Valorization of Lignin as a Sustainable Component of Structural Materials and Composites: Advances from 2011 to 2019


Abstract
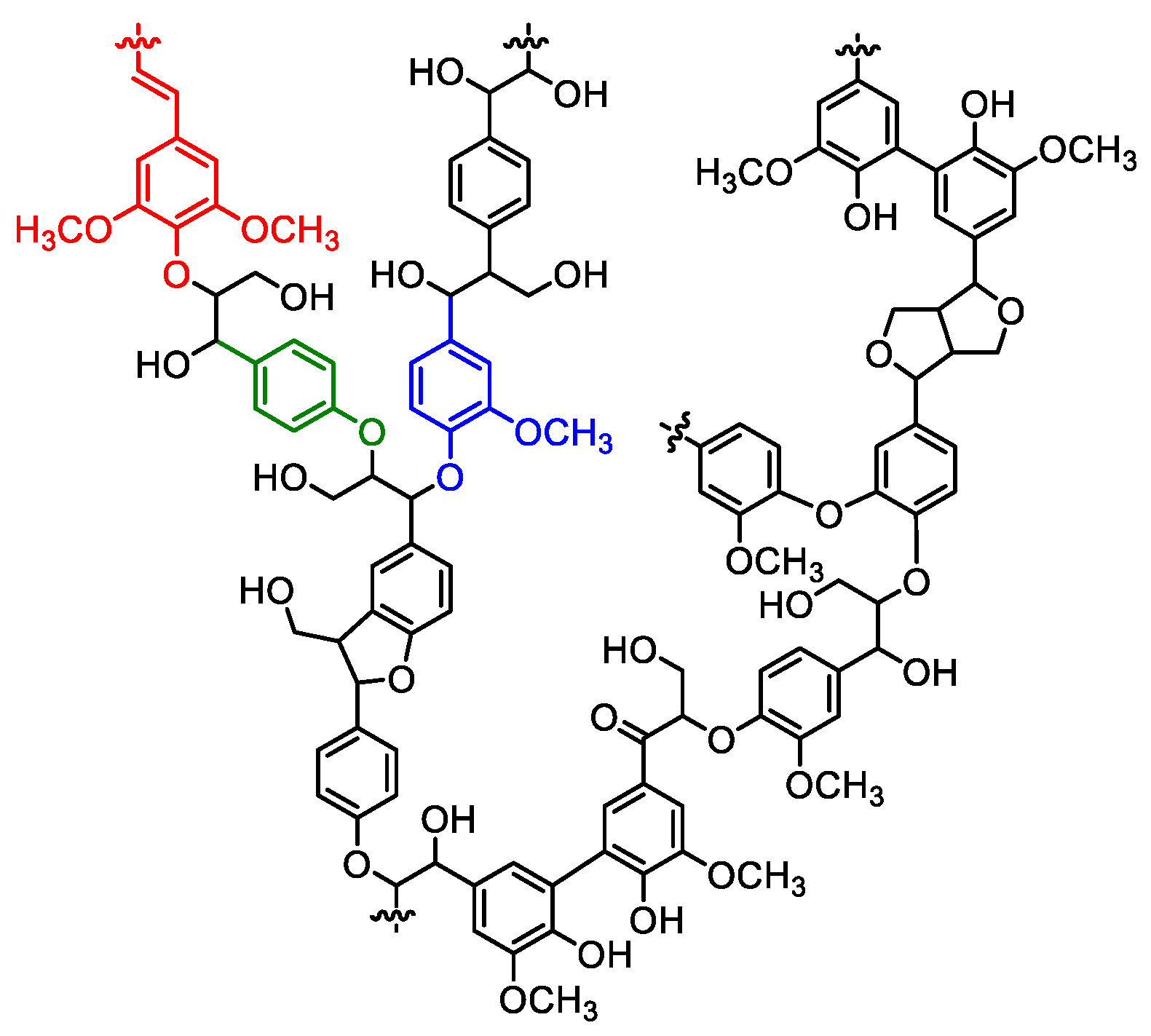
1. Introduction
2. Lignin Derivatives for Polyurethane Synthesis
2.1. Polyurethanes Prepared from Unmodified Lignin
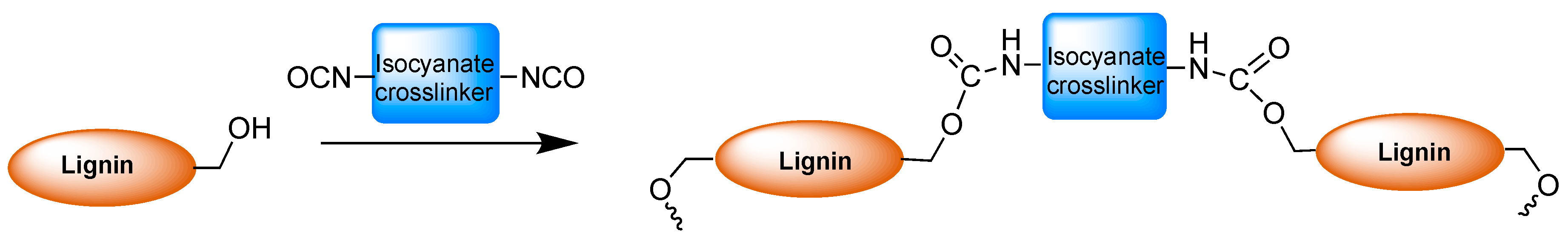
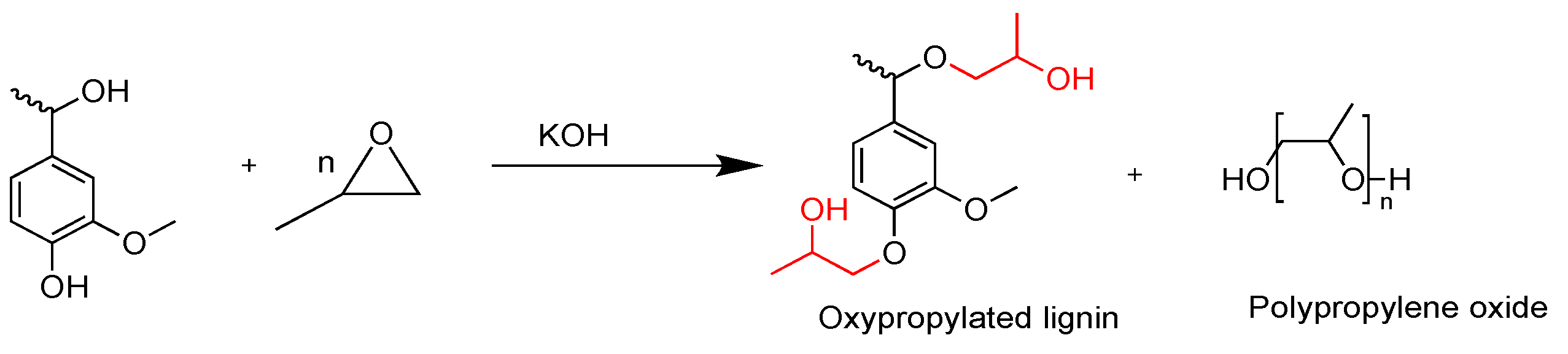
2.2. Polyurethanes Prepared from Modified Lignin
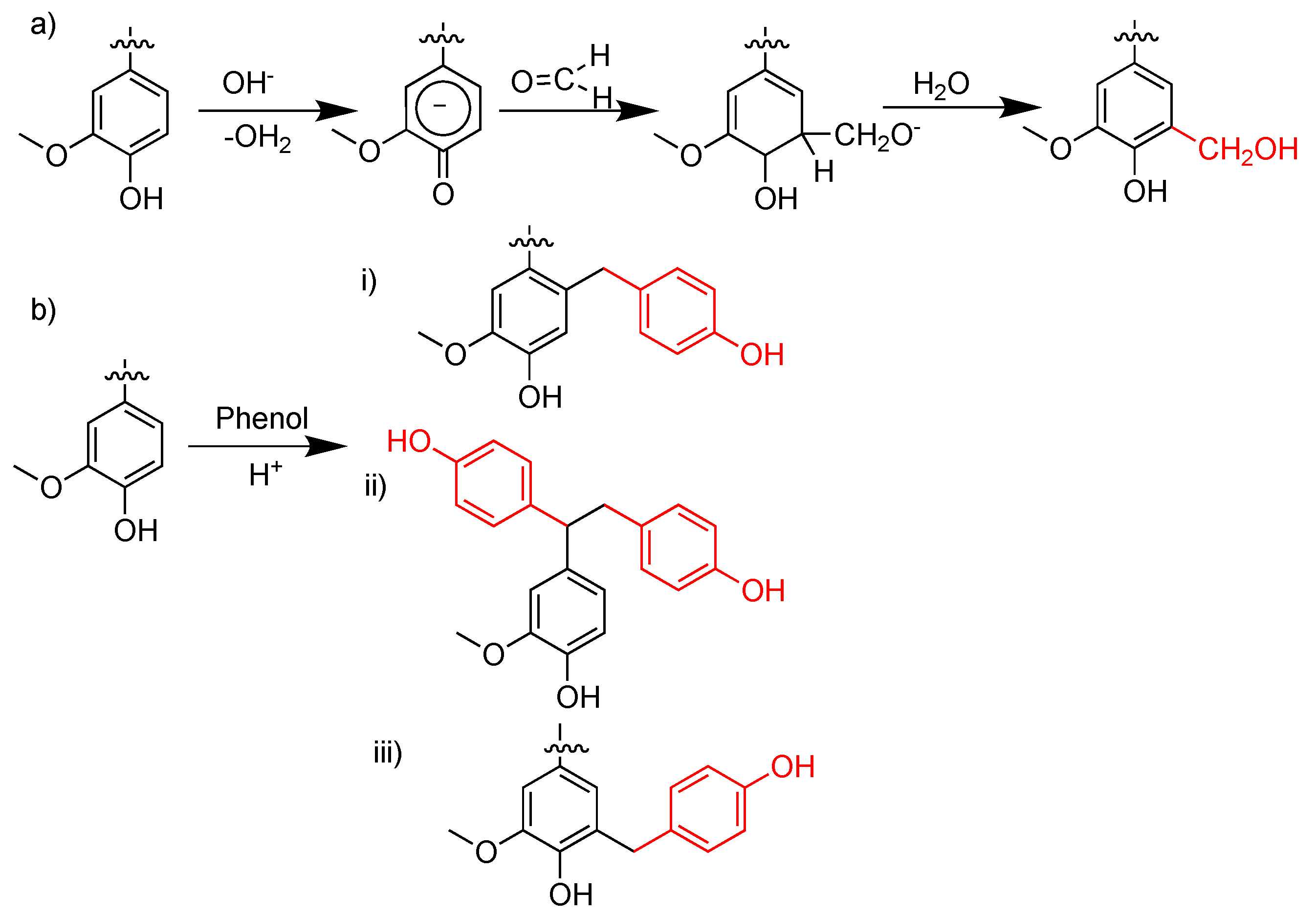
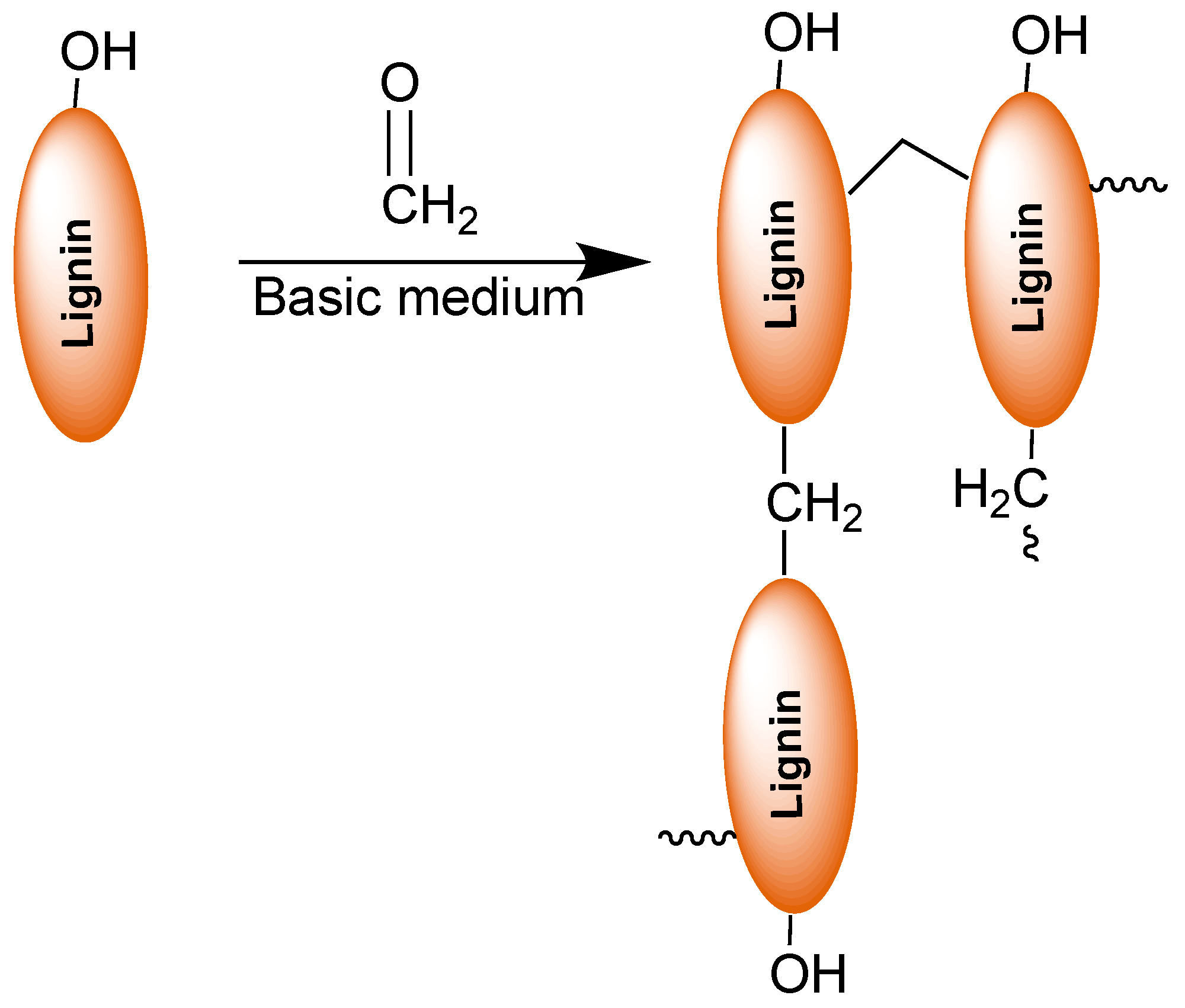
3. Lignin in Phenol-Formaldehyde (PF) Resins
4. Lignin-Derived Carbon Fibers
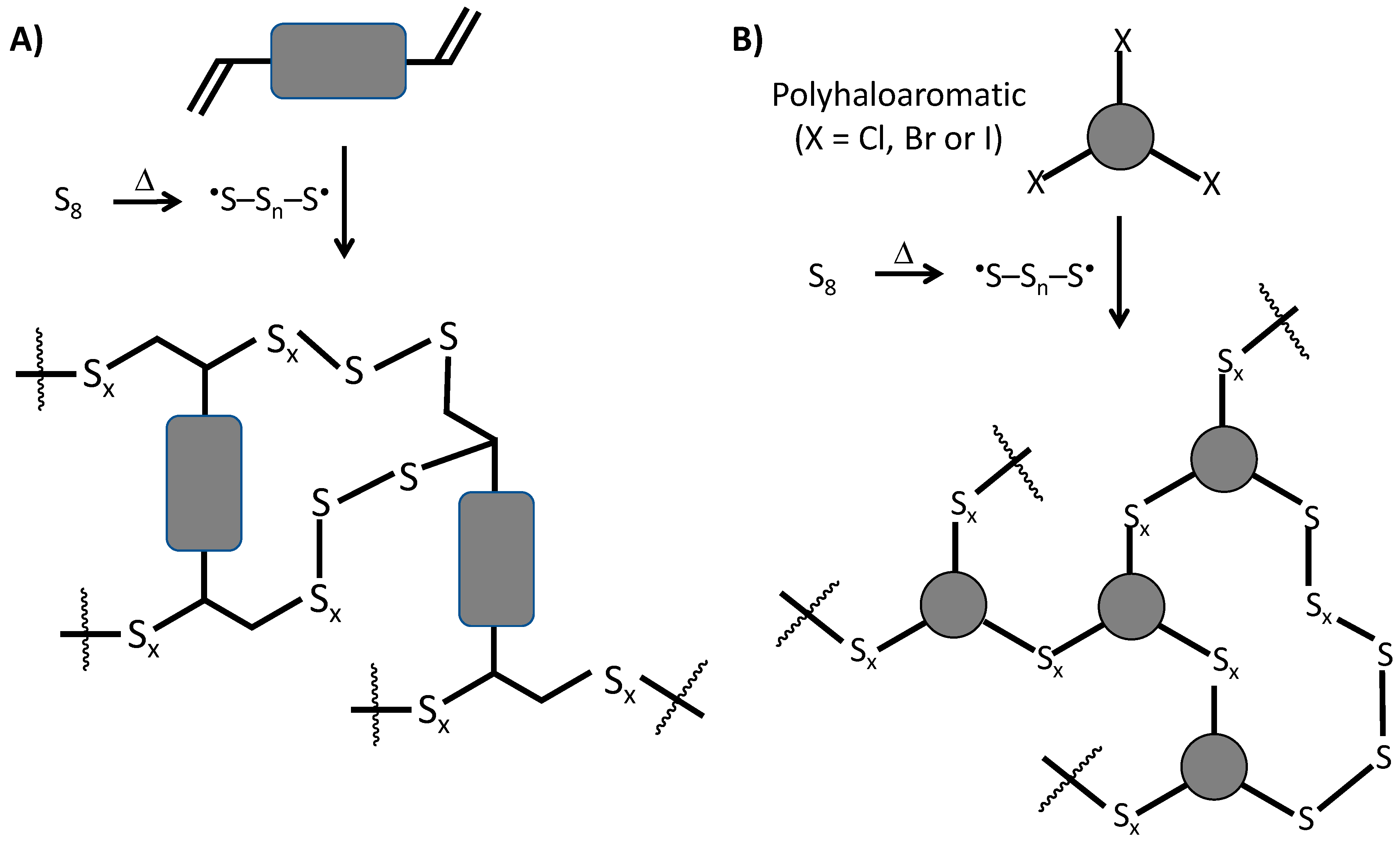
5. Lignin in High Sulfur-Content Materials
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crop. Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Tribot, A.; Amer, G.; Alio, M.A.; de Baynast, H.; Delattre, C.; Pons, A.; Mathias, J.-D.; Callois, J.-M.; Vial, C.; Michaud, P. Wood-lignin: Supply, extraction processes and use as bio-based material. Eur. Polym. J. 2019, 112, 228–240. [Google Scholar] [CrossRef]
- Doherty, W.O.S.; Mousavioun, P.; Fellows, C.M. Value-adding to cellulosic ethanol: Lignin polymers. Ind. Crop. Prod. 2011, 33, 259–276. [Google Scholar] [CrossRef]
- Morandim-Giannetti, A.A.; Agnelli, J.A.M.; Lanças, B.Z.; Magnabosco, R.; Casarin, S.A.; Bettini, S.H.P. Lignin as additive in polypropylene/coir composites: Thermal, mechanical and morphological properties. Carbohydr. Polym. 2012, 87, 2563–2568. [Google Scholar] [CrossRef]
- Zoia, L.; Salanti, A.; Frigerio, P.; Orlandi, M. Exploring allylation and Claisen rearrangement as a novel chemical modification of lignin. BioResources 2014, 9, 6540–6561. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Stevens, J.; Gardner, D.J. Enhancing the fuel value of wood pellets with the addition of lignin. Wood Fiber Sci. 2010, 42, 439–443. [Google Scholar]
- Wang, Y.-Y.; Cai, C.M.; Ragauskas, A.J. Recent advances in lignin-based polyurethanes. Tappi J. 2017, 16, 1376496. [Google Scholar] [CrossRef]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C. Depolymerization of lignins and their applications for the preparation of polyols and rigid polyurethane foams: A review. Renew. Sustain. Energy Rev. 2016, 60, 317–329. [Google Scholar] [CrossRef]
