Luteolin Targets the Toll-Like Receptor Signaling Pathway in Prevention of Hepatic and Adipocyte Fibrosis and Insulin Resistance in Diet-Induced Obese Mice

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Diets 17.65 82.35
2.2. Blood Glucose, IPGTT, and Plasma Biomarkers
2.3. Plasma and Hepatic Free Fatty Acid (FFA) Measurements
2.4. Histopathological Analysis
2.5. Hepatic Enzyme Activities and Lipotoxicity Markers
2.6. RNA Preparation and Quality Control
2.7. RT-qPCR
2.8. Microarray Analysis
2.9. Statistical Analysis
3. Results
3.1. Luteolin Lowered Body Fat in Diet-Induced Obese (DIO) Mice
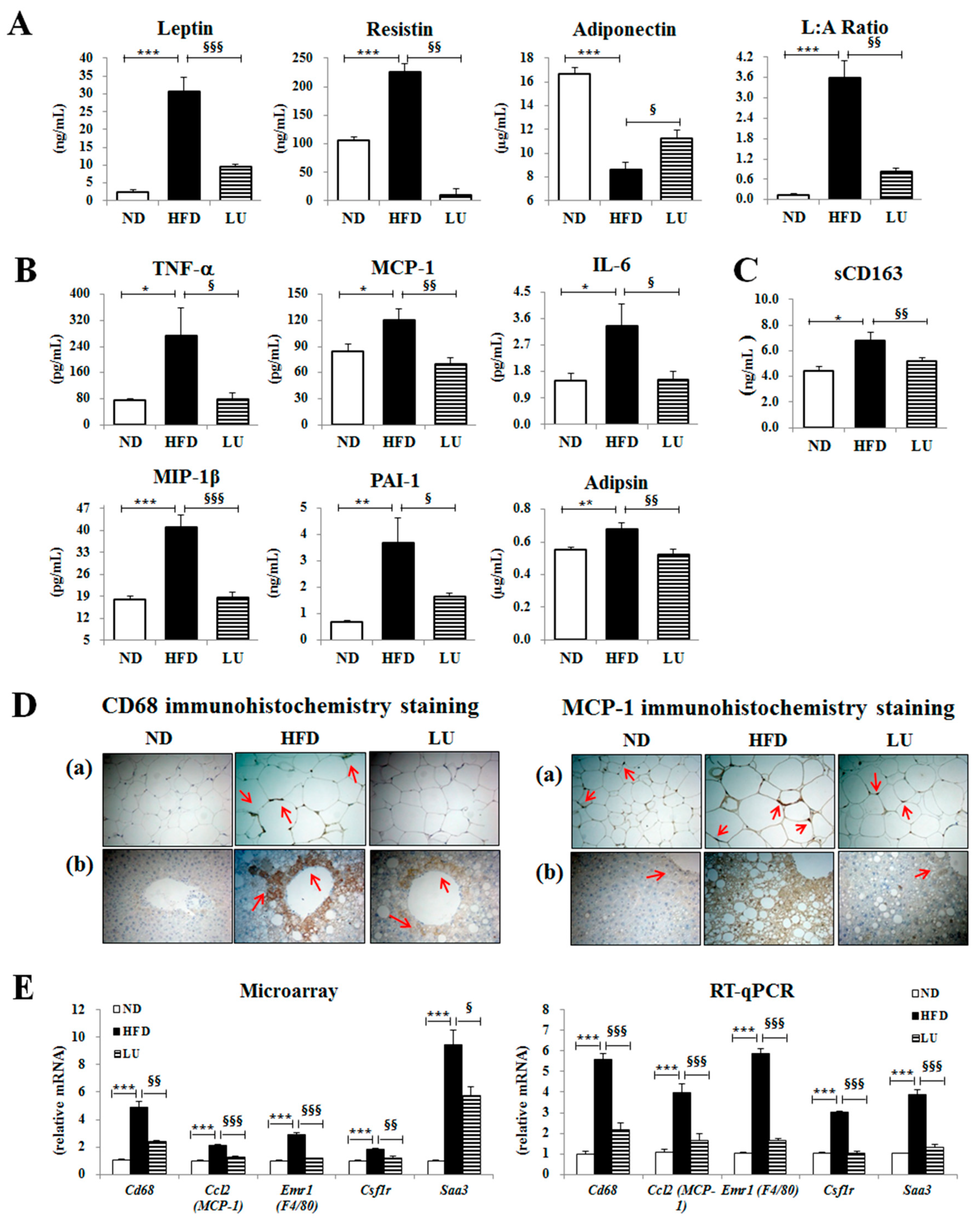
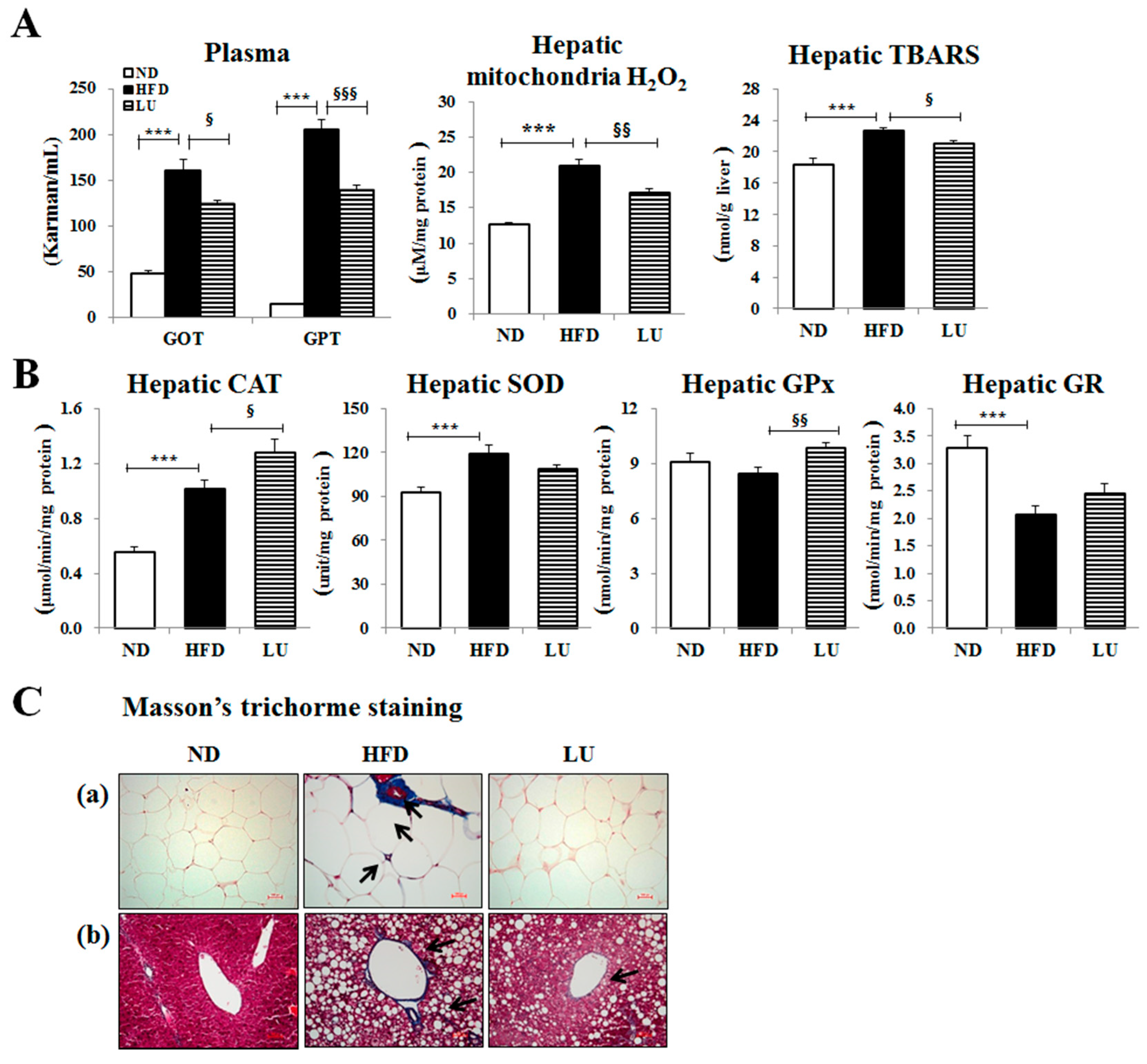
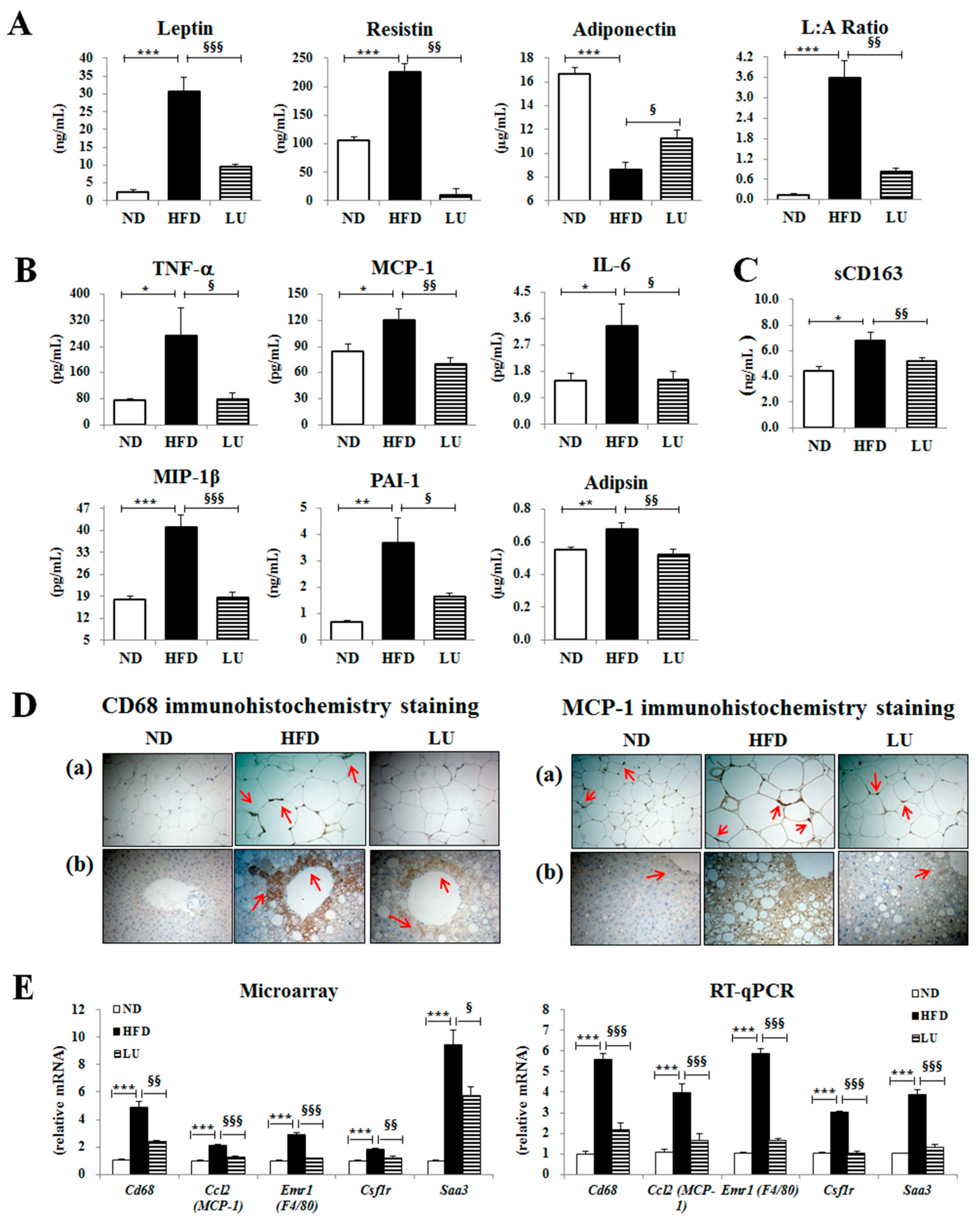
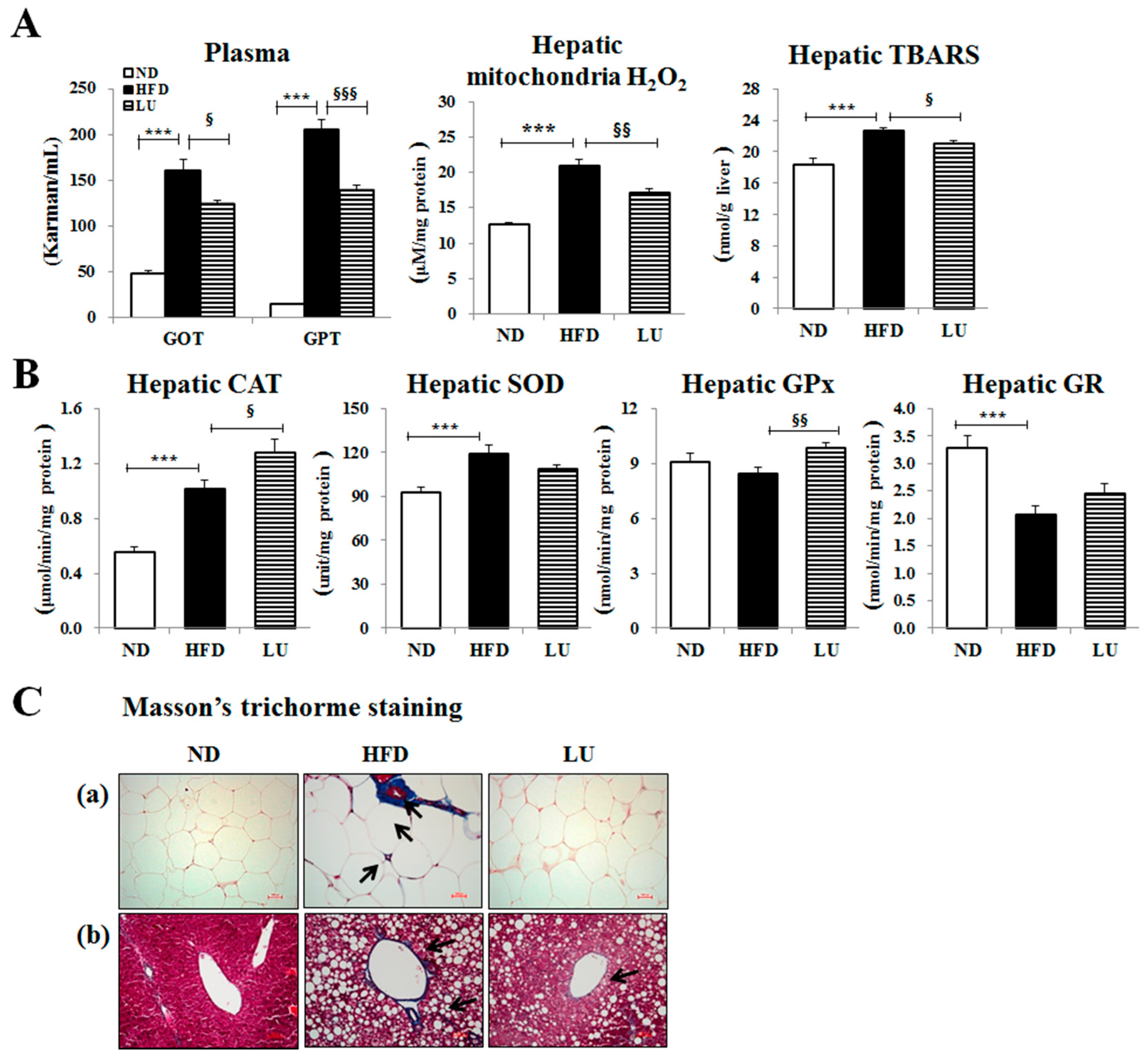
3.2. Luteolin Reduced Macrophage Infiltration and Fibrosis by Modulating the Inflammatory Response and Activities of Hepatic Antioxidant Enzymes in DIO Mice
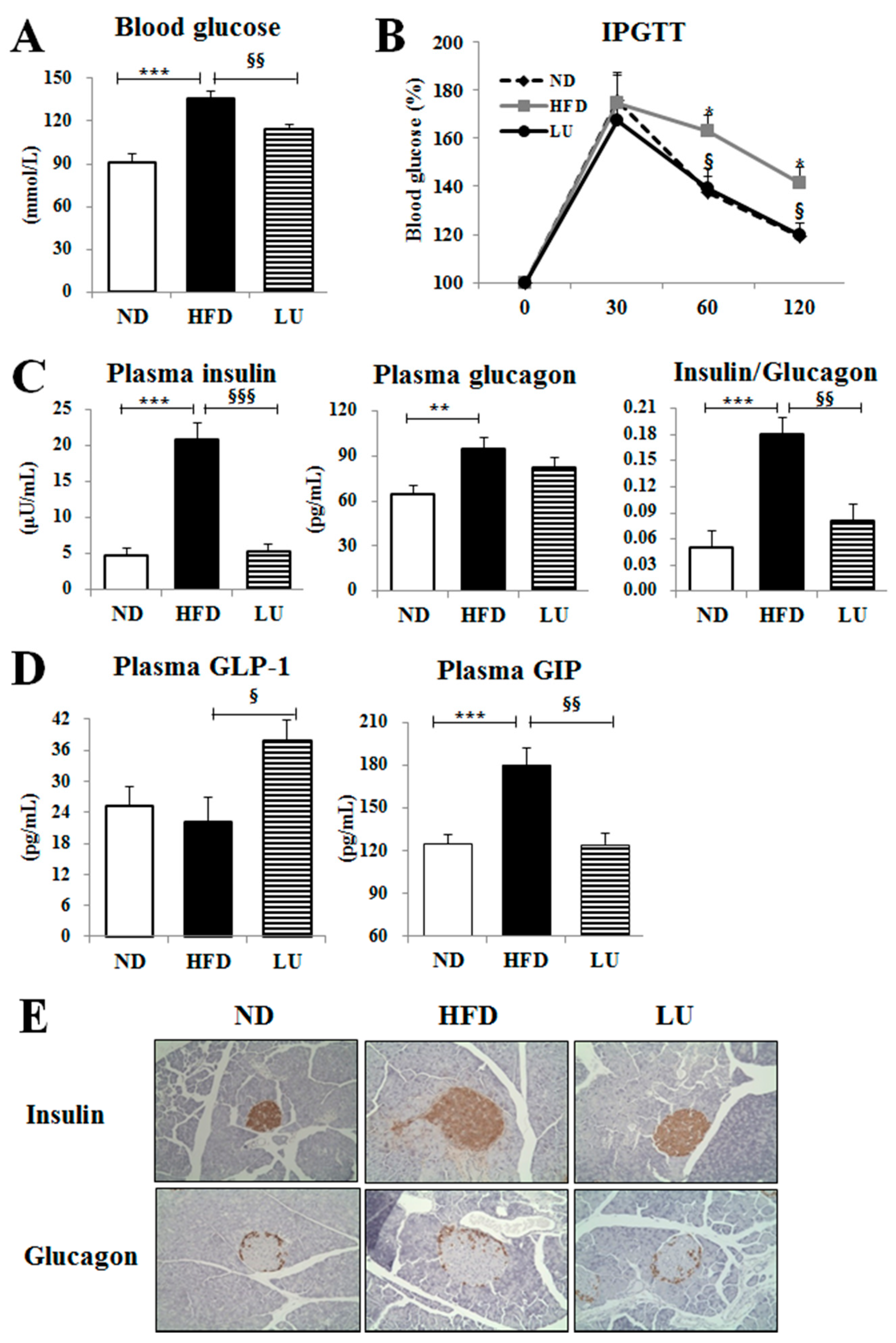
3.3. Luteolin Reduced IR and Normalized the Pancreatic Beta-Cell Mass in DIO Mice
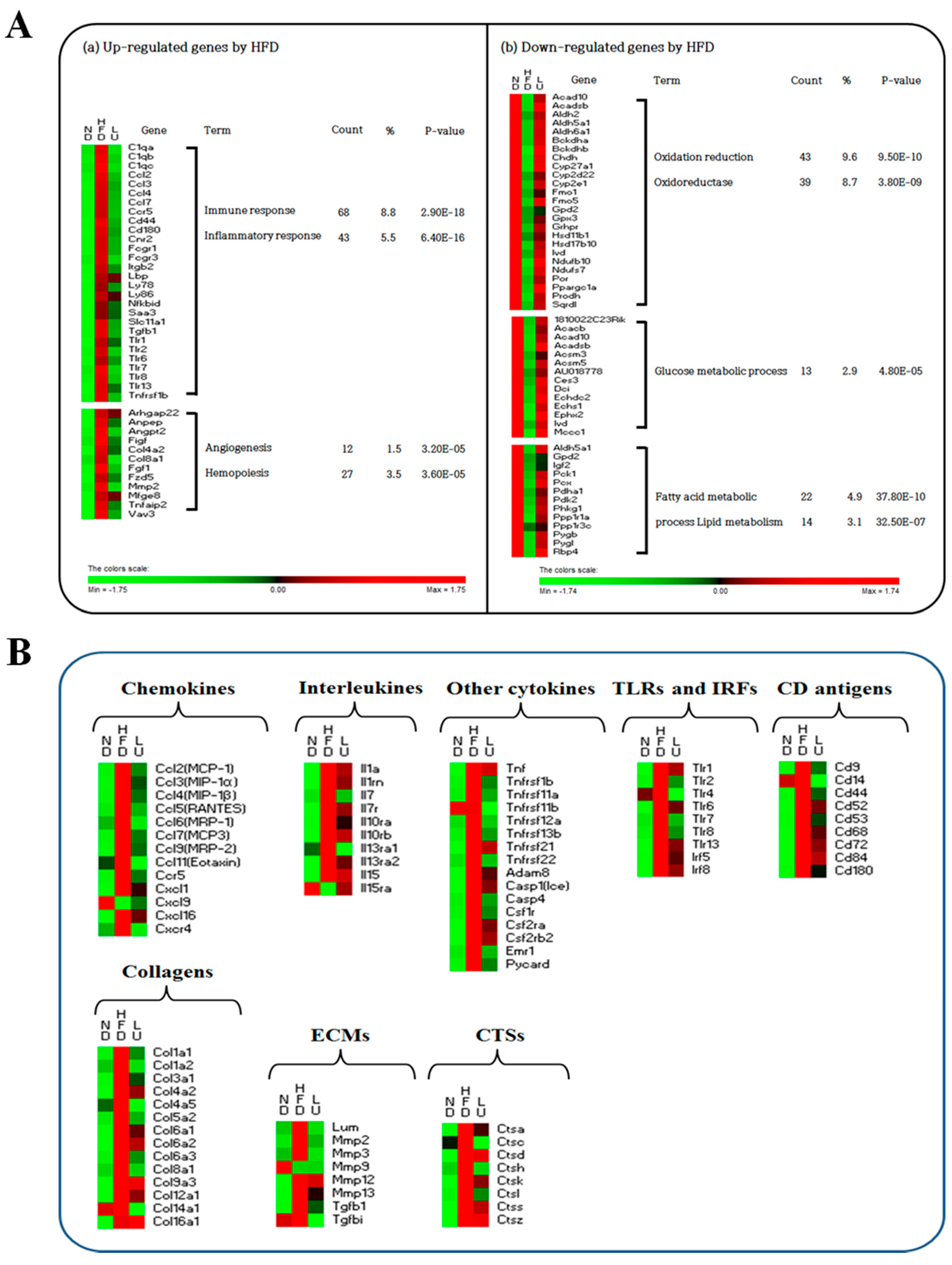
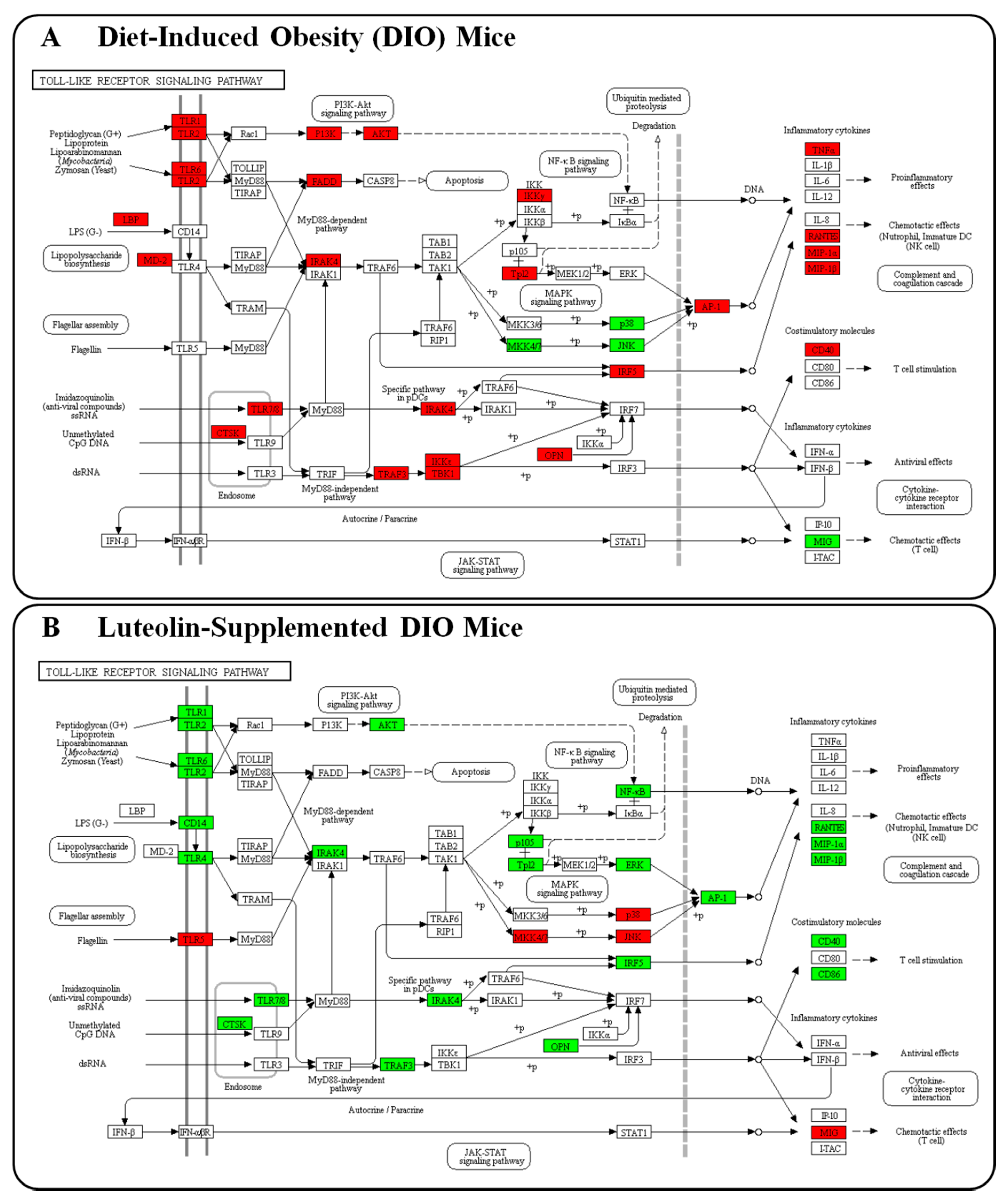
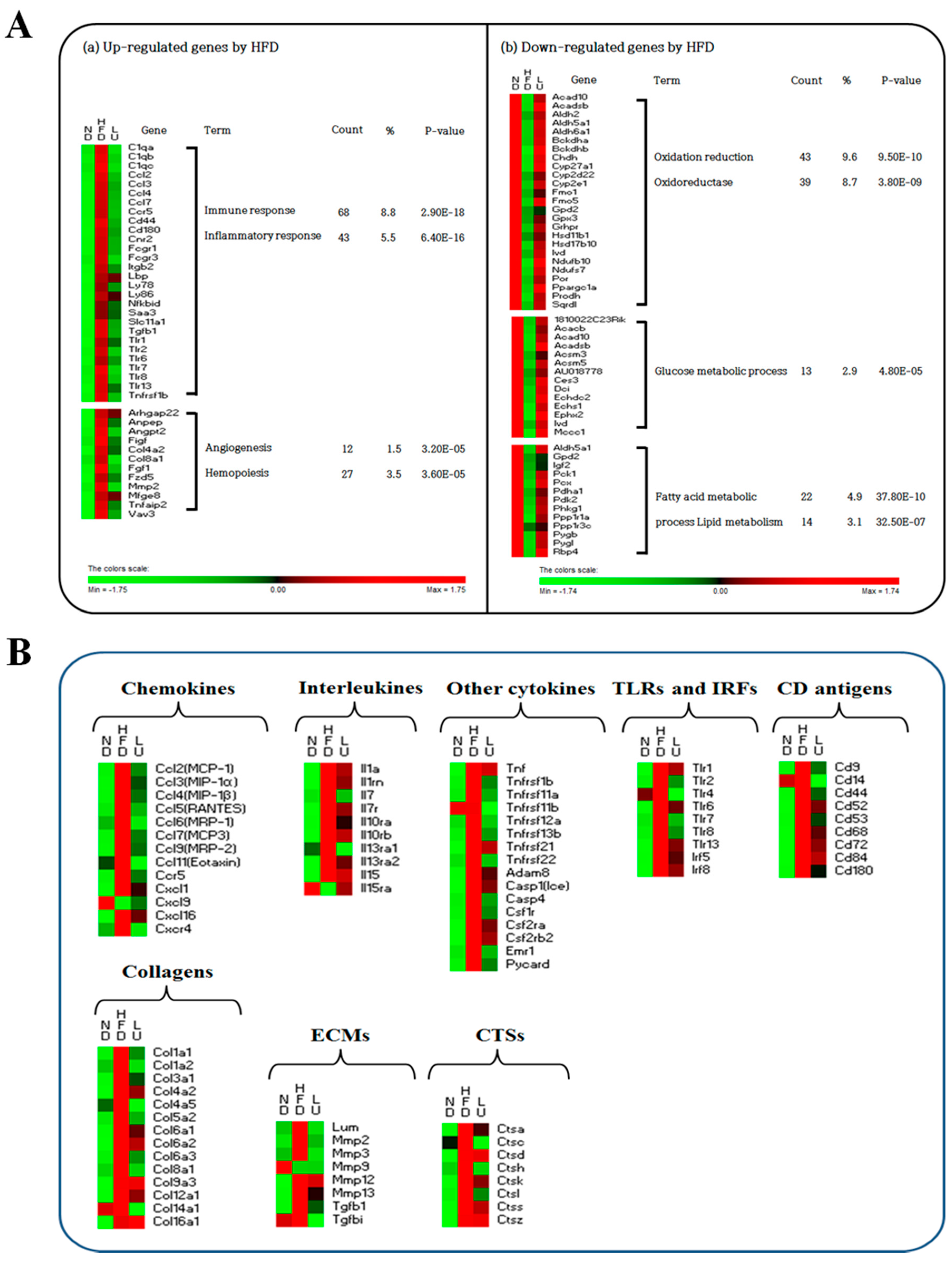
3.4. Luteolin Altered the Transcriptional Responses in the Epididymal WAT of DIO Mice
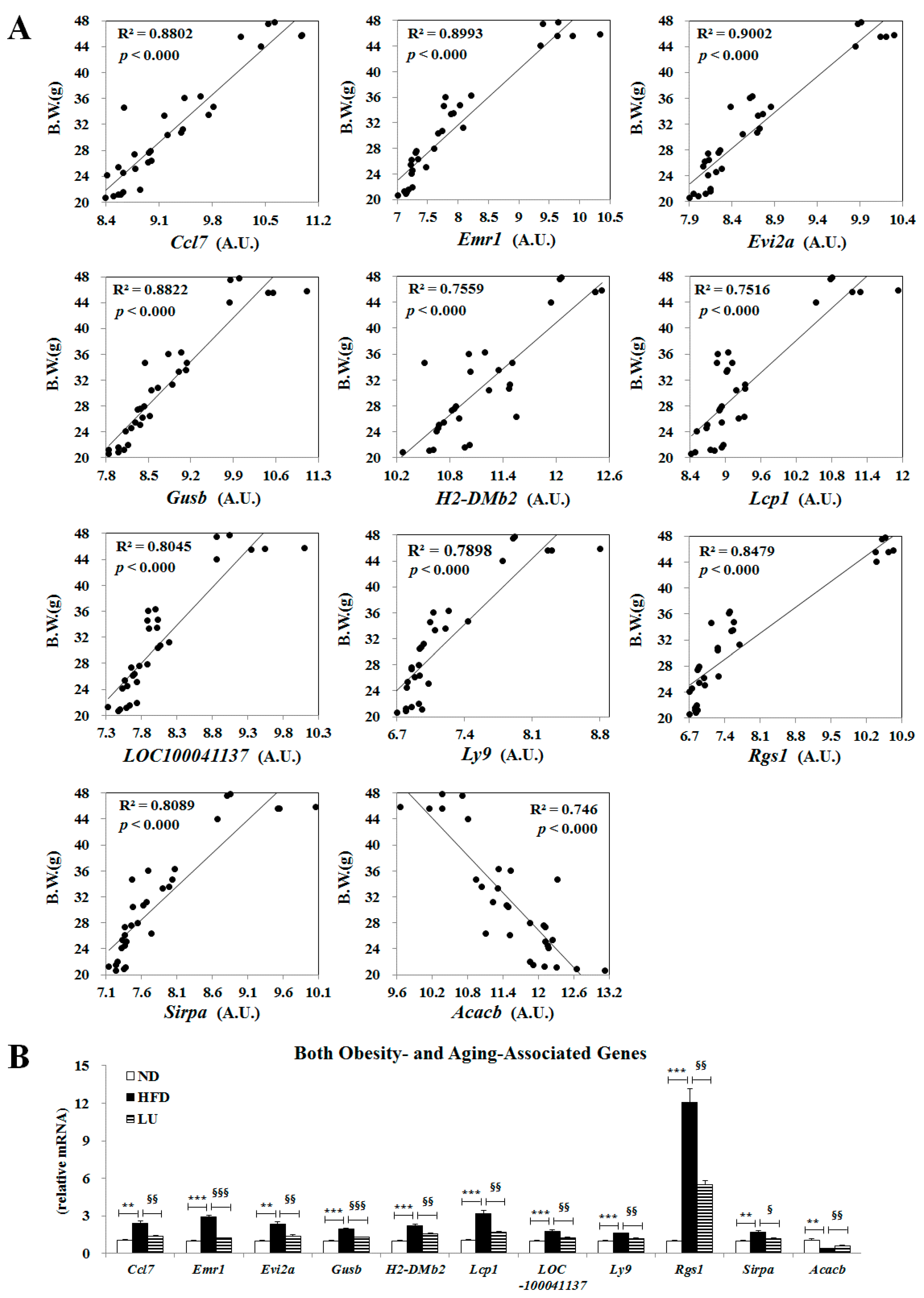
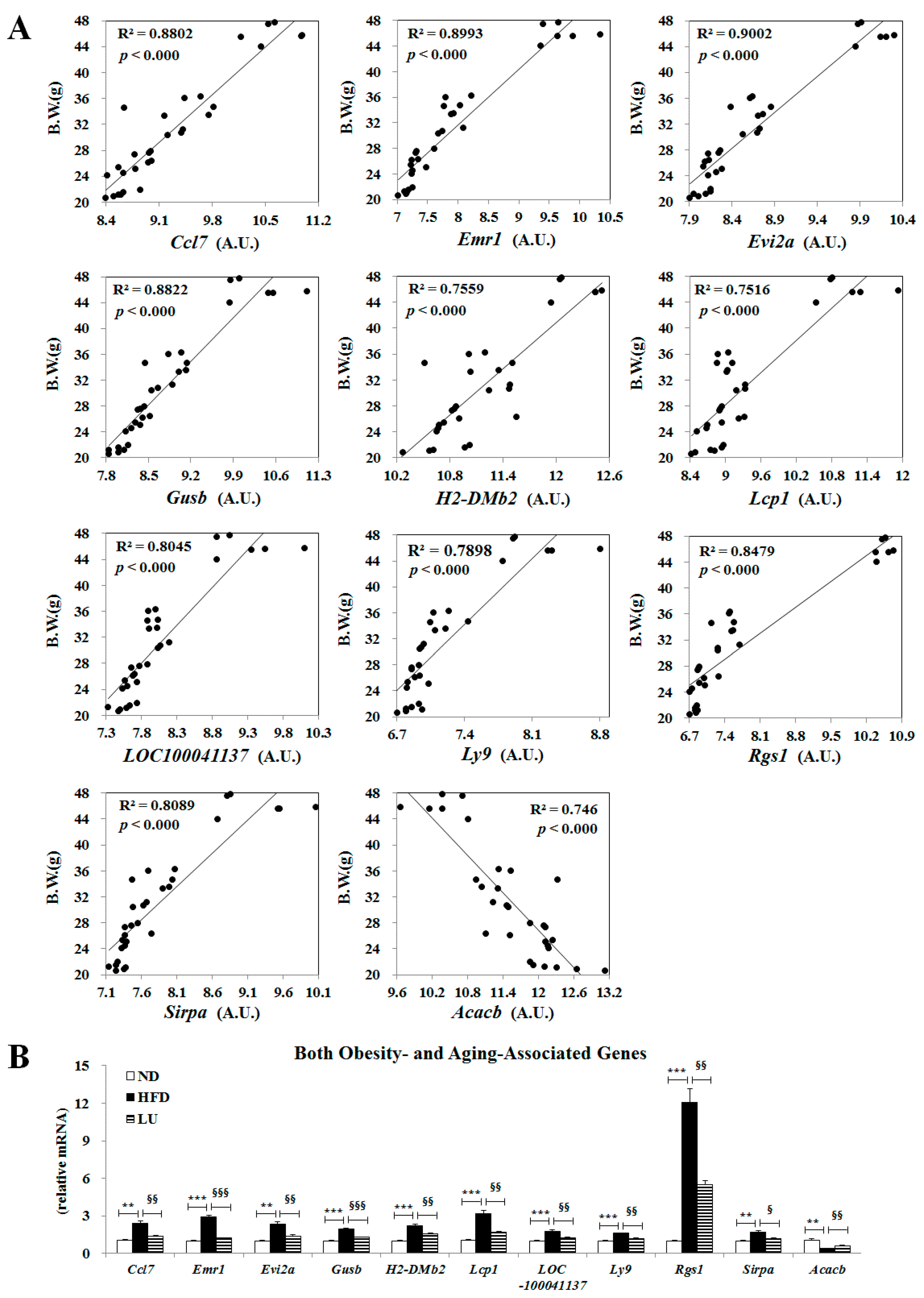
