Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues


Abstract
:1. Introduction
2. De Novo Lipogenesis (DNL)
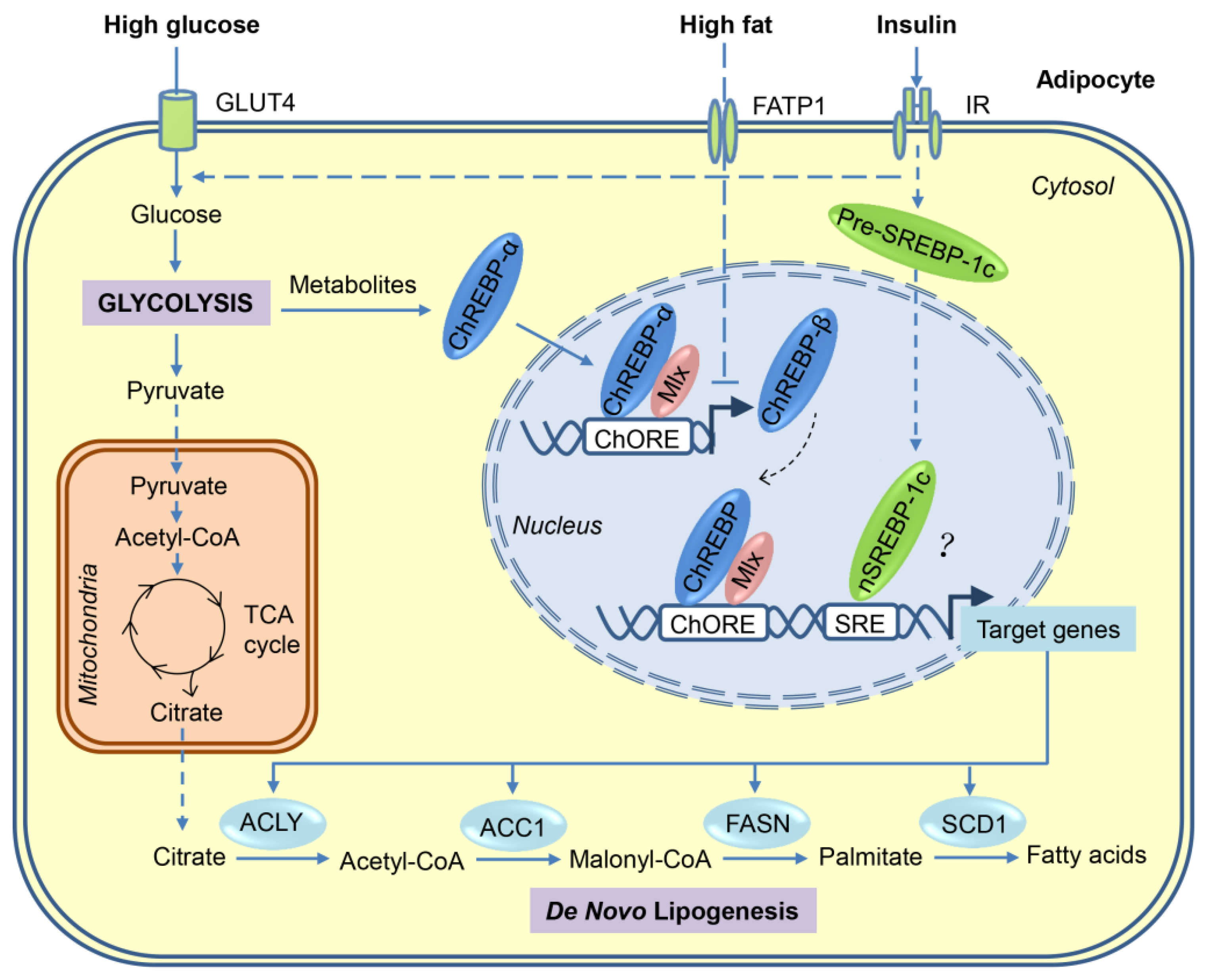
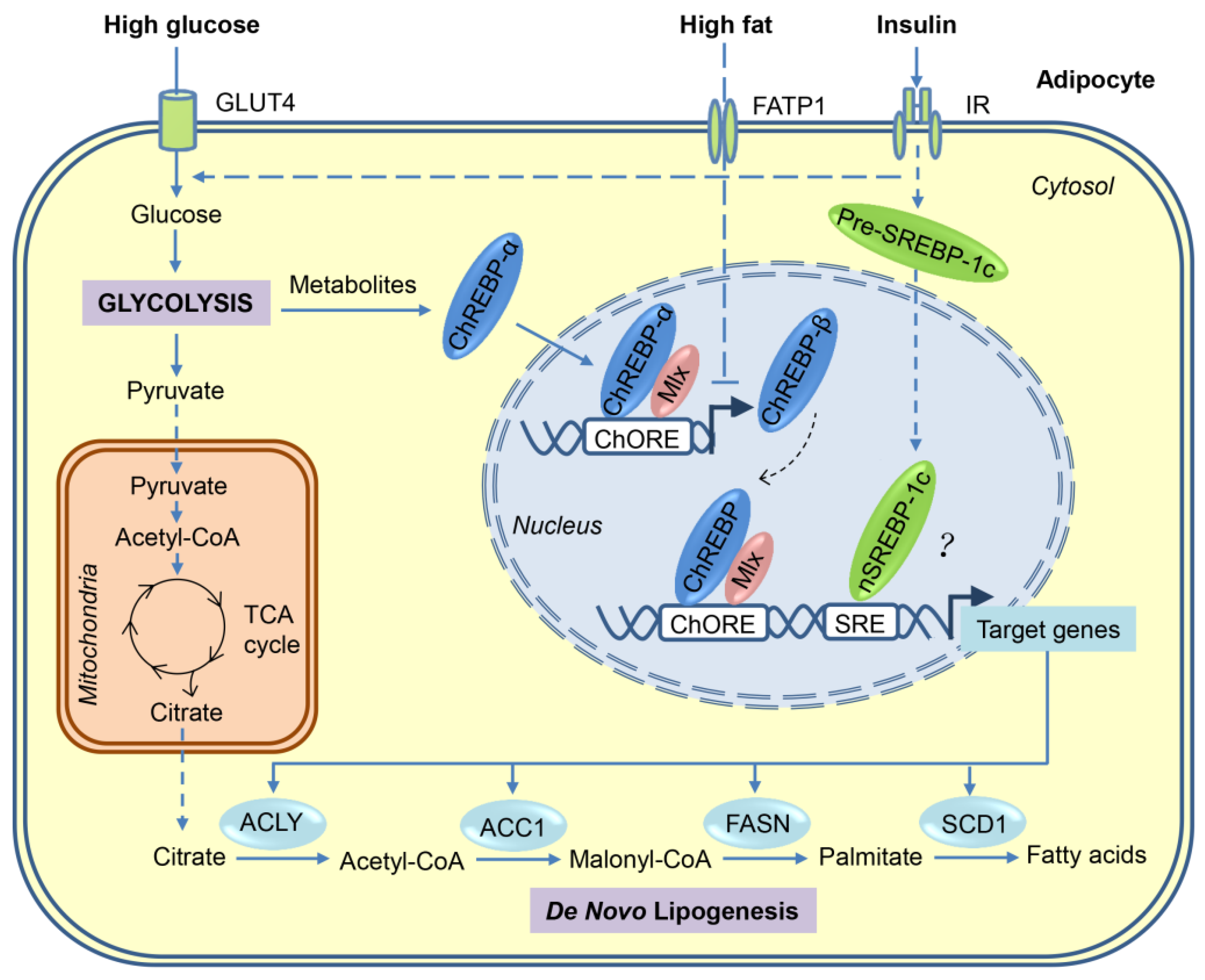
3. Transcriptional Regulation of DNL in Adipocytes
3.1. SREBP-1
3.2. ChREBP
3.3. LXRs
4. Post-Translational Regulation of DNL in Adipocytes
5. Central Regulation of DNL in Adipocytes
6. Role of Adipocyte DNL in Insulin Resistance
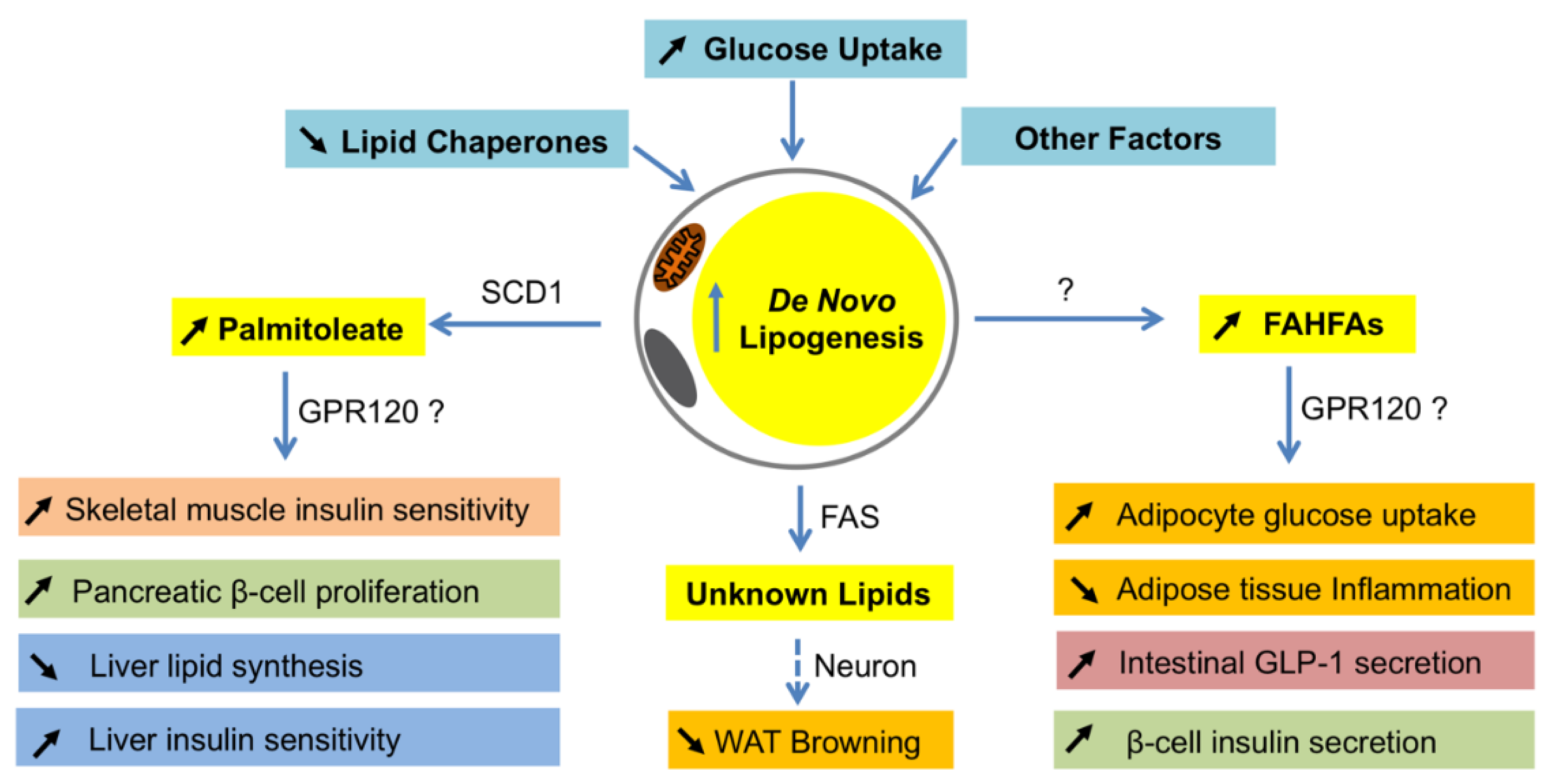
6.1. DNL in White Adipocytes
6.2. DNL in Brown Adipocytes
7. Role of Adipocyte DNL in Thermogenesis
7.1. DNL in Thermogenesis of BAT
7.2. DNL in WAT Browning
8. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DNL | de novo lipogenesis |
| AT | adipose tissue |
| WAT | white adipose tissue |
| TG | Triglyceride |
| FAHFA | fatty acid ester of hydroxyl fatty acid |
| PAHSA | palmitic acid ester of hydroxyl stearic acid |
| T2DM | type 2 diabetes |
| CVD | cardiovascular disease |
| NAFLD | non-alcoholic fatty liver disease |
| VLDL | very low density lipoproteins |
| NEFA | non-esterified fatty acids |
| LPL | lipoprotein lipase |
| FATP1 | fatty acid transport protein-1 |
| ATGL | adipose triglyceride lipase |
| DAG | diacylglycerol |
| HSL | hormone-sensitive lipase |
| MAG | monoacylglycerol |
| MGL | monoacylglycerol lipase |
| BAT | brown adipose tissue |
| TCA | tricarboxylic acid |
| ACLY | ATP-citrate lyase |
| ACC1 | acetyl-CoA carboxylase-1 |
| FASN | fatty acid synthase |
| SREBP | sterol regulatory element-binding protein |
| ChREBP | carbohydrate response element-binding protein |
| LXR | liver X receptor |
| ER | endoplasmic reticulum |
| SCAP | SREBP cleavage-activating protein |
| mTORC | mammalian target of rapamycin complex |
| SCD1 | stearoyl-CoA desaturase-1 |
| PPAR | peroxisome proliferator-activated receptor |
| BCKDH | branched-chain α-ketoacid dehydrogenase |
| BCAA | branched-chain amino acids |
| AMPK | AMP-activated protein kinase |
| O-GlcNAc | O-Linked N-Acetylglucosamine |
| OGT | O-Linked N-Acetylglucosamine transferase |
| OGA | O-GlcNAcase |
| USP2A | ubiquitin-specific protease-2a |
| MBH | media basal hypothalamus |
| NPGL | neurosecretory protein GL |
| GPR120 | G protein-coupled receptor 120 |
References
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, A.; Magnuson, A.; Fouts, J.; Foster, M.T. Adipose tissue: An endocrine organ playing a role in metabolic regulation. Horm. Mol. Biol. Clin. Investig. 2016, 26, 25–42. [Google Scholar] [CrossRef] [PubMed]
- McGown, C.; Birerdinc, A.; Younossi, Z.M. Adipose tissue as an endocrine organ. Clin. Liver Dis. 2014, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Diaz, A.; Romero, M.; Thaller, S.; Blomberg, B.B. Secretion of autoimmune antibodies in the human subcutaneous adipose tissue. PLoS ONE 2018, 13, e0197472. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Claiborn, K.C.; Hotamisligil, G.S. De novo lipogenesis products and endogenous lipokines. Diabetes 2016, 65, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Indias, I.; Tinahones, F.J. Impaired adipose tissue expandability and lipogenic capacities as ones of the main causes of metabolic disorders. J. Diabetes Res. 2015, 2015, 970375. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, F.; Dashty, M. Role of adipose tissue in metabolic system disorders adipose tissue is the initiator of metabolic diseases. J. Diabetes Metab. 2013, S13. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erion, D.M.; Park, H.J.; Lee, H.Y. The role of lipids in the pathogenesis and treatment of type 2 diabetes and associated co-morbidities. BMB Rep. 2016, 49, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.H.; Sul, H.S. Insulin signaling in fatty acid and fat synthesis: A transcriptional perspective. Curr. Opin. Pharmacol. 2010, 10, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Merkel, M.; Eckel, R.H.; Goldberg, I.J. Lipoprotein lipase: Genetics, lipid uptake, and regulation. J. Lipid Res. 2002, 43, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. Cd36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [PubMed]
- Wu, Q.; Ortegon, A.M.; Tsang, B.; Doege, H.; Feingold, K.R.; Stahl, A. Fatp1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol. Cell. Biol. 2006, 26, 3455–3467. [Google Scholar] [CrossRef] [PubMed]