- Li, Y.; Ragauskas, A.J. Kraft Lignin-Based Rigid Polyurethane Foam. J. Wood Chem. Technol. 2012, 32, 210–224. [Google Scholar] [CrossRef]
- Nacas, A.M.; Ito, N.M.; Sousa, R.R.D.; Spinacé, M.A.; Dos Santos, D.J. Effects of NCO:OH ratio on the mechanical properties and chemical structure of Kraft lignin–based polyurethane adhesive. J. Adhes. 2017, 93, 18–29. [Google Scholar] [CrossRef]
- Jia, Z.; Lu, C.; Zhou, P.; Wang, L. Preparation and characterization of high boiling solvent lignin-based polyurethane film with lignin as the only hydroxyl group provider. RSC Adv. 2015, 5, 53949–53955. [Google Scholar] [CrossRef]
- Griffini, G.; Passoni, V.; Suriano, R.; Levi, M.; Turri, S. Polyurethane Coatings Based on Chemically Unmodified Fractionated Lignin. ACS Sustain. Chem. Eng. 2015, 3, 1145–1154. [Google Scholar] [CrossRef]
- Wu, S.; Zhan, H. Characteristics of demethylated wheat straw soda lignin and its utilization in lignin-based phenolic formaldehyde resins. Cellul. Chem. Technol. 2001, 35, 253–262. [Google Scholar]
- Chung, H.; Washburn, N.R. Improved Lignin Polyurethane Properties with Lewis Acid Treatment. ACS Appl. Mater. Interfaces 2012, 4, 2840–2846. [Google Scholar] [CrossRef]
- Cateto, C.A.; Barreiro, M.F.; Rodrigues, A.E.; Belgacem, M.N. Optimization Study of Lignin Oxypropylation in View of the Preparation of Polyurethane Rigid Foams. Ind. Eng. Chem. Res. 2009, 48, 2583–2589. [Google Scholar] [CrossRef]
- Sadeghifar, H.; Cui, C.; Argyropoulos, D.S. Toward Thermoplastic Lignin Polymers. Part 1. Selective Masking of Phenolic Hydroxyl Groups in Kraft Lignins via Methylation and Oxypropylation Chemistries. Ind. Eng. Chem. Res. 2012, 51, 16713–16720. [Google Scholar] [CrossRef]
- Ahvazi, B.; Wojciechowicz, O.; Ton-That, T.-M.; Hawari, J. Preparation of Lignopolyols from Wheat Straw Soda Lignin. J. Agric. Food Chem. 2011, 59, 10505–10516. [Google Scholar] [CrossRef]
- Pavier, C.; Gandini, A. Oxypropylation of sugar beet pulp. 2. Separation of the grafted pulp from the propylene oxide homopolymer. Carbohydr. Polym. 2000, 42, 13–17. [Google Scholar] [CrossRef]
- Wu, L.C.F.; Glasser, W.G. Engineering plastics from lignin. I. Synthesis of hydroxypropyl lignin. J. Appl. Polym. Sci. 1984, 29, 1111–1123. [Google Scholar] [CrossRef]