3.5. Luteolin Normalized Epididymal WAT Depot Genes Altered by Advanced Age and Diet-Induced Obesity
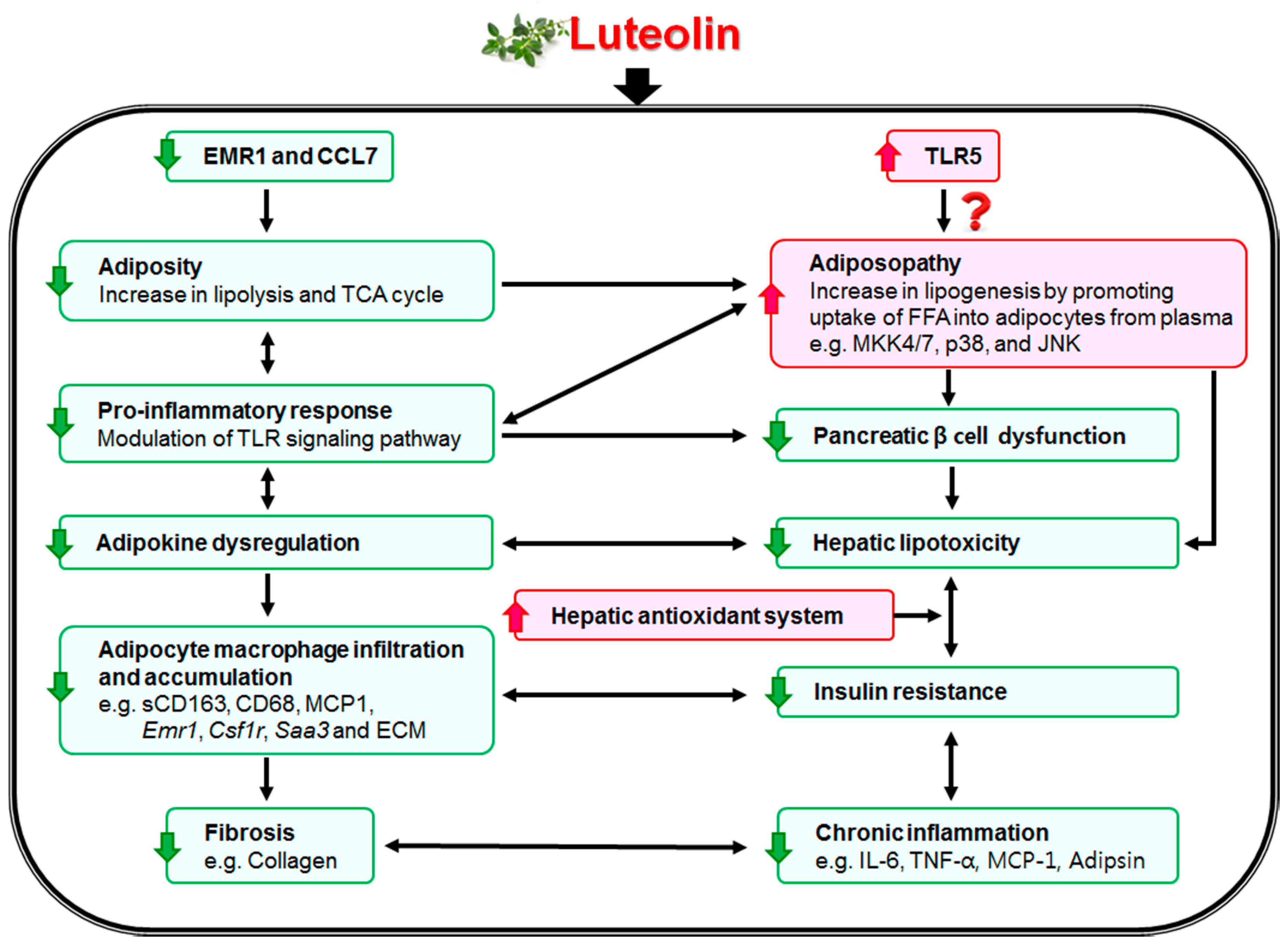
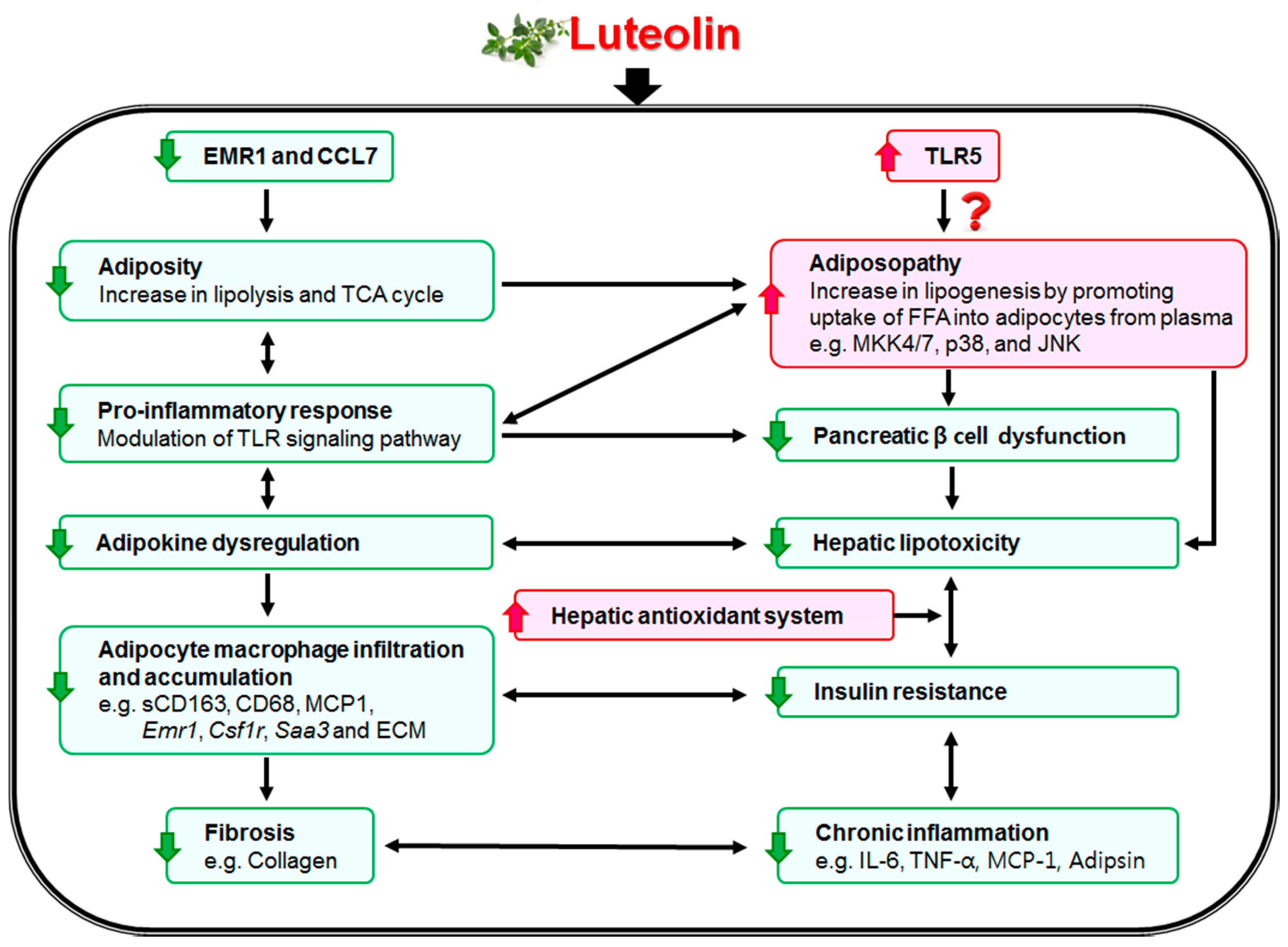
4. Discussions and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAT | catalase |
| DIO | diet-induced obesity |
| ECM | extracellular matrix |
| FFA | free fatty acid |
| GIP | gastric inhibitory polypeptide |
| GLP-1 | glucagon-like peptide-1 |
| GOT | glutamic oxaloacetic transaminase |
| GPT | glutamic pyruvic transaminase |
| GPx | glutathione peroxidase |
| GR | glutathione reductase |
| HFD | high-fat diet |
| IR | insulin resistance |
| MCP-1 | monocyte chemoattractant protein-1 |
| MIP-1β | macrophage inflammatory protein 1 beta |
| PAI-1 | plasminogen activator inhibitor-1 |
| sCD163 | soluble CD antigen 163 |
| SOD | superoxide dismutase |
| TBARS | thiobarbituric acid reactive substances |
| TLR | Toll-like receptor |
| WAT | white adipose tissue |
References
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönherr, E.; Hausser, H.J. Extracellular matrix and cytokines: A functional unit. J. Immunol. Res. 2000, 7, 89–101. [Google Scholar] [CrossRef]
- McGrath, M.E. The lysosomal cysteine proteases. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Manchanda, M.; Das, P.; Gahlot, G.P.S.; Singh, R.; Roeb, E.; Roderfeld, M.; Datta Gupta, S.; Saraya, A.; Pandey, R.M.; Chauhan, S.S. Cathepsin L and B as Potential Markers for Liver Fibrosis: Insights from Patients and Experimental Models. Transl. Gastroenterol. 2017, 8, e99. [Google Scholar] [CrossRef] [PubMed]
