746. [Google Scholar] [CrossRef]
- Cao, W.H.; Morrison, S.F. Disinhibition of rostral raphe pallidus neurons increases cardiac sympathetic nerve activity and heart rate. Brain Res. 2003, 980, 1–10. [Google Scholar] [CrossRef]
- Simonds, S.E.; Cowley, M.A. Hypertension in obesity: Is leptin the culprit? Trends Neurosci. 2013, 36, 121–132. [Google Scholar] [CrossRef]
- Zhang, Y.; Kerman, I.A.; Laque, A.; Nguyen, P.; Faouzi, M.; Louis, G.W.; Jones, J.C.; Rhodes, C.; Munzberg, H. Leptin-receptor-expressing neurons in the dorsomedial hypothalamus and median preoptic area regulate sympathetic brown adipose tissue circuits. J. Neurosci. 2011, 31, 1873–1884. [Google Scholar] [CrossRef]
- Nakamura, K.; Morrison, S.F. Preoptic mechanism for cold-defensive responses to skin cooling. J. Physiol. 2008, 586, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K. Central circuitries for body temperature regulation and fever. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1207–R1228. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Li, X.; Cano, G.; Lazarus, M.; Saper, C.B. Parallel preoptic pathways for thermoregulation. J. Neurosci. 2009, 29, 11954–11964. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Matsumura, K.; Kobayashi, S.; Kaneko, T. Sympathetic premotor neurons mediating thermoregulatory functions. Neurosci. Res. 2005, 51, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Morgan, D.A.; Kuburas, A.; Thedens, D.R.; Russo, A.F.; Rahmouni, K. Neuronal receptor activity-modifying protein 1 promotes energy expenditure in mice. Diabetes 2011, 60, 1063–1071. [Google Scholar] [CrossRef]
- Song, C.K.; Vaughan, C.H.; Keen-Rhinehart, E.; Harris, R.B.; Richard, D.; Bartness, T.J. Melanocortin-4 receptor mRNA expressed in sympathetic outflow neurons to brown adipose tissue: Neuroanatomical and functional evidence. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R417–R428. [Google Scholar] [CrossRef]
- Morrison, S.F.; Madden, C.J.; Tupone, D. An orexinergic projection from perifornical hypothalamus to raphe pallidus increases rat brown adipose tissue thermogenesis. Adipocyte 2012, 1, 116–120. [Google Scholar] [CrossRef]
- Morrison, S.F.; Madden, C.J.; Tupone, D. Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab. 2014, 19, 741–756. [Google Scholar] [CrossRef]
- Madden, C.J.; Morrison, S.F. Neurons in the paraventricular nucleus of the hypothalamus inhibit sympathetic outflow to brown adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R831–R843. [Google Scholar] [CrossRef]
- Pandit, R.; Beerens, S.; Adan, R.A.H. Role of leptin in energy expenditure: The hypothalamic perspective. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 312, R938–R947. [Google Scholar] [CrossRef]
- Mogenson, G.J.; Swanson, L.W.; Wu, M. Neural projections from nucleus accumbens to globus pallidus, substantia innominata, and lateral preoptic-lateral hypothalamic area: An anatomical and electrophysiological investigation in the rat. J. Neurosci. 1983, 3, 189–202. [Google Scholar] [CrossRef] [PubMed]
- ter Horst, G.J.; Luiten, P.G. The projections of the dorsomedial hypothalamic nucleus in the rat. Brain Res. Bull 1986, 16, 231–248. [Google Scholar] [CrossRef]
- Thompson, R.H.; Canteras, N.S.; Swanson, L.W. Organization of projections from the dorsomedial nucleus of the hypothalamus: A PHA-L study in the rat. J. Comp. Neurol. 1996, 376, 143–173. [Google Scholar] [CrossRef]
- Satoh, N.; Ogawa, Y.; Katsuura, G.; Numata, Y.; Tsuji, T.; Hayase, M.; Ebihara, K.; Masuzaki, H.; Hosoda, K.; Yoshimasa, Y.; et al. Sympathetic activation of leptin via the ventromedial hypothalamus: Leptin-induced increase in catecholamine secretion. Diabetes 1999, 48, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Haque, M.S.; Shimazu, T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes 1999, 48, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Marsh, A.J.; Fontes, M.A.; Killinger, S.; Pawlak, D.B.; Polson, J.W.; Dampney, R.A. Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension 2003, 42, 488–493. [Google Scholar] [CrossRef]