- Tian, T.; Hu, R.; Tang, B.Z. Room Temperature One-Step Conversion from Elemental Sulfur to Functional Polythioureas through Catalyst-Free Multicomponent Polymerizations. J. Am. Chem. Soc. 2018, 140, 6156–6163. [Google Scholar] [CrossRef] [PubMed]
- Pierre, L.E.S.; Price, C.C. The room temperature polymerization of propylene oxide. J. Am. Chem. Soc. 1956, 78, 3432–3436. [Google Scholar] [CrossRef]
- Li, Y.; Ragauskas, A.J. Ethanol organosolv lignin-based rigid polyurethane foam reinforced with cellulose nanowhiskers. RSC Adv. 2012, 2, 3347–3351. [Google Scholar] [CrossRef]
- Faria, F.A.C.; Evtuguin, D.V.; Rudnitskaya, A.; Gomes, M.T.S.R.; Oliveira, J.A.B.P.; Graca, M.P.F.; Costa, L.C. Lignin-based polyurethane doped with carbon nanotubes for sensor applications. Polym. Int. 2012, 61, 788–794. [Google Scholar] [CrossRef]
- El Mansouri, N.E.; Yuan, Q.; Huang, F. Preparation and Characterization of Phenol-Formaldehyde Resins Modified with Alkaline Rice Straw Lignin. Bioresources 2018, 13, 8061–8075. [Google Scholar] [CrossRef]
- Pizzi, A.; Cameron, F.-A.; van der Klashorst, G.H. Soda Bagasse Lignin Adhesives for Particleboard. In Adhesives from Renewable Resources; American Chemical Society: Washington, DC, USA, 1989; Volume 385, pp. 82–95. [Google Scholar]
- Doherty, W.; Halley, P.; Edye, L.; Rogers, D.; Cardona, F.; Park, Y.; Woo, T. Studies on polymers and composites from lignin and fiber derived from sugar cane. Polym. Adv. Technol. 2007, 18, 673–678. [Google Scholar] [CrossRef]
- Hu, L.; Pan, H.; Zhou, Y.; Zhang, M. Methods to improve lignin’s reactivity as a phenol substitute and as replacement for other phenolic compounds: A brief review. Bioresources 2011, 6, 3515–3525. [Google Scholar]
- Jiang, X.; Liu, J.; Du, X.; Hu, Z.; Chang, H.-M.; Jameel, H. Phenolation to Improve Lignin Reactivity toward Thermosets Application. ACS Sustain. Chem. Eng. 2018, 6, 5504–5512. [Google Scholar] [CrossRef]
- Çetin, N.S.; Özmen, N. Studies on lignin-based adhesives for particleboard panels. Turk. J. Agric. For. 2003, 27, 183–189. [Google Scholar]
- Solt, P.; van Herwijnen, H.W.G.; Konnerth, J. Thermoplastic and moisture-dependent behavior of lignin phenol formaldehyde resins. J. Appl. Polym. Sci. 2019, 136, 48011. [Google Scholar] [CrossRef]