- Bühling, F.; Röcken, C.; Brasch, F.; Hartig, R.; Yasuda, Y.; Saftig, P.; Brömme, D.; Welte, T. Pivotal role of cathepsin K in lung fibrosis. Am. J. Pathol. 2004, 164, 2203–2216. [Google Scholar] [CrossRef]
- Fiebiger, E.; Maehr, R.; Villadangos, J.; Weber, E.; Erickson, A.; Bikoff, E.; Ploegh, H.L.; Lennon-Duménil, A.M. Invariant chain controls the activity of extracellular cathepsin L. J. Exp. Med. 2002, 196, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.; Lacasa, D.; Bastard, J.P.; Poitou, C.; Cancello, R.; Pelloux, V.; Viguerie, N.; Benis, A.; Zucker, J.D.; Bouillot, J.L.; et al. Cathepsin S, a novel biomarker of adiposity: Relevance to atherogenesis. FASEB J. 2005, 19, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Davidson, A.L.; Arion, W.J. Factors underlying significant underestimations of glucokinase activity in crude liver extracts: Physiological implications of higher cellular activity. Biochem. Biophys. 1987, 253, 156–167. [Google Scholar] [CrossRef]
- Aebi, H. Catalase. In Methods of Enzymatic Analysis, 2nd ed.; Bergmeyer, H.U., Ed.; Academic Press: New York, NY, USA, 1974; pp. 673–680. ISBN 978-0-12-091302-2. [Google Scholar]
