
MicroRNAs: From Junk RNA to Life Regulators and Their Role in Cardiovascular Disease

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