- Grossman, A.; Vermerris, W. Lignin-based polymers and nanomaterials. Curr. Opin. Biotechnol. 2019, 56, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Forss, K.G.; Fuhrmann, A. Finnish plywood, particleboard, and fiberboard made with a lignin-base adhesive. For. Prod. J. 1979, 29, 39–43. [Google Scholar]
- Zhao, M.; Jing, J.; Zhu, Y.; Yang, X.; Wang, X.; Wang, Z. Preparation and performance of lignin–phenol–formaldehyde adhesives. Int. J. Adhes. Adhes. 2016, 64, 163–167. [Google Scholar] [CrossRef]
- Younesi-Kordkheili, H.; Pizzi, A.; Niyatzade, G. Reduction of Formaldehyde Emission from Particleboard by Phenolated Kraft Lignin. J. Adhes. 2016, 92, 485–497. [Google Scholar] [CrossRef]
- Kadla, J.F.; Kubo, S.; Venditti, R.A.; Gilbert, R.D.; Compere, A.L.; Griffith, W. Lignin-based carbon fibers for composite fiber applications. Carbon 2002, 40, 2913–2920. [Google Scholar] [CrossRef]
- Lim, T.H.; Yeo, S.Y. Investigation of the degradation of pitch-based carbon fibers properties upon insufficient or excess thermal treatment. Sci. Rep. 2017, 7, 4733. [Google Scholar] [CrossRef] [PubMed]
- Souto, F.; Calado, V.; Pereira, N. Lignin-based carbon fiber: A current overview. Mater. Res. Express 2018, 5, 072001. [Google Scholar] [CrossRef]
- Baker, D.A.; Gallego, N.C.; Baker, F.S. On the characterization and spinning of an organic-purified lignin toward the manufacture of low-cost carbon fiber. J. Appl. Polym. Sci. 2012, 124, 227–234. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Xiang, H.; Zhou, Z.; Chang, T.; Zhu, M. Low cost carbon fibers from bio-renewable lignin/poly (lactic acid)(PLA) blends. Compos. Sci. Technol. 2015, 119, 20–25. [Google Scholar] [CrossRef]
- Li, Q.; Xie, S.; Serem, W.K.; Naik, M.T.; Liu, L.; Yuan, J.S. Quality carbon fibers from fractionated lignin. Green Chem. 2017, 19, 1628–1634. [Google Scholar] [CrossRef]
- Jin, J.; Ding, J.; Klett, A.; Thies, M.C.; Ogale, A.A. Carbon Fibers Derived from Fractionated–Solvated Lignin Precursors for Enhanced Mechanical Performance. ACS Sustain. Chem. Eng. 2018, 6, 14135–14142. [Google Scholar] [CrossRef]
- Karhunen, P.; Rummakko, P.; Sipilä, J.; Brunow, G.; Kilpeläinen, I. Dibenzodioxocins; a novel type of linkage in softwood lignins. Tetrahedron Lett. 1995, 36, 169–170. [Google Scholar] [CrossRef]
- Brodin, I. Chemical Properties and Thermal Behaviour of Kraft Lignins. Licentiate Thesis, KTH Royal Institute of Technology, Stockholm, Sweden, 2009. [Google Scholar]