- Marklund, S.; Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar] [PubMed]
- Pinto, R.E.; Bartley, W. The effect of age and sex on glutathione reductase and glutathione peroxidase activities and on aerobic glutathione oxidation in rat liver homogenates. Biochem. J. 1969, 112, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, S.P. Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 1994, 233, 182–189. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Do, G.M.; Kwon, E.Y.; Kim, E.; Kim, H.S.; Choi, M.S. Hepatic transcription response to high-fat treatment in mice: Microarray comparison of individual vs. pooled RNA samples. Biotechnol. J. 2010, 5, 970–973. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.Y.; Shin, S.K.; Cho, Y.Y.; Jung, U.J.; Kim, E.; Park, T.; Park, J.H.; Yun, J.W.; McGregor, R.A.; Park, Y.B.; et al. Time-course microarrays reveal early activation of the immune transcriptome and adipokine dysregulation leads to fibrosis in visceral adipose depots during diet-induced obesity. BMC Genom. 2012, 13, 450. [Google Scholar] [CrossRef] [PubMed]
- Bourlier, V.; Zakaroff-Girard, A.; Miranville, A.; De Barros, S.; Maumus, M.; Sengenes, C.; Galitzky, J.; Lafontan, M.; Karpe, F.; Frayn, K.N.; et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation 2008, 117, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008, 9, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, H.J.; Grønbaek, H.; Schiødt, F.V.; Holland-Fischer, P.; Schilsky, M.; Munoz, S.; Hassanein, T.; Lee, W.M. Soluble CD163 from activated macrophages predicts mortality in acute liver failure. J. Hepatol. 2007, 47, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Parkner, T.; Sørensen, L.P.; Nielsen, A.R.; Fischer, C.P.; Bibby, B.M.; Nielsen, S.; Pedersen, B.K.; Møller, H.J. Soluble CD163: A biomarker linking macrophages and insulin resistance. Diabetologia 2010, 55, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Møller, H.J.; Frikke-Schmidt, R.; Moestrup, S.K.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Serum soluble CD163 predicts risk of type 2 diabetes in the general population. Clin. Chem. 2011, 57, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Zanni, M.V.; Burdo, T.H.; Makimura, H.; Williams, K.C.; Grinspoon, S.K. Relationship between monocyte/macrophage activation marker soluble CD163 and insulin resistance in obese and normal-weight subjects. Clin. Endocrinol. 2012, 77, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, N.M.; Nelson, Y.B.; Michailidou, Z.; Di Rollo, E.M.; Ramage, L.; Hadoke, P.W.; Seckl, J.R.; Bunger, L.; Horvat, S.; Kenyon, C.J.; et al. A stratified transcriptomics analysis of polygenic fat and lean mouse adipose tissues identifies novel candidate obesity genes. PLoS ONE 2011, 6, e23944. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Lisanti, M.P.; Scherer, P.E. Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J. Biol. Chem. 1998, 273, 32111–32120. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Berg, A.H.; Lewis, R.Y.; Lin, A.; Lisanti, M.P.; Scherer, P.E. Constitutively active mitogen-activated protein kinase kinase 6 (MKK6) or salicylate induces spontaneous 3T3-L1 adipogenesis. J. Biol. Chem. 1999, 274, 35630–35638. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.Y.; Jung, U.J.; Park, T.; Yun, J.W.; Choi, M.S. Luteolin attenuates hepatic steatosis and insulin resistance through the interplay between the liver and adipose tissue in mice with diet-induced obesity. Diabetes 2015, 64, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadler, S.T.; Stoehr, J.P.; Schueler, K.L.; Tanimoto, G.; Yandell, B.S.; Attie, A.D. The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc. Natl. Acad. Sci. USA 2000, 97, 11371–11376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geronikaki, A.A.; Gavalas, A.M. Antioxidants and inflammatory disease: Synthetic and natural antioxidants with anti-inflammatory activity. Comb. Chem. High Throughput Screen. 2006, 9, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Baker, S.S.; Baker, R.D.; Zhu, L. Antioxidant Mechanisms in Nonalcoholic Fatty Liver Disease. Curr. Drug Target 2015, 16, 1301–1314. [Google Scholar] [CrossRef]