- Montanaro, M.S.; Allen, A.M.; Oldfield, B.J. Structural and functional evidence supporting a role for leptin in central neural pathways influencing blood pressure in rats. Exp. Physiol. 2005, 90, 689–696. [Google Scholar] [CrossRef]
- Ikeda, Y.; Luo, X.; Abbud, R.; Nilson, J.H.; Parker, K.L. The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol. Endocrinol. 1995, 9, 478–486. [Google Scholar] [CrossRef]
- Elias, C.F.; Kelly, J.F.; Lee, C.E.; Ahima, R.S.; Drucker, D.J.; Saper, C.B.; Elmquist, J.K. Chemical characterization of leptin-activated neurons in the rat brain. J. Comp. Neurol. 2000, 423, 261–281. [Google Scholar] [CrossRef]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A.; et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef]
- Kim, K.W.; Zhao, L.; Donato, J., Jr.; Kohno, D.; Xu, Y.; Elias, C.F.; Lee, C.; Parker, K.L.; Elmquist, J.K. Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc. Natl. Acad. Sci. USA 2011, 108, 10673–10678. [Google Scholar] [CrossRef] [PubMed]
- Ovesjo, M.L.; Gamstedt, M.; Collin, M.; Meister, B. GABAergic nature of hypothalamic leptin target neurones in the ventromedial arcuate nucleus. J. Neuroendocrinol. 2001, 13, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q.; Ye, C.; McCrimmon, R.J.; Dhillon, H.; Choi, B.; Kramer, M.D.; Yu, J.; Yang, Z.; Christiansen, L.M.; Lee, C.E.; et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007, 5, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, D.; Chen, P.; Li, C. Conditional viral tracing reveals that steroidogenic factor 1-positive neurons of the dorsomedial subdivision of the ventromedial hypothalamus project to autonomic centers of the hypothalamus and hindbrain. J. Comp. Neurol. 2013, 521, 3167–3190. [Google Scholar] [CrossRef] [PubMed]
- Kenny, D.A.; Jurata, L.W.; Saga, Y.; Gill, G.N. Identification and characterization of LMO4, an LMO gene with a novel pattern of expression during embryogenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 11257–11262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Gomez-Smith, M.; Qin, Z.; Duquette, P.M.; Cardenas-Blanco, A.; Rai, P.S.; Harper, M.E.; Tsai, E.C.; Anisman, H.; Chen, H.H. Ablation of LMO4 in glutamatergic neurons impairs leptin control of fat metabolism. Cell. Mol. Life Sci. 2012, 69, 819–828. [Google Scholar] [CrossRef]
- Cardinal, P.; Andre, C.; Quarta, C.; Bellocchio, L.; Clark, S.; Elie, M.; Leste-Lasserre, T.; Maitre, M.; Gonzales, D.; Cannich, A.; et al. CB1 cannabinoid receptor in SF1-expressing neurons of the ventromedial hypothalamus determines metabolic responses to diet and leptin. Mol. Metab. 2014, 3, 705–716. [Google Scholar] [CrossRef]
- Kim, K.W.; Donato, J., Jr.; Berglund, E.D.; Choi, Y.H.; Kohno, D.; Elias, C.F.; Depinho, R.A.; Elmquist, J.K. FOXO1 in the ventromedial hypothalamus regulates energy balance. J. Clin. Investig. 2012, 122, 2578–2589. [Google Scholar] [CrossRef]
- Seamon, M.; Ahn, W.; Li, A.J.; Ritter, S.; Harris, R.B.S. Leptin receptor-expressing neurons in ventromedial nucleus of the hypothalamus contribute to weight loss caused by fourth ventricle leptin infusions. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E586–E596. [Google Scholar] [CrossRef]
- Rahmouni, K.; Morgan, D.A.; Morgan, G.M.; Liu, X.; Sigmund, C.D.; Mark, A.L.; Haynes, W.G. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J. Clin. Investig. 2004, 114, 652–658. [Google Scholar] [CrossRef]
- Rahmouni, K.; Morgan, D.A. Hypothalamic arcuate nucleus mediates the sympathetic and arterial pressure responses to leptin. Hypertension 2007, 49, 647–652. [Google Scholar] [CrossRef]