- Fang, W.; Yang, S.; Wang, X.-L.; Yuan, T.-Q.; Sun, R.-C. Manufacture and application of lignin-based carbon fibers (LCFs) and lignin-based carbon nanofibers (LCNFs). Green Chem. 2017, 19, 1794–1827. [Google Scholar] [CrossRef]
- Hosseinaei, O.; Harper, D.P.; Bozell, J.J.; Rials, T.G. Improving Processing and Performance of Pure Lignin Carbon Fibers through Hardwood and Herbaceous Lignin Blends. Int. J. Mol. Sci. 2017, 18, 1410. [Google Scholar] [CrossRef]
- Akpan, E.I. Stabilization of Lignin Fibers. In Sustainable Lignin for Carbon Fibers: Principles, Techniques, and Applications; Springer: Berlin/Heidelberg, Germany, 2019; pp. 325–352. [Google Scholar]
- Eckert, R.C.; Abdullah, Z. Carbon Fibers from Kraft Softwood Lignin. U.S. Patent 20080317661, 25 December 2008. [Google Scholar]
- Zhang, M.; Ogale, A. Carbon fibers from dry-spinning of acetylated softwood kraft lignin. Carbon 2014, 69, 626–629. [Google Scholar] [CrossRef]
- Chung, W.J.; Griebel, J.J.; Kim, E.T.; Yoon, H.; Simmonds, A.G.; Ji, H.J.; Dirlam, P.T.; Glass, R.S.; Wie, J.J.; Nguyen, N.A.; et al. The use of elemental sulfur as an alternative feedstock for polymeric materials. Nat. Chem. 2013, 5, 518–524. [Google Scholar] [CrossRef]
- Griebel, J.J.; Glass, R.S.; Char, K.; Pyun, J. Polymerizations with elemental sulfur: A novel route to high sulfur content polymers for sustainability, energy and defense. Prog. Polym. Sci. 2016, 58, 90–125. [Google Scholar] [CrossRef]
- Griebel, J.J.; Li, G.; Glass, R.S.; Char, K.; Pyun, J. Kilogram scale inverse vulcanization of elemental sulfur to prepare high capacity polymer electrodes for Li-S batteries. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 173–177. [Google Scholar] [CrossRef]
- Westerman, C.R.; Jenkins, C.L. Dynamic Sulfur Bonds Initiate Polymerization of Vinyl and Allyl Ethers at Mild Temperatures. Macromolecules 2018, 51, 7233–7238. [Google Scholar] [CrossRef]
- Griebel, J.J.; Namnabat, S.; Kim, E.T.; Himmelhuber, R.; Moronta, D.H.; Chung, W.J.; Simmonds, A.G.; Kim, K.-J.; van der Laan, J.; Nguyen, N.A.; et al. New Infrared Transmitting Material via Inverse Vulcanization of Elemental Sulfur to Prepare High Refractive Index Polymers. Adv. Mater. 2014, 26, 3014–3018. [Google Scholar] [CrossRef]