- Yang, J.; Kang, J.; Guan, Y. The mechanisms linking adiposopathy to type 2 diabetes. Front. Med. 2013, 7, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Divella, R.; De Luca, R.; Abbate, I.; Naglieri, E.; Daniele, A. Obesity and cancer: The role of adipose tissue and adipo-cytokines-induced chronic inflammation. J. Cancer 2016, 7, 2346–2359. [Google Scholar] [CrossRef] [PubMed]
- Golson, M.L.; Misfeldt, A.A.; Kopsombut, U.G.; Petersen, C.P.; Gannon, M. High Fat Diet Regulation of β-Cell Proliferation and β-Cell Mass. Open Endocrinol. J. 2010, 4. [Google Scholar] [CrossRef]
- Irwin, N.; Flatt, P.R. Evidence for beneficial effects of compromised gastric inhibitory polypeptide action in obesity-related diabetes and possible therapeutic implications. Diabetologia 2009, 52, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Creutzfeldt, W.; Ebert, R.; Willms, B.; Frerichs, H.; Brown, J.C. Gastric inhibitory polypeptide (GIP) and insulin in obesity: Increased response to stimulation and defective feedback control of serum levels. Diabetologia 1978, 14, 15–24. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ND | HFD | LU | |
|---|---|---|---|
| Initial Body Weight (g) | 19.33 ± 0.25 | 19.33 ± 0.32 | 19.33 ± 0.29 |
| Final Body Weight (g) | 30.32 ± 0.78 | 42.17 ± 0.89 *** | 37.48 ± 0.79 §§§ |