- Harlan, S.M.; Morgan, D.A.; Agassandian, K.; Guo, D.F.; Cassell, M.D.; Sigmund, C.D.; Mark, A.L.; Rahmouni, K. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ. Res. 2011, 108, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.D. The Central Melanocortin System and Energy Homeostasis. Trends Endocrinol. Metab. 1999, 10, 211–216. [Google Scholar] [CrossRef]
- Shi, Y.C.; Lau, J.; Lin, Z.; Zhang, H.; Zhai, L.; Sperk, G.; Heilbronn, R.; Mietzsch, M.; Weger, S.; Huang, X.F.; et al. Arcuate NPY controls sympathetic output and BAT function via a relay of tyrosine hydroxylase neurons in the PVN. Cell Metab. 2013, 17, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.C.; Hollopeter, G.; Palmiter, R.D. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 1996, 274, 1704–1707. [Google Scholar] [CrossRef]
- Billington, C.J.; Briggs, J.E.; Grace, M.; Levine, A.S. Effects of intracerebroventricular injection of neuropeptide Y on energy metabolism. Am. J. Physiol. 1991, 260, R321–R327. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Valpuesta, F.J.; Nyce, J.W.; Griffin-Biggs, T.A.; Ice, J.C.; Myers, R.D. Antisense to NPY-Y1 demonstrates that Y1 receptors in the hypothalamus underlie NPY hypothermia and feeding in rats. Proc. Biol. Sci. 1996, 263, 881–886. [Google Scholar] [CrossRef]
- Stanley, B.G.; Kyrkouli, S.E.; Lampert, S.; Leibowitz, S.F. Neuropeptide Y chronically injected into the hypothalamus: A powerful neurochemical inducer of hyperphagia and obesity. Peptides 1986, 7, 1189–1192. [Google Scholar] [CrossRef]
- Cusin, I.; Rohner-Jeanrenaud, F.; Stricker-Krongrad, A.; Jeanrenaud, B. The weight-reducing effect of an intracerebroventricular bolus injection of leptin in genetically obese fa/fa rats. Reduced sensitivity compared with lean animals. Diabetes 1996, 45, 1446–1450. [Google Scholar] [CrossRef][Green Version]
- Sahu, A. Leptin decreases food intake induced by melanin-concentrating hormone (MCH), galanin (GAL) and neuropeptide Y (NPY) in the rat. Endocrinology 1998, 139, 4739–4742. [Google Scholar] [CrossRef]
- Jang, M.; Mistry, A.; Swick, A.G.; Romsos, D.R. Leptin rapidly inhibits hypothalamic neuropeptide Y secretion and stimulates corticotropin-releasing hormone secretion in adrenalectomized mice. J. Nutr. 2000, 130, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bing, C.; Al-Barazanji, K.; Mossakowaska, D.E.; Wang, X.M.; McBay, D.L.; Neville, W.A.; Taddayon, M.; Pickavance, L.; Dryden, S.; et al. Interactions between leptin and hypothalamic neuropeptide Y neurons in the control of food intake and energy homeostasis in the rat. Diabetes 1997, 46, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Valpuesta, F.J.; Nyce, J.W.; Myers, R.D. NPY-Y1 receptor antisense injected centrally in rats causes hyperthermia and feeding. Neuroreport 1996, 7, 2781–2784. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.M.; Kleopoulos, S.P.; Bergen, H.T.; Roberts, J.L.; Priest, C.A.; Mobbs, C.V. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting in ob/ob and db/db mice, but is stimulated by leptin. Diabetes 1998, 47, 294–297. [Google Scholar] [CrossRef]
- do Carmo, J.M.; da Silva, A.A.; Cai, Z.; Lin, S.; Dubinion, J.H.; Hall, J.E. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension 2011, 57, 918–926. [Google Scholar] [CrossRef]
- Huo, L.; Grill, H.J.; Bjorbaek, C. Divergent regulation of proopiomelanocortin neurons by leptin in the nucleus of the solitary tract and in the arcuate hypothalamic nucleus. Diabetes 2006, 55, 567–573. [Google Scholar] [CrossRef]
- Kishi, T.; Aschkenasi, C.J.; Lee, C.E.; Mountjoy, K.G.; Saper, C.B.; Elmquist, J.K. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J. Comp. Neurol. 2003, 457, 213–235. [Google Scholar] [CrossRef]
- Begriche, K.; Sutton, G.M.; Butler, A.A. Homeostastic and non-homeostatic functions of melanocortin-3 receptors in the control of energy balance and metabolism. Physiol. Behav. 2011, 104, 546–554. [Google Scholar] [CrossRef]