- Parker, D.J.; Jones, H.A.; Petcher, S.; Cervini, L.; Griffin, J.M.; Akhtar, R.; Hasell, T. Low cost and renewable sulfur-polymers by inverse vulcanization, and their potential for mercury capture. J. Mater. Chem. A Mater. Energy Sustain. 2017, 5, 11682–11692. [Google Scholar] [CrossRef]
- Hasell, T.; Smith Jessica, A.; Green Sarah, J.; Petcher, S.; Parker Douglas, J.; Zhang, B.; Wu, X.; Kelly Catherine, A.; Baker, T.; Jenkins Mike, J.; et al. Crosslinker co-polymerisation for property control in inverse vulcanisation. Chemistry 2019. [Google Scholar] [CrossRef]
- Mann, M.; Kruger, J.E.; Andari, F.; McErlean, J.; Gascooke, J.R.; Smith, J.A.; Worthington, M.J.H.; McKinley, C.C.C.; Campbell, J.A.; Lewis, D.A.; et al. Sulfur polymer composites as controlled-release fertilizers. Org. Biomol. Chem. 2019, 17, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, N.A.; Worthington, M.J.H.; Adamson, N.; Gibson, C.T.; Johnston, M.R.; Ellis, A.V.; Chalker, J.M. Polysulfides made from re-purposed waste are sustainable materials for removing iron from water. RSC Adv. 2018, 8, 1232–1236. [Google Scholar] [CrossRef]
- Hoefling, A.; Lee, Y.J.; Theato, P. Sulfur-Based Polymer Composites from Vegetable Oils and Elemental Sulfur: A Sustainable Active Material for Li-S Batteries. Macromol. Chem. Phys. 2017, 218, 1600303. [Google Scholar] [CrossRef]
- Qin, X.; He, Y.; Khan, S.; Zhang, B.; Chen, F.; Dong, D.; Wang, Z.; Zhang, L. Controllable Synthesis and Characterization of Soybean-Oil-Based Hyperbranched Polymers via One-Pot Method. ACS Sustain. Chem. Eng. 2018, 6, 12865–12871. [Google Scholar] [CrossRef]
- Valle, S.F.; Giroto, A.S.; Klaic, R.; Guimaraes, G.G.F.; Ribeiro, C. Sulfur fertilizer based on inverse vulcanization process with soybean oil. Polym. Degrad. Stab. 2019, 162, 102–105. [Google Scholar] [CrossRef]
- Worthington, M.J.H.; Shearer, C.J.; Esdaile, L.J.; Campbell, J.A.; Gibson, C.T.; Legg, S.K.; Yin, Y.; Lundquist, N.A.; Gascooke, J.R.; Albuquerque, I.S.; et al. Sustainable Polysulfides for Oil Spill Remediation: Repurposing Industrial Waste for Environmental Benefit. Adv. Sustain. Syst. 2018, 2, 1800024. [Google Scholar] [CrossRef]
- Worthington, M.J.H.; Kucera, R.L.; Albuquerque, I.S.; Gibson, C.T.; Sibley, A.; Slattery, A.D.; Campbell, J.A.; Alboaiji, S.F.K.; Muller, K.A.; Young, J.; et al. Laying Waste to Mercury: Inexpensive Sorbents Made from Sulfur and Recycled Cooking Oils. Chem. Eur. J. 2017, 23, 16219–16230. [Google Scholar] [CrossRef]