- Bolsoni-Lopes, A.; Alonso-Vale, M.I. Lipolysis and lipases in white adipose tissue—An update. Arch. Endocrinol. Metab 2015, 59, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The subtle balance between lipolysis and lipogenesis: A critical point in metabolic homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Nasr, A.; Tfayli, H.; Bacha, F.; Michaliszyn, S.F.; Arslanian, S. Increased lipolysis, diminished adipose tissue insulin sensitivity and impaired β-cell function relative to adipose tissue insulin sensitivity in obese youth with impaired glucose tolerance (IGT). Diabetes 2017, 66, 3085–3090. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Aryal, P.; Wen, J.; Syed, I.; Vazirani, R.P.; Moraes-Vieira, P.M.; Camporez, J.P.; Gallop, M.R.; Perry, R.J.; Peroni, O.D.; et al. Absence of carbohydrate response element binding protein in adipocytes causes systemic insulin resistance and impairs glucose transport. Cell Rep. 2017, 21, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.S.; Jessen, N.; Jorgensen, J.O.; Moller, N.; Lund, S. Dissecting adipose tissue lipolysis: Molecular regulation and implications for metabolic disease. J. Mol. Endocrinol. 2014, 52, R199–R222. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeck, G.; Mendez-Gimenez, L.; Fernandez-Formoso, J.A.; Fernandez, S.; Rodriguez, A. Regulation of adipocyte lipolysis. Nutr. Res. Rev. 2014, 27, 63–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Boren, J.; Dulloo, A.G. De novo lipogenesis in metabolic homeostasis: More friend than foe? Mol. Metab. 2015, 4, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Hollands, M.A.; Cawthorne, M.A. Important sites of lipogenesis in the mouse other than liver and white adipose tissue. Biochem. J. 1981, 196, 645–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarsland, A.; Chinkes, D.; Wolfe, R.R. Hepatic and whole-body fat synthesis in humans during carbohydrate overfeeding. Am. J. Clin. Nutr. 1997, 65, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, I.J.; Wei, X.; Semenkovich, C.F. Lipoexpediency: De novo lipogenesis as a metabolic signal transmitter. Trends Endocrinol. Metab. 2011, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Smith, U.; Kahn, B.B. Adipose tissue regulates insulin sensitivity: Role of adipogenesis, de novo lipogenesis and novel lipids. J. Intern. Med. 2016, 280, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Funai, K.; Song, H.; Yin, L.; Lodhi, I.J.; Wei, X.; Yoshino, J.; Coleman, T.; Semenkovich, C.F. Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling. J. Clin. Investig. 2013, 123, 1229–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumanov, S.; Bulusu, V.; Kamphorst, J.J. Analysis of fatty acid metabolism using stable isotope tracers and mass spectrometry. Methods Enzymol. 2015, 561, 197–217. [Google Scholar] [PubMed]
- Herath, K.B.; Zhong, W.; Yang, J.; Mahsut, A.; Rohm, R.J.; Shah, V.; Castro-Perez, J.; Zhou, H.; Attygalle, A.B.; Kang, L.; et al. Determination of low levels of 2h-labeling using high-resolution mass spectrometry: Application in studies of lipid flux and beyond. Rapid Commun. Mass Spectrom. 2014, 28, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. Ampk phosphorylates and inhibits srebp activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Osatomi, K.; Yamashita, H.; Kabashima, T.; Uyeda, K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: Regulation of carbohydrate-responsive element-binding protein by amp-activated protein kinase. J. Biol. Chem. 2002, 277, 3829–3835. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Shyy, J.Y. Sterol regulatory element-binding protein 1 is negatively modulated by pka phosphorylation. Am. J. Physiol. Cell Physiol. 2006, 290, C1477–C1486. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and camp regulate the l-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gurmaches, J.; Tang, Y.; Jespersen, N.Z.; Wallace, M.; Martinez Calejman, C.; Gujja, S.; Li, H.; Edwards, Y.J.K.; Wolfrum, C.; Metallo, C.M.; et al. Brown fat akt2 is a cold-induced kinase that stimulates chrebp-mediated de novo lipogenesis to optimize fuel storage and thermogenesis. Cell Metab. 2018, 27, 195–209.e6. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wallace, M.; Sanchez-Gurmaches, J.; Hsiao, W.Y.; Li, H.; Lee, P.L.; Vernia, S.; Metallo, C.M.; Guertin, D.A. Adipose tissue mtorc2 regulates chrebp-driven de novo lipogenesis and hepatic glucose metabolism. Nat. Commun. 2016, 7, 11365. [Google Scholar] [CrossRef] [PubMed]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vazquez-Martin, A.; Ortega, F.J.; Fernandez-Real, J.M. Fatty acid synthase: Association with insulin resistance, type 2 diabetes, and cancer. Clin. Chem. 2009, 55, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. Srebp transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Horton, J.D.; Shimomura, I.; Hammer, R.E.; Brown, M.S.; Goldstein, J.L. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J. Clin. Investig. 1997, 99, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Wu, J.; Goldstein, J.L.; Brown, M.S.; Hobbs, H.H. Structure of the human gene encoding sterol regulatory element binding protein-1 (srebf1) and localization of srebf1 and srebf2 to chromosomes 17p11.2 and 22q13. Genomics 1995, 25, 667–673. [Google Scholar] [CrossRef]
- Czech, M.P.; Tencerova, M.; Pedersen, D.J.; Aouadi, M. Insulin signalling mechanisms for triacylglycerol storage. Diabetologia 2013, 56, 949–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, T.F. Sterol regulatory element-binding proteins (srebps): Key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000, 275, 32379–32382. [Google Scholar] [CrossRef] [PubMed]
- Yellaturu, C.R.; Deng, X.; Cagen, L.M.; Wilcox, H.G.; Mansbach, C.M., 2nd; Siddiqi, S.A.; Park, E.A.; Raghow, R.; Elam, M.B. Insulin enhances post-translational processing of nascent srebp-1c by promoting its phosphorylation and association with copii vesicles. J. Biol. Chem. 2009, 284, 7518–7532. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of lxrs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (srebp-1c) by oxysterol receptors, lxralpha and lxrbeta. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Bakan, I.; Laplante, M. Connecting mtorc1 signaling to srebp-1 activation. Curr. Opin. Lipidol. 2012, 23, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Romero, D.; Swiatek, W.I.; Dorweiler, I.; Kikani, C.K.; Sabic, H.; Zweifel, B.S.; McKearn, J.; Blitzer, J.T.; Nickols, G.A.; et al. Pas kinase drives lipogenesis through srebp-1 maturation. Cell Rep. 2014, 8, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Bengoechea-Alonso, M.T.; Ye, X.; Lukiyanchuk, V.; Jin, J.; Harper, J.W.; Ericsson, J. Control of lipid metabolism by phosphorylation-dependent degradation of the srebp family of transcription factors by scf(fbw7). Cell Metab. 2005, 1, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N. Mtor complex 1 regulates lipin 1 localization to control the srebp pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Feng, D.; Wang, Q.; Abdulla, A.; Xie, X.J.; Zhou, J.; Sun, Y.; Yang, E.S.; Liu, L.P.; Vaitheesvaran, B.; et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of srebp-1. J. Clin. Investig. 2012, 122, 2417–2427. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. Srebp-1c, regulated by the insulin and ampk signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased levels of nuclear srebp-1c associated with fatty livers in two mouse models of diabetes mellitus. J. Biol. Chem. 1999, 274, 30028–30032. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Köllmer, C.; Nitzgen, U.; Muller–Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active srebp-1c is associated with fatty liver and increased visceral fat mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Hasty, A.H.; Matsuzaka, T.; Ide, T.; Yoshikawa, T.; Amemiya-Kudo, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Absence of sterol regulatory element-binding protein-1 (srebp-1) ameliorates fatty livers but not obesity or insulin resistance in lep(ob)/lep(ob) mice. J. Biol. Chem. 2002, 277, 19353–19357. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The scap/srebp pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Spiegelman, B.M. Add1/srebp1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Shimomura, I.; Hammer, R.E.; Herz, J.; Goldstein, J.L.; Brown, M.S.; Horton, J.D. Elevated levels of srebp-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the srebp-1 gene. J. Clin. Investig. 