- Seeley, R.J.; Yagaloff, K.A.; Fisher, S.L.; Burn, P.; Thiele, T.E.; van Dijk, G.; Baskin, D.G.; Schwartz, M.W. Melanocortin receptors in leptin effects. Nature 1997, 390, 349. [Google Scholar] [CrossRef]
- Haynes, W.G.; Morgan, D.A.; Djalali, A.; Sivitz, W.I.; Mark, A.L. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension 1999, 33, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L.; Bowers, R.R.; Bartness, T.J.; Kaplan, J.M.; Grill, H.J. Brainstem melanocortin 3/4 receptor stimulation increases uncoupling protein gene expression in brown fat. Endocrinology 2003, 144, 4692–4697. [Google Scholar] [CrossRef] [PubMed]
- Ste Marie, L.; Miura, G.I.; Marsh, D.J.; Yagaloff, K.; Palmiter, R.D. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 12339–12344. [Google Scholar] [CrossRef] [PubMed]
- Rahmouni, K.; Haynes, W.G.; Morgan, D.A.; Mark, A.L. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J. Neurosci. 2003, 23, 5998–6004. [Google Scholar] [CrossRef] [PubMed]
- Voss-Andreae, A.; Murphy, J.G.; Ellacott, K.L.; Stuart, R.C.; Nillni, E.A.; Cone, R.D.; Fan, W. Role of the central melanocortin circuitry in adaptive thermogenesis of brown adipose tissue. Endocrinology 2007, 148, 1550–1560. [Google Scholar] [CrossRef]
- Lute, B.; Jou, W.; Lateef, D.M.; Goldgof, M.; Xiao, C.; Pinol, R.A.; Kravitz, A.V.; Miller, N.R.; Huang, Y.G.; Girardet, C.; et al. Biphasic effect of melanocortin agonists on metabolic rate and body temperature. Cell Metab. 2014, 20, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Baran, K.; Preston, E.; Wilks, D.; Cooney, G.J.; Kraegen, E.W.; Sainsbury, A. Chronic central melanocortin-4 receptor antagonism and central neuropeptide-Y infusion in rats produce increased adiposity by divergent pathways. Diabetes 2002, 51, 152–158. [Google Scholar] [CrossRef]
- Geerling, J.C.; Loewy, A.D. Aldosterone-sensitive neurons in the nucleus of the solitary tract: Bidirectional connections with the central nucleus of the amygdala. J. Comp. Neurol. 2006, 497, 646–657. [Google Scholar] [CrossRef]
- Grill, H.J.; Schwartz, M.W.; Kaplan, J.M.; Foxhall, J.S.; Breininger, J.; Baskin, D.G. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology 2002, 143, 239–246. [Google Scholar] [CrossRef]
- Hermann, G.E.; Barnes, M.J.; Rogers, R.C. Leptin and thyrotropin-releasing hormone: Cooperative action in the hindbrain to activate brown adipose thermogenesis. Brain Res. 2006, 1117, 118–124. [Google Scholar] [CrossRef]
- Mark, A.L.; Agassandian, K.; Morgan, D.A.; Liu, X.; Cassell, M.D.; Rahmouni, K. Leptin signaling in the nucleus tractus solitarii increases sympathetic nerve activity to the kidney. Hypertension 2009, 53, 375–380. [Google Scholar] [CrossRef]
- Rogers, R.C.; Barnes, M.J.; Hermann, G.E. Leptin “gates” thermogenic action of thyrotropin-releasing hormone in the hindbrain. Brain Res. 2009, 1295, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.C.; McDougal, D.H.; Hermann, G.E. Leptin amplifies the action of thyrotropin-releasing hormone in the solitary nucleus: An in vitro calcium imaging study. Brain Res. 2011, 1385, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Matheny, M.; Scarpace, P.J. beta 3-Adrenergic-mediated suppression of leptin gene expression in rats. Am. J. Physiol. 1997, 272, E1031–E1036. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Duncan, J.S.; Rayner, D.V. Acute cold-induced suppression of ob (obese) gene expression in white adipose tissue of mice: Mediation by the sympathetic system. Biochem. J. 1995, 311, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 2015, 163, 84–94. [Google Scholar] [CrossRef]
- Plum, L.; Rother, E.; Munzberg, H.; Wunderlich, F.T.; Morgan, D.A.; Hampel, B.; Shanabrough, M.; Janoschek, R.; Konner, A.C.; Alber, J.; et al. Enhanced leptin-stimulated Pi3k activation in the CNS promotes white adipose tissue transdifferentiation. Cell Metab. 2007, 6, 431–445. [Google Scholar] [CrossRef]