| Body Weight Gain (g/weeks) | 0.70 ± 0.07 | 1.47 ± 0.07 *** | 1.12 ± 0.07 §§§ |
| Liver (g/100 g BW) | 3.54 ± 0.13 | 6.57 ± 0.20 *** | 5.87 ± 0.30 |
| Visceral WAT (g/100 g BW) | 5.83 ± 0.31 | 9.63 ± 0.19 *** | 7.33 ± 0.24 §§§ |
| Subcutaneous WAT (g/100 g BW) | 1.62 ± 0.10 | 3.65 ± 0.10 *** | 2.51 ± 0.05 §§§ |
| Total WAT (g/100 g BW) | 8.62 ± 0.52 | 15.31 ± 0.27 *** | 11.68 ± 0.20 §§§ |
| Plasma FFA (mmol/L) | 1.13 ± 0.07 | 1.43 ± 0.04 ** | 1.15 ± 0.02 §§§ |
| Hepatic FFA (mEq/g liver) | 0.014 ± 0.001 | 0.028 ± 0.001 *** | 0.020 ± 0.001 §§§ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, E.-Y.; Choi, M.-S. Luteolin Targets the Toll-Like Receptor Signaling Pathway in Prevention of Hepatic and Adipocyte Fibrosis and Insulin Resistance in Diet-Induced Obese Mice. Nutrients 2018, 10, 1415. https://doi.org/10.3390/nu10101415
Kwon E-Y, Choi M-S. Luteolin Targets the Toll-Like Receptor Signaling Pathway in Prevention of Hepatic and Adipocyte Fibrosis and Insulin Resistance in Diet-Induced Obese Mice. Nutrients. 2018; 10(10):1415. https://doi.org/10.3390/nu10101415
Chicago/Turabian StyleKwon, Eun-Young, and Myung-Sook Choi. 2018. "Luteolin Targets the Toll-Like Receptor Signaling Pathway in Prevention of Hepatic and Adipocyte Fibrosis and Insulin Resistance in Diet-Induced Obese Mice" Nutrients 10, no. 10: 1415. https://doi.org/10.3390/nu10101415