1997, 100, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Narita, T.; Okita, N.; Kobayashi, M.; Furuta, Y.; Chujo, Y.; Sakai, M.; Yamada, A.; Takeda, K.; Konishi, T. Sterol regulatory element-binding protein-1c orchestrates metabolic remodeling of white adipose tissue by caloric restriction. Aging Cell 2017, 16, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, M.; Yahagi, N.; Matsuzaka, T.; Takeuchi, Y.; Nakagawa, Y.; Takahashi, H.; Okazaki, H.; Iizuka, Y.; Ohashi, K.; Gotoda, T.; et al. Srebp-1-independent regulation of lipogenic gene expression in adipocytes. J. Lipid Res. 2007, 48, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Shimomura, I.; Ikemoto, S.; Bashmakov, Y.; Hammer, R.E. Overexpression of sterol regulatory element-binding protein-1a in mouse adipose tissue produces adipocyte hypertrophy, increased fatty acid secretion, and fatty liver. J. Biol. Chem. 2003, 278, 36652–36660. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Hammer, R.E.; Richardson, J.A.; Ikemoto, S.; Bashmakov, Y.; Goldstein, J.L.; Brown, M.S. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear srebp-1c in adipose tissue: Model for congenital generalized lipodystrophy. Genes Dev. 1998, 12, 3182–3194. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Boord, J.B.; Maeda, K.; Babaev, V.R.; Uysal, K.T.; Morgan, M.A.; Parker, R.A.; Suttles, J.; Fazio, S.; Hotamisligil, G.S.; et al. Lack of macrophage fatty-acid-binding protein ap2 protects mice deficient in apolipoprotein e against atherosclerosis. Nat. Med. 2001, 7, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Cereijo, R.; Villarroya, J.; Gavalda-Navarro, A.; Giralt, M. Toward an understanding of how immune cells control brown and beige adipobiology. Cell Metab. 2018, 27, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (chrebp) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, K.; Yamashita, H.; Kawaguchi, T. Carbohydrate responsive element-binding protein (chrebp): A key regulator of glucose metabolism and fat storage. Biochem. Pharmacol. 2002, 63, 2075–2080. [Google Scholar] [CrossRef]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schon, M.R.; Abumrad, N.A.; Bluher, M.; Klein, S.; Kahn, B.B. A novel chrebp isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (chrebp). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.P.; et al. Metabolite regulation of nuclear localization of carbohydrate-response element-binding protein (chrebp): Role of amp as an allosteric inhibitor. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef] [PubMed]
- Stoeckman, A.K.; Ma, L.; Towle, H.C. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J. Biol. Chem. 2004, 279, 15662–15669. [Google Scholar] [CrossRef] [PubMed]
- Denechaud, P.D.; Dentin, R.; Girard, J.; Postic, C. Role of chrebp in hepatic steatosis and insulin resistance. FEBS Lett. 2008, 582, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Hurtado del Pozo, C.; Vesperinas-Garcia, G.; Rubio, M.A.; Corripio-Sanchez, R.; Torres-Garcia, A.J.; Obregon, M.J.; Calvo, R.M. Chrebp expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. Biochim. Biophys. Acta 2011, 1811, 1194–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, N.; Muenzner, M.; Rietscher, J.; Knauer, M.; Heidenreich, S.; Nuotio-Antar, A.M.; Graef, F.A.; Fedders, R.; Tolkachov, A.; Goehring, I.; et al. The glucose sensor chrebp links de novo lipogenesis to ppargamma activity and adipocyte differentiation. Endocrinology 2015, 156, 4008–4019. [Google Scholar] [CrossRef] [PubMed]
- Nuotio-Antar, A.M.; Poungvarin, N.; Li, M.; Schupp, M.; Mohammad, M.; Gerard, S.; Zou, F.; Chan, L. Fabp4-cre mediated expression of constitutively active chrebp protects against obesity, fatty liver, and insulin resistance. Endocrinology 2015, 156, 4020–4032. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.M.; Calejman, C.M.; Sanchez-Gurmaches, J.; Li, H.; Clish, C.B.; Hettmer, S.; Wagers, A.J.; Guertin, D.A. Rictor/mtorc2 loss in the myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep. 2014, 8, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Weigelt, C.; Cherradi, M.L.; Niemeier, A.; Todter, K.; Heeren, J.; Scheja, L. Effects of adipocyte lipoprotein lipase on de novo lipogenesis and white adipose tissue browning. Biochim. Biophys. Acta 2013, 1831, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.