- Dodd, G.T.; Andrews, Z.B.; Simonds, S.E.; Michael, N.J.; DeVeer, M.; Bruning, J.C.; Spanswick, D.; Cowley, M.A.; Tiganis, T. A Hypothalamic Phosphatase Switch Coordinates Energy Expenditure with Feeding. Cell Metab. 2017, 26, 577. [Google Scholar] [CrossRef]
- Ruan, H.B.; Dietrich, M.O.; Liu, Z.W.; Zimmer, M.R.; Li, M.D.; Singh, J.P.; Zhang, K.; Yin, R.; Wu, J.; Horvath, T.L.; et al. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 2014, 159, 306–317. [Google Scholar] [CrossRef]
- Berglund, E.D.; Liu, T.; Kong, X.; Sohn, J.W.; Vong, L.; Deng, Z.; Lee, C.E.; Lee, S.; Williams, K.W.; Olson, D.P.; et al. Melanocortin 4 receptors in autonomic neurons regulate thermogenesis and glycemia. Nat. Neurosci. 2014, 17, 911–913. [Google Scholar] [CrossRef]
- Lidell, M.E.; Seifert, E.L.; Westergren, R.; Heglind, M.; Gowing, A.; Sukonina, V.; Arani, Z.; Itkonen, P.; Wallin, S.; Westberg, F.; et al. The adipocyte-expressed forkhead transcription factor Foxc2 regulates metabolism through altered mitochondrial function. Diabetes 2011, 60, 427–435. [Google Scholar] [CrossRef]
- Cederberg, A.; Gronning, L.M.; Ahren, B.; Tasken, K.; Carlsson, P.; Enerback, S. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell 2001, 106, 563–573. [Google Scholar] [CrossRef]
- Rahman, S.; Lu, Y.; Czernik, P.J.; Rosen, C.J.; Enerback, S.; Lecka-Czernik, B. Inducible brown adipose tissue, or beige fat, is anabolic for the skeleton. Endocrinology 2013, 154, 2687–2701. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Liu, Z.; Feng, F.; Wu, T.; Luo, D.; Hu, C.; Sun, C. Foxc2 coordinates inflammation and browning of white adipose by leptin-STAT3-PRDM16 signal in mice. Int. J. Obes. 2018, 42, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ge, J.; Cao, H.; Zhang, X.; Guo, Y.; Li, X.; Xia, B.; Yang, G.; Shi, X. Leptin Promotes White Adipocyte Browning by Inhibiting the Hh Signaling Pathway. Cells 2019, 8. [Google Scholar] [CrossRef]
- Tartaglia, L.A. The leptin receptor. J. Biol. Chem. 1997, 272, 6093–6096. [Google Scholar] [CrossRef]
- Zhang, Y.; Kilroy, G.E.; Henagan, T.M.; Prpic-Uhing, V.; Richards, W.G.; Bannon, A.W.; Mynatt, R.L.; Gettys, T.W. Targeted deletion of melanocortin receptor subtypes 3 and 4, but not CART, alters nutrient partitioning and compromises behavioral and metabolic responses to leptin. FASEB J. 2005, 19, 1482–1491. [Google Scholar] [CrossRef]
- Siegrist-Kaiser, C.A.; Pauli, V.; Juge-Aubry, C.E.; Boss, O.; Pernin, A.; Chin, W.W.; Cusin, I.; Rohner-Jeanrenaud, F.; Burger, A.G.; Zapf, J.; et al. Direct effects of leptin on brown and white adipose tissue. J. Clin. Investig. 1997, 100, 2858–2864. [Google Scholar] [CrossRef]
- Simonds, S.E.; Pryor, J.T.; Ravussin, E.; Greenway, F.L.; Dileone, R.; Allen, A.M.; Bassi, J.; Elmquist, J.K.; Keogh, J.M.; Henning, E.; et al. Leptin mediates the increase in blood pressure associated with obesity. Cell 2014, 159, 1404–1416. [Google Scholar] [CrossRef]
- Dulloo, A.G.; Stock, M.J.; Solinas, G.; Boss, O.; Montani, J.P.; Seydoux, J. Leptin directly stimulates thermogenesis in skeletal muscle. FEBS Lett. 2002, 515, 109–113. [Google Scholar] [CrossRef]
- Paracchini, V.; Pedotti, P.; Taioli, E. Genetics of leptin and obesity: A HuGE review. Am. J. Epidemiol. 2005, 162, 101–114. [Google Scholar] [CrossRef]
- Mizuta, E.; Kokubo, Y.; Yamanaka, I.; Miyamoto, Y.; Okayama, A.; Yoshimasa, Y.; Tomoike, H.; Morisaki, H.; Morisaki, T. Leptin gene and leptin receptor gene polymorphisms are associated with sweet preference and obesity. Hypertens. Res. 2008, 31, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Casamassimi, A.; Ciccodicola, A. Nutritional genomics era: Opportunities toward a genome-tailored nutritional regimen. J. Nutr. Biochem. 2010, 21, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Ghalandari, H.; Hosseini-Esfahani, F.; Mirmiran, P. The Association of Polymorphisms in Leptin/Leptin Receptor Genes and Ghrelin/Ghrelin Receptor Genes With Overweight/Obesity and the Related Metabolic Disturbances: A Review. Int. J. Endocrinol. Metab. 2015, 13, e19073. [Google Scholar] [CrossRef]