- Crockett, M.P.; Evans, A.M.; Worthington, M.J.H.; Albuquerque, I.S.; Slattery, A.D.; Gibson, C.T.; Campbell, J.A.; Lewis, D.A.; Bernardes, G.J.L.; Chalker, J.M. Sulfur-Limonene Polysulfide: A Material Synthesized Entirely from Industrial By-Products and Its Use in Removing Toxic Metals from Water and Soil. Angew. Chem. Int. Ed. 2016, 55, 1714–1718. [Google Scholar] [CrossRef]
- Fu, C.; Li, G.; Zhang, J.; Cornejo, B.; Piao, S.S.; Bozhilov, K.N.; Haddon, R.C.; Guo, J. Electrochemical Lithiation of Covalently Bonded Sulfur in Vulcanized Polyisoprene. ACS Energy Lett. 2016, 1, 115–120. [Google Scholar] [CrossRef]
- Hoefling, A.; Nguyen, D.T.; Lee, Y.J.; Song, S.-W.; Theato, P. A sulfur-eugenol allyl ether copolymer: A material synthesized via inverse vulcanization from renewable resources and its application in Li-S batteries. Mater. Chem. Front. 2017, 1, 1818–1822. [Google Scholar] [CrossRef]
- Thiounn, T.; Tennyson, A.G.; Smith, R.C. Durable, Acid-Resistant Copolymers from Industrial By-Product Sulfur and Microbially-Produced Tyrosine. RSC Adv. 2019, 9, 31460–31465. [Google Scholar] [CrossRef]
- Smith, A.D.; Thiounn, T.; Lyles, E.W.; Kibler, E.K.; Smith, R.C.; Tennyson, A.G. Combining Agriculture and Energy Industry Waste Products to Yield Recyclable, Thermally Healable Copolymers of Elemental Sulfur and Oleic Acid. J. Polym. Sci. Part A 2019, 57, 1704–1710. [Google Scholar] [CrossRef]
- Lauer, M.K.; Estrada-Mendoza, T.A.; McMillen, C.D.; Chumanov, G.; Tennyson, A.G.; Smith, R.C. Durable, Remeltable Materials from Agricultural and Petrochemical Wastes. Adv. Sustain. Syst. 2019. [Google Scholar] [CrossRef]
- Thiounn, T.; Lauer, M.K.; Bedford, M.S.; Smith, R.C.; Tennyson, A.G. Thermally-Healable Network Solids of Sulfur-Crosslinked Poly(4-allyloxystyrene). RCS Adv. 2018, 8, 39074–39082. [Google Scholar] [CrossRef]
- Karunarathna, M.; Lauer, M.K.; Thiounn, T.; Smith, R.C.; Tennyson, A.G. Valorisation of Waste to Yield Recyclable Composites of Elemental Sulfur and Lignin. J. Mater. Chem. A 2019, 7, 15683–15690. [Google Scholar] [CrossRef]