-M.A.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor lxrα. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- Alberti, S.; Steffensen, K.R.; Gustafsson, J.-Å. Structural characterisation of the mouse nuclear oxysterol receptor genes lxrα and lxrβ. Gene 2000, 243, 93–103. [Google Scholar] [CrossRef]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. Lxr, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Kokontis, J.M.; Hiipakka, R.A.; Liao, S. Ubiquitous receptor: A receptor that modulates gene activation by retinoic acid and thyroid hormone receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 10809–10813. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, T.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. Liver x receptor biology and pharmacology: New pathways, challenges and opportunities. Trends Pharmacol. Sci. 2012, 33, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Shimano, H.; Inoue, N.; Nakagawa, Y.; Matsuzaka, T.; Takahashi, A.; Yahagi, N.; Sone, H.; Suzuki, H.; Toyoshima, H.; et al. Protein kinase a suppresses sterol regulatory element-binding protein-1c expression via phosphorylation of liver x receptor in the liver. J. Biol. Chem. 2007, 282, 11687–11695. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Xu, Z.; Zhang, Z.; Yin, X.; Lin, X.; Li, H.; Zheng, F. Impaired adipose expansion caused by liver x receptor activation is associated with insulin resistance in mice fed a high-fat diet. J. Mol. Endocrinol. 2017, 58, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Beaven, S.W.; Matveyenko, A.; Wroblewski, K.; Chao, L.; Wilpitz, D.; Hsu, T.W.; Lentz, J.; Drew, B.; Hevener, A.L.; Tontonoz, P. Reciprocal regulation of hepatic and adipose lipogenesis by liver x receptors in obesity and insulin resistance. Cell Metab. 2013, 18, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Jois, T.; Chen, W.; Howard, V.; Harvey, R.; Youngs, K.; Thalmann, C.; Saha, P.; Chan, L.; Cowley, M.A.; Sleeman, M.W. Deletion of hepatic carbohydrate response element binding protein (chrebp) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab. 2017, 6, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Dib, L.; Bugge, A.; Collins, S. Lxralpha fuels fatty acid-stimulated oxygen consumption in white adipocytes. J. Lipid Res. 2014, 55, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Horton, J.D.; Hammer, R.E.; Shimomura, I.; Brown, M.S.; Goldstein, J.L. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated srebp-1a. J. Clin. Investig. 1996, 98, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor chrebp dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef] [PubMed]
- White, P.J.; McGarrah, R.W.; Grimsrud, P.A.; Tso, S.C.; Yang, W.H.; Haldeman, J.M.; Grenier-Larouche, T.; An, J.; Lapworth, A.L.; Astapova, I.; et al. The bckdh kinase and phosphatase integrate bcaa and lipid metabolism via regulation of atp-citrate lyase. Cell Metab. 2018, 27, 1281–1293. [Google Scholar] [CrossRef] [PubMed]
- Daval, M.; Foufelle, F.; Ferre, P. Functions of amp-activated protein kinase in adipose tissue. J. Physiol. 2006, 574, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Loh, K.; Murray-Segal, L.; Steinberg, G.R.; Andrews, Z.B.; Kemp, B.E. Ampk signaling to acetyl-coa carboxylase is required for fasting-and cold-induced appetite but not thermogenesis. eLife 2018, 7, e32656. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Regulation of fatty acid synthesis via phosphorylation of acetyl-coa carboxylase. Prog. Lipid Res. 1989, 28, 117–146. [Google Scholar] [CrossRef]
- An, Z.; Wang, H.; Song, P.; Zhang, M.; Geng, X.; Zou, M.H. Nicotine-induced activation of amp-activated protein kinase inhibits fatty acid synthase in 3t3l1 adipocytes: A role for oxidant stress. J. Biol. Chem. 2007, 282, 26793–26801. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.P.N.; Hart, G.W. O-glcnac transferase. In Handbook of Glycosyltransferases and Related Genes; Springer-Verlag: Tokyo, Japan, 2002; pp. 158–163. [Google Scholar]
- Dong, D.L.; Hart, G.W. Purification and characterization of an o-glcnac selective n-acetyl-beta-d-glucosaminidase from rat spleen cytosol. J. Biol. Chem. 1994, 269, 19321–19330. [Google Scholar] [PubMed]