- Murugesan, D.; Arunachalam, T.; Ramamurthy, V.; Subramanian, S. Association of polymorphisms in leptin receptor gene with obesity and type 2 diabetes in the local population of Coimbatore. Indian J. Hum. Genet. 2010, 16, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, T.; Naka, I.; Yamauchi, T.; Natsuhara, K.; Kimura, R.; Nakazawa, M.; Ishida, T.; Inaoka, T.; Matsumura, Y.; Ataka, Y.; et al. The Q223R polymorphism in LEPR is associated with obesity in Pacific Islanders. Hum. Genet. 2010, 127, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Mahendran, Y.; Dwivedi, O.P.; Chauhan, G.; Ghosh, S.; Marwaha, R.K.; Tandon, N.; Bharadwaj, D. Common variants of IL6, LEPR, and PBEF1 are associated with obesity in Indian children. Diabetes 2012, 61, 626–631. [Google Scholar] [CrossRef]
- Boumaiza, I.; Omezzine, A.; Rejeb, J.; Rebhi, L.; Ben Rejeb, N.; Nabli, N.; Ben Abdelaziz, A.; Bouslama, A. Association between four resistin polymorphisms, obesity, and metabolic syndrome parameters in Tunisian volunteers. Genet. Test Mol. Biomark. 2012, 16, 1356–1362. [Google Scholar] [CrossRef]
- Etemad, A.; Ramachandran, V.; Pishva, S.R.; Heidari, F.; Aziz, A.F.; Yusof, A.K.; Pei, C.P.; Ismail, P. Analysis of Gln223Agr polymorphism of Leptin Receptor Gene in type II diabetic mellitus subjects among Malaysians. Int. J. Mol. Sci. 2013, 14, 19230–19244. [Google Scholar] [CrossRef]
- Lu, J.; Zou, D.; Zheng, L.; Chen, G.; Lu, J.; Feng, Z. Synergistic effect of LEP and LEPR gene polymorphism on body mass index in a Chinese population. Obes. Res. Clin. Pract. 2013, 7, e445–e449. [Google Scholar] [CrossRef]
- Heo, M.; Leibel, R.L.; Fontaine, K.R.; Boyer, B.B.; Chung, W.K.; Koulu, M.; Karvonen, M.K.; Pesonen, U.; Rissanen, A.; Laakso, M.; et al. A meta-analytic investigation of linkage and association of common leptin receptor (LEPR) polymorphisms with body mass index and waist circumference. Int. J. Obes. 2002, 26, 640–646. [Google Scholar] [CrossRef]
- Komsu-Ornek, Z.; Demirel, F.; Dursun, A.; Ermis, B.; Piskin, E.; Bideci, A. Leptin receptor gene Gln223Arg polymorphism is not associated with obesity and metabolic syndrome in Turkish children. Turk. J. Pediatr. 2012, 54, 20–24. [Google Scholar] [PubMed]
- Pyrzak, B.; Wisniewska, A.; Kucharska, A.; Wasik, M.; Demkow, U. No association of LEPR Gln223Arg polymorphism with leptin, obesity or metabolic disturbances in children. Eur. J. Med. Res. 2009, 14, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Oral, E.A.; Simha, V.; Ruiz, E.; Andewelt, A.; Premkumar, A.; Snell, P.; Wagner, A.J.; DePaoli, A.M.; Reitman, M.L.; Taylor, S.I.; et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 2002, 346, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Welt, C.K.; Chan, J.L.; Bullen, J.; Murphy, R.; Smith, P.; DePaoli, A.M.; Karalis, A.; Mantzoros, C.S. Recombinant human leptin in women with hypothalamic amenorrhea. N. Engl. J. Med. 2004, 351, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Chamberland, J.P.; Liu, X.; Matarese, G.; Gao, C.; Stefanakis, R.; Brinkoetter, M.T.; Gong, H.; Arampatzi, K.; Mantzoros, C.S. Leptin is an effective treatment for hypothalamic amenorrhea. Proc. Natl. Acad. Sci. USA 2011, 108, 6585–6590. [Google Scholar] [CrossRef] [PubMed]
- Paz-Filho, G.; Mastronardi, C.A.; Licinio, J. Leptin treatment: Facts and expectations. Metabolism 2015, 64, 146–156. [Google Scholar] [CrossRef]