- Karunarathna, M.; Tennyson, A.G.; Smith, R.C. Facile New Approach to High Sulfur-Content Materials and Preparation of Sulfur-Lignin Copolymers. J. Mater. Chem. A 2019, 8, 548–553. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lignin type | Application | NCO/OH Ratio | Optimal Lignin wt.% | Figure of Merit Most Important for Target Application | Ref. |
|---|---|---|---|---|---|
| Spruce wood meal lignin | Coating | 1.7 | 47 | Tensile strength 41.6 Mpa | 12 |
| Kraft lignin | Coatings and adhesives | 1.3 | 70 | Elastic modulus 3.70 Gpa | 13 |
| Kraft lignin | Adhesives | 1.2 | 46.5 | Lap shear strength 6.3 Mpa | 14 |
| Organosolve lignin, modified with cellulose nanowhiskers | Rigid foam | NA | 33 | Compressive strength 0.52 Mpa, modulus 12.8 Mpa | 24 |
| Oxypropylated kraft lignin | Rigid foam | 1.7 | 100 | Compressive strength 0.14 Mpa, modulus 3.41 Mpa | 10 |
| Lignin Type | Incorporated Lignin % of the Best Sample | Formaldehyde/Phenol Mass Ratio | Free Formaldehyde % | Gelation Time/Min | Mechanical Properties/Mpa | Ref. |
|---|---|---|---|---|---|---|
| Hydroxymethylated organosolve lignin | 20 | 1.07 | 0.25 | 29 | Tensile strength 1.02 | 31 |
| Hydroxymethylated alkaline rice straw lignin | 35 | 1.8 | 0.01 | 10.2 | NA | 26 |
| Phenolated wheat straw steam explosion lignin | 40 | 1.5 | 0.43 | 7 | Tensile strength 1.11 | 35 |
| Unmodified kraft lignin | 50 | NA | 0.40 | 20 | Lap shear strength 3.4 | 32 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karunarathna, M.S.; Smith, R.C. Valorization of Lignin as a Sustainable Component of Structural Materials and Composites: Advances from 2011 to 2019. Sustainability 2020, 12, 734. https://doi.org/10.3390/su12020734
Karunarathna MS, Smith RC. Valorization of Lignin as a Sustainable Component of Structural Materials and Composites: Advances from 2011 to 2019. Sustainability. 2020; 12(2):734. https://doi.org/10.3390/su12020734
Chicago/Turabian StyleKarunarathna, Menisha S., and Rhett C. Smith. 2020. "Valorization of Lignin as a Sustainable Component of Structural Materials and Composites: Advances from 2011 to 2019" Sustainability 12, no. 2: 734. https://doi.org/10.3390/su12020734
APA StyleKarunarathna, M. S., & Smith, R. C. (2020). Valorization of Lignin as a Sustainable Component of Structural Materials and Composites: Advances from 2011 to 2019. Sustainability, 12(2), 734. https://doi.org/10.3390/su12020734