- Baldini, S.F.; Wavelet, C.; Hainault, I.; Guinez, C.; Lefebvre, T. The nutrient-dependent o-glcnac modification controls the expression of liver fatty acid synthase. J. Mol. Biol. 2016, 428, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Graner, E.; Tang, D.; Rossi, S.; Baron, A.; Migita, T.; Weinstein, L.J.; Lechpammer, M.; Huesken, D.; Zimmermann, J.; Signoretti, S. The isopeptidase usp2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 2004, 5, 253–261. [Google Scholar] [CrossRef]
- Teo, C.F.; Wollaston-Hayden, E.E.; Wells, L. Hexosamine flux, the o-glcnac modification, and the development of insulin resistance in adipocytes. Mol. Cell. Endocrinol. 2010, 318, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, K.; Wells, L. Multiple tissue-specific roles for the o-glcnac post-translational modification in the induction of and complications arising from type ii diabetes. J. Biol. Chem. 2014, 289, 34466–34471. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Olson, D.P. Central nervous system control of metabolism. Nature 2012, 491, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Nogueiras, R.; Wiedmer, P.; Perez-Tilve, D.; Veyrat-Durebex, C.; Keogh, J.M.; Sutton, G.M.; Pfluger, P.T.; Castaneda, T.R.; Neschen, S.; Hofmann, S.M.; et al. The central melanocortin system directly controls peripheral lipid metabolism. J. Clin. Investig. 2007, 117, 3475–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, T.; Buettner, C. Yin and yang of hypothalamic insulin and leptin signaling in regulating white adipose tissue metabolism. Rev. Endocr. Metab. Disord. 2011, 12, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Scherer, T.; O’Hare, J.; Diggs-Andrews, K.; Schweiger, M.; Cheng, B.; Lindtner, C.; Zielinski, E.; Vempati, P.; Su, K.; Dighe, S.; et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 2011, 13, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Buettner, C.; Muse, E.D.; Cheng, A.; Chen, L.; Scherer, T.; Pocai, A.; Su, K.; Cheng, B.; Li, X.; Harvey-White, J.; et al. Leptin controls adipose tissue lipogenesis via central, stat3-independent mechanisms. Nat. Med. 2008, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Batkai, S.; Harvey-White, J.; Mackie, K.; Offertaler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic cb1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi-Ukena, E.; Shikano, K.; Kondo, K.; Taniuchi, S.; Furumitsu, M.; Ochi, Y.; Sasaki, T.; Okamoto, S.; Bentley, G.E.; Kriegsfeld, L.J. Neurosecretory protein gl stimulates food intake, de novo lipogenesis, and onset of obesity. eLife 2017, 6, e28527. [Google Scholar] [CrossRef] [PubMed]
- Eissing, L.; Scherer, T.; Todter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to chrebp-beta and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia-and insulin-resistance-induced heart disease. Diabetologia 2018, 61, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Poungvarin, N.; Lee, J.; Yechoor, V.; Li, M.; Assavapokee, T.; Suksaranjit, P.; Thepsongwajja, J.; Saha, P.; Oka, K.; Chan, L. Carbohydrate response element-binding protein (chrebp) plays a pivotal role in beta cell glucotoxicity. Diabetologia 2012, 55, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Pedersen, D.J.; Henchey, E.; Henriques, F.S.; Danai, L.V.; Shen, Y.; Yenilmez, B.; Jung, D.; Kim, J.K.; Lodhi, I.J.; et al. Adipocyte lipid synthesis coupled to neuronal control of thermogenic programming. Mol. Metab. 2017, 6, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Auguet, T.; Guiu-Jurado, E.; Berlanga, A.; Terra, X.; Martinez, S.; Porras, J.A.; Ceausu, A.; Sabench, F.; Hernandez, M.; Aguilar, C. Downregulation of lipogenesis and fatty acid oxidation in the subcutaneous adipose tissue of morbidly obese women. Obesity 2014, 22, 2032–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allister, C.A.; Liu, L.F.; Lamendola, C.A.; Craig, C.M.; Cushman, S.W.; Hellerstein, M.K.; McLaughlin, T.L. In vivo 2h2o administration reveals impaired triglyceride storage in adipose tissue of insulin-resistant humans. J. Lipid Res. 2015, 56, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.; Hodson, L.; Dennis, A.; Neville, M.; Humphreys, S.; Harnden, K.; Micklem, K.; Frayn, K. Markers of de novo lipogenesis in adipose tissue: Associations with small adipocytes and insulin sensitivity in humans. Diabetologia 2009, 52, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Yore, M.M.; Syed, I.; Moraes-Vieira, P.M.; Zhang, T.; Herman, M.A.; Homan, E.A.; Patel, R.T.; Lee, J.; Chen, S.; Peroni, O.D. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 2014, 159, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Ntambi, J.M.; Miyazaki, M. Regulation of stearoyl-coa desaturases and role in metabolism. Prog. Lipid Res. 2004, 43, 91–104. [Google Scholar] [CrossRef]