- Frederich, R.C.; Hamann, A.; Anderson, S.; Lollmann, B.; Lowell, B.B.; Flier, J.S. Leptin levels reflect body lipid content in mice: Evidence for diet-induced resistance to leptin action. Nat. Med. 1995, 1, 1311–1314. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Heymsfield, S.B.; Haft, C.; Kahn, B.B.; Laughlin, M.; Leibel, R.L.; Tschop, M.H.; Yanovski, J.A. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012, 15, 150–156. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Greenberg, A.S.; Fujioka, K.; Dixon, R.M.; Kushner, R.; Hunt, T.; Lubina, J.A.; Patane, J.; Self, B.; Hunt, P.; et al. Recombinant leptin for weight loss in obese and lean adults: A randomized, controlled, dose-escalation trial. JAMA 1999, 282, 1568–1575. [Google Scholar] [CrossRef]
- Westerterp-Plantenga, M.S.; Saris, W.H.; Hukshorn, C.J.; Campfield, L.A. Effects of weekly administration of pegylated recombinant human OB protein on appetite profile and energy metabolism in obese men. Am. J. Clin. Nutr. 2001, 74, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Hukshorn, C.J.; van Dielen, F.M.; Buurman, W.A.; Westerterp-Plantenga, M.S.; Campfield, L.A.; Saris, W.H. The effect of pegylated recombinant human leptin (PEG-OB) on weight loss and inflammatory status in obese subjects. Int. J. Obes. 2002, 26, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Hukshorn, C.J.; Westerterp-Plantenga, M.S.; Saris, W.H. Pegylated human recombinant leptin (PEG-OB) causes additional weight loss in severely energy-restricted, overweight men. Am. J. Clin. Nutr. 2003, 77, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Zelissen, P.M.; Stenlof, K.; Lean, M.E.; Fogteloo, J.; Keulen, E.T.; Wilding, J.; Finer, N.; Rossner, S.; Lawrence, E.; Fletcher, C.; et al. Effect of three treatment schedules of recombinant methionyl human leptin on body weight in obese adults: A randomized, placebo-controlled trial. Diabetes Obes. Metab. 2005, 7, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Korner, J.; Conroy, R.; Febres, G.; McMahon, D.J.; Conwell, I.; Karmally, W.; Aronne, L.J. Randomized double-blind placebo-controlled study of leptin administration after gastric bypass. Obesity 2013, 21, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Shetty, G.K.; Matarese, G.; Magkos, F.; Moon, H.S.; Liu, X.; Brennan, A.M.; Mylvaganam, G.; Sykoutri, D.; Depaoli, A.M.; Mantzoros, C.S. Leptin administration to overweight and obese subjects for 6 months increases free leptin concentrations but does not alter circulating hormones of the thyroid and IGF axes during weight loss induced by a mild hypocaloric diet. Eur. J. Endocrinol. 2011, 165, 249–254. [Google Scholar] [CrossRef]
- Quarta, C.; Sanchez-Garrido, M.A.; Tschop, M.H.; Clemmensen, C. Renaissance of leptin for obesity therapy. Diabetologia 2016, 59, 920–927. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Murphy, E.M.; Heymsfield, S.B.; Matthews, D.E.; Leibel, R.L. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J. Clin. Endocrinol. Metab. 2002, 87, 2391–2394. [Google Scholar] [CrossRef]
- Munzberg, H.; Myers, M.G., Jr. Molecular and anatomical determinants of central leptin resistance. Nat. Neurosci. 2005, 8, 566–570. [Google Scholar] [CrossRef]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef]
- Banks, W.A. The blood-brain barrier as a cause of obesity. Curr. Pharm. Des. 2008, 14, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.L.; Findlay, P.A. Decreased blood-brain leptin transfer in an ovine model of obesity and weight loss: Resolving the cause of leptin resistance. Int. J. Obes. 2010, 34, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.D.; Roland, B.L.; Cole, R.L.; Trevaskis, J.L.; Weyer, C.; Koda, J.E.; Anderson, C.M.; Parkes, D.G.; Baron, A.D. Leptin responsiveness restored by amylin agonism in diet-induced obesity: Evidence from nonclinical and clinical studies. Proc. Natl. Acad. Sci. USA 2008, 105, 7257–7262. [Google Scholar] [CrossRef]
- Ravussin, E.; Smith, S.R.; Mitchell, J.A.; Shringarpure, R.; Shan, K.; Maier, H.; Koda, J.E.; Weyer, C. Enhanced weight loss with pramlintide/metreleptin: An integrated neurohormonal approach to obesity pharmacotherapy. Obesity 2009, 17, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, S.; Watkins, J.; Young, A. Synergy between amylin and cholecystokinin for inhibition of food intake in mice. Physiol. Behav. 1998, 64, 557–561. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Lei, C.; Koda, J.E.; Weyer, C.; Parkes, D.G.; Roth, J.D. Interaction of leptin and amylin in the long-term maintenance of weight loss in diet-induced obese rats. Obesity 2010, 18, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L.; Baskin, D.G.; Schwartz, M.W. Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes 2006, 55, 3387–3393. [Google Scholar] [CrossRef]
- Bojanowska, E.; Nowak, A. Interactions between leptin and exendin-4, a glucagon-like peptide-1 agonist, in the regulation of food intake in the rat. J. Physiol. Pharmacol. 2007, 58, 349–360. [Google Scholar]
- Zhao, S.; Kanoski, S.E.; Yan, J.; Grill, H.J.; Hayes, M.R. Hindbrain leptin and glucagon-like-peptide-1 receptor signaling interact to suppress food intake in an additive manner. Int. J. Obes. 2012, 36, 1522–1528. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Ong, Z.Y.; Fortin, S.M.; Schlessinger, E.S.; Grill, H.J. Liraglutide, leptin and their combined effects on feeding: Additive intake reduction through common intracellular signalling mechanisms. Diabetes Obes. Metab. 2015, 17, 285–293. [Google Scholar] [CrossRef]
- Muller, T.D.; Sullivan, L.M.; Habegger, K.; Yi, C.X.; Kabra, D.; Grant, E.; Ottaway, N.; Krishna, R.; Holland, J.; Hembree, J.; et al. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. J. Pept. Sci. 2012, 18, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Clemmensen, C.; Chabenne, J.; Finan, B.; Sullivan, L.; Fischer, K.; Kuchler, D.; Sehrer, L.; Ograjsek, T.; Hofmann, S.M.; Schriever, S.C.; et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes 2014, 63, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Yang, Y.; Saito, K.; Xu, P.; Wang, C.; Hinton, A.O., Jr.; Yan, X.; Wu, Q.; Tong, Q.; Elmquist, J.K.; et al. Meta-chlorophenylpiperazine enhances leptin sensitivity in diet-induced obese mice. Br. J. Pharmacol. 2015, 172, 3510–3521. [Google Scholar] [CrossRef] [PubMed]
- Labyb, M.; Chretien, C.; Caillon, A.; Rohner-Jeanrenaud, F.; Altirriba, J. Oxytocin Administration Alleviates Acute but Not Chronic Leptin Resistance of Diet-Induced Obese Mice. Int. J. Mol. Sci. 2018, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Folgueira, C.; Beiroa, D.; Gonzalez-Rellan, M.J.; Porteiro, B.; Milbank, E.; Castelao, C.; Garcia-Palacios, M.; Casanueva, F.F.; Lopez, M.; Dieguez, C.; et al. Uroguanylin Improves Leptin Responsiveness in Diet-Induced Obese Mice. Nutrients 2019, 11, 752. [Google Scholar] [CrossRef]
- Saladin, R.; De Vos, P.; Guerre-Millo, M.; Leturque, A.; Girard, J.; Staels, B.; Auwerx, J. Transient increase in obese gene expression after food intake or insulin administration. Nature 1995, 377, 527–529. [Google Scholar] [CrossRef]
- Kiess, W.; Englaro, P.; Hanitsch, S.; Rascher, W.; Attanasio, A.; Blum, W.F. High leptin concentrations in serum of very obese children are further stimulated by dexamethasone. Horm. Metab. Res. 1996, 28, 708–710. [Google Scholar] [CrossRef]
- Zhao, Y.; Dunbar, J.D.; Kharitonenkov, A. FGF21 as a therapeutic reagent. Adv. Exp. Med. Biol. 2012, 728, 214–228. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seoane-Collazo, P.; Martínez-Sánchez, N.; Milbank, E.; Contreras, C. Incendiary Leptin. Nutrients 2020, 12, 472. https://doi.org/10.3390/nu12020472
Seoane-Collazo P, Martínez-Sánchez N, Milbank E, Contreras C. Incendiary Leptin. Nutrients. 2020; 12(2):472. https://doi.org/10.3390/nu12020472
Chicago/Turabian StyleSeoane-Collazo, Patricia, Noelia Martínez-Sánchez, Edward Milbank, and Cristina Contreras. 2020. "Incendiary Leptin" Nutrients 12, no. 2: 472. https://doi.org/10.3390/nu12020472
APA StyleSeoane-Collazo, P., Martínez-Sánchez, N., Milbank, E., & Contreras, C. (2020). Incendiary Leptin. Nutrients, 12(2), 472. https://doi.org/10.3390/nu12020472