- De Souza, C.O.; Vannice, G.K.; Rosa Neto, J.C.; Calder, P.C. Is palmitoleic acid a plausible nonpharmacological strategy to prevent or control chronic metabolic and inflammatory disorders? Mol. Nutr. Food Res. 2018, 62, 1700504. [Google Scholar] [CrossRef] [PubMed]
- Frigolet, M.E.; Gutierrez-Aguilar, R. The role of the novel lipokine palmitoleic acid in health and disease. Adv. Nutr. 2017, 8, 173S–181S. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Kantartzis, K.; Celebi, N.; Staiger, H.; Machann, J.; Schick, F.; Cegan, A.; Elcnerova, M.; Schleicher, E.; Fritsche, A.; et al. Circulating palmitoleate strongly and independently predicts insulin sensitivity in humans. Diabetes Care 2010, 33, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F.; Su, X.; Abumrad, N.A.; Nejedly, N.; Coughlin, C.C.; Okunade, A.L.; Patterson, B.W.; Klein, S. Insulin sensitivity is not associated with palmitoleate availability in obese humans. J. Lipid Res. 2011, 52, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, B.C.; Howard, D.; Schauer, I.E.; Maahs, D.M.; Snell-Bergeon, J.K.; Clement, T.W.; Eckel, R.H.; Perreault, L.; Rewers, M. The importance of palmitoleic acid to adipocyte insulin resistance and whole-body insulin sensitivity in type 1 diabetes. J. Clin. Endocrinol. Metab. 2013, 98, E40–E50. [Google Scholar] [CrossRef] [PubMed]
- Moraes-Vieira, P.M.; Saghatelian, A.; Kahn, B.B. Glut4 expression in adipocytes regulates de novo lipogenesis and levels of a novel class of lipids with antidiabetic and anti-inflammatory effects. Diabetes 2016, 65, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.I.; Lynes, M.D.; Takahashi, H.; Baer, L.A.; Arts, P.J.; May, F.J.; Lehnig, A.C.; Middelbeek, R.J.W.; Richard, J.J.; So, K.; et al. 12,13-dihome: An exercise-induced lipokine that increases skeletal muscle fatty acid uptake. Cell Metab. 2018, 27, 1111–1120.e3. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.D.; Leiria, L.O.; Lundh, M.; Bartelt, A.; Shamsi, F.; Huang, T.L.; Takahashi, H.; Hirshman, M.F.; Schlein, C.; Lee, A.; et al. The cold-induced lipokine 12,13-dihome promotes fatty acid transport into brown adipose tissue. Nat. Med. 2017, 23, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Peirce, V.; Vidal-Puig, A. Regulation of glucose homoeostasis by brown adipose tissue. Lancet Diabetes Endocrinol. 2013, 1, 353–360. [Google Scholar] [CrossRef]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Van der Lans, A.A.; Hoeks, J.; Brans, B.; Vijgen, G.H.; Visser, M.G.; Vosselman, M.J.; Hansen, J.; Jorgensen, J.A.; Wu, J.; Mottaghy, F.M.; et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J. Clin. Investig. 2013, 123, 3395–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, L.P.; Koza, R.A.; Anunciado-Koza, R. Brown fat thermogenesis and body weight regulation in mice: Relevance to humans. Int. J. Obes. 2010, 34 (Suppl. 1), S23–S27. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Pedersen, D.J.; Henriques, F.; Bedard, A.H.; Henchey, E.; Kelly, M.; Morgan, D.A.; Rahmouni, K.; Czech, M.P. Neuronal modulation of brown adipose activity through perturbation of white adipocyte lipogenesis. Mol. Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Diwoky, C.; Schoiswohl, G.; Feiler, U.; Wongsiriroj, N.; Abdellatif, M.; Kolb, D.; Hoeks, J.; Kershaw, E.E.; Sedej, S.; et al. Cold-induced thermogenesis depends on atgl-mediated lipolysis in cardiac muscle, but not brown adipose tissue. Cell Metab. 2017, 26, 753–763.e7. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Ma, Y.; Chanturiya, T.; Cao, Q.; Wang, Y.; Kadegowda, A.K.G.; Jackson, R.; Rumore, D.; Xue, B.; Shi, H.; et al. Lipolysis in brown adipocytes is not essential for cold-induced thermogenesis in mice. Cell Metab. 2017, 26, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, I.J.; Yin, L.; Jensen-Urstad, A.P.; Funai, K.; Coleman, T.; Baird, J.H.; El Ramahi, M.K.; Razani, B.; Song, H.; Fu-Hsu, F.; et al. Inhibiting adipose tissue lipogenesis reprograms thermogenesis and ppargamma activation to decrease diet-induced obesity. Cell Metab. 2012, 16, 189–201. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| TF | Loss of Function | Phenotypes | References |
|---|---|---|---|
| SREBP-1 | Global | Decreased hepatic lipogenesis, while increased hepatic cholesterol synthesis due to elevated SREBP-2 in liver; No effect on adiposity and lipogenic enzymes expression in WAT. | Shimano et al., 1997 [60] |
| Adipose tissues | Not available | Not available | |
| Liver | Decreased hepatic lipogenesis, abolished sucrose-induced hypertriglyceridemia, and prevented hepatic steatosis in ob/ob mice and HFD-fed mice, despite persistent obesity, hyperinsulinemia, and hyperglycemia. | Moon, et al., 2012 [58] | |
| ChREBP | Global | Decreased hepatic lipogenesis and glycolysis; Increased hepatic glycogen level; Reduced adiposity; Impaired insulin sensitivity and glucose tolerance. | Iizuka et al., 2004 [68] |
| Adipose tissues | Decreased sucrose-induced lipogenesis in adipose tissue but not in the liver; Decreased PAHSAs level in serum; Impaired insulin sensitivity and glucose tolerance. | Vijayakumar et al., 2017 [19] | |
| Liver | No effects on hepatic lipogenesis, but altered expression of lipogenic genes in liver, WAT and BAT; Protected from high-carbohydrate diet induced hepatic steatosis, but with increased hepatic glucose production and impaired hepatic insulin sensitivity and systemic glucose tolerance; Reduced WAT mass and adipocyte size. | Jois et al., 2017 [89] | |
| LXRs | Global | Decreased hepatic lipogenesis and protected from hepatic steatosis; Impaired β-cell expansion and glucose tolerance; Improved insulin sensitivity due to increased WAT lipogenesis and WAT mass. | Beaven et al., 2013 [88] |
| Adipose tissues | Increased adipocyte size and adiposity by decreasing WAT lipolytic and oxidative capacities. | Dib et al., 2014 [90] | |
| Liver | Not available | Not available |
| TF | Gain of Function | Phenotypes | References |
|---|---|---|---|
| SREBP-1c | Adipose tissues | Impaired adipocytes differentiation, markedly reduced adiposity; Increased fatty liver development; Impaired insulin sensitivity and glucose tolerance. | Shimomura et al., 1998 [64] |
| Liver | Increased hepatic lipogenesis and fatty liver development; Increased visceral adipose tissue mass; Impaired insulin sensitivity. | Knebel et al., 2012 [56] | |
| SREBP-1a | Adipose tissues | Increased adipose tissue lipogenesis and adipocyte hypertrophy; Enhanced fatty acid secretion and fatty liver development. | Horton et al., 2003 [63] |
| Liver | Increased hepatic lipogenesis and cholesterol synthesis, and enhanced fatty liver development. | Shimano et al., 1996 [91] | |
| ChREBP | Adipose tissues | Increased adipose tissue lipogenesis; Reduced adiposity; Protected from HFD-diet induced fatty liver; Improved insulin sensitivity and glucose tolerance. | Nuotio-Antar et al., 2015 [78] |
| Liver | Increased hepatic glycolysis and lipogenesis, enhanced fatty liver development; Decreased visceral adipose tissue mass; Improved hepatic insulin signaling and systemic glucose tolerance. | Benhamed et al., 2012 [92] | |
| LXRs | Global | Increased hepatic lipogenesis and enhanced fatty liver development; Increased WAT lipolysis and apoptosis, and decreased fat mass; Impaired insulin sensitivity but not glucose tolerance. pharmacological treatment | Dong et al., 2017 [87] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients 2018, 10, 1383. https://doi.org/10.3390/nu10101383
Song Z, Xiaoli AM, Yang F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients. 2018; 10(10):1383. https://doi.org/10.3390/nu10101383
Chicago/Turabian StyleSong, Ziyi, Alus M. Xiaoli, and Fajun Yang. 2018. "Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues" Nutrients 10, no. 10: 1383. https://doi.org/10.3390/nu10101383
APA StyleSong, Z., Xiaoli, A. M., & Yang, F. (2018). Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients, 10(10), 1383. https://doi.org/10.3390/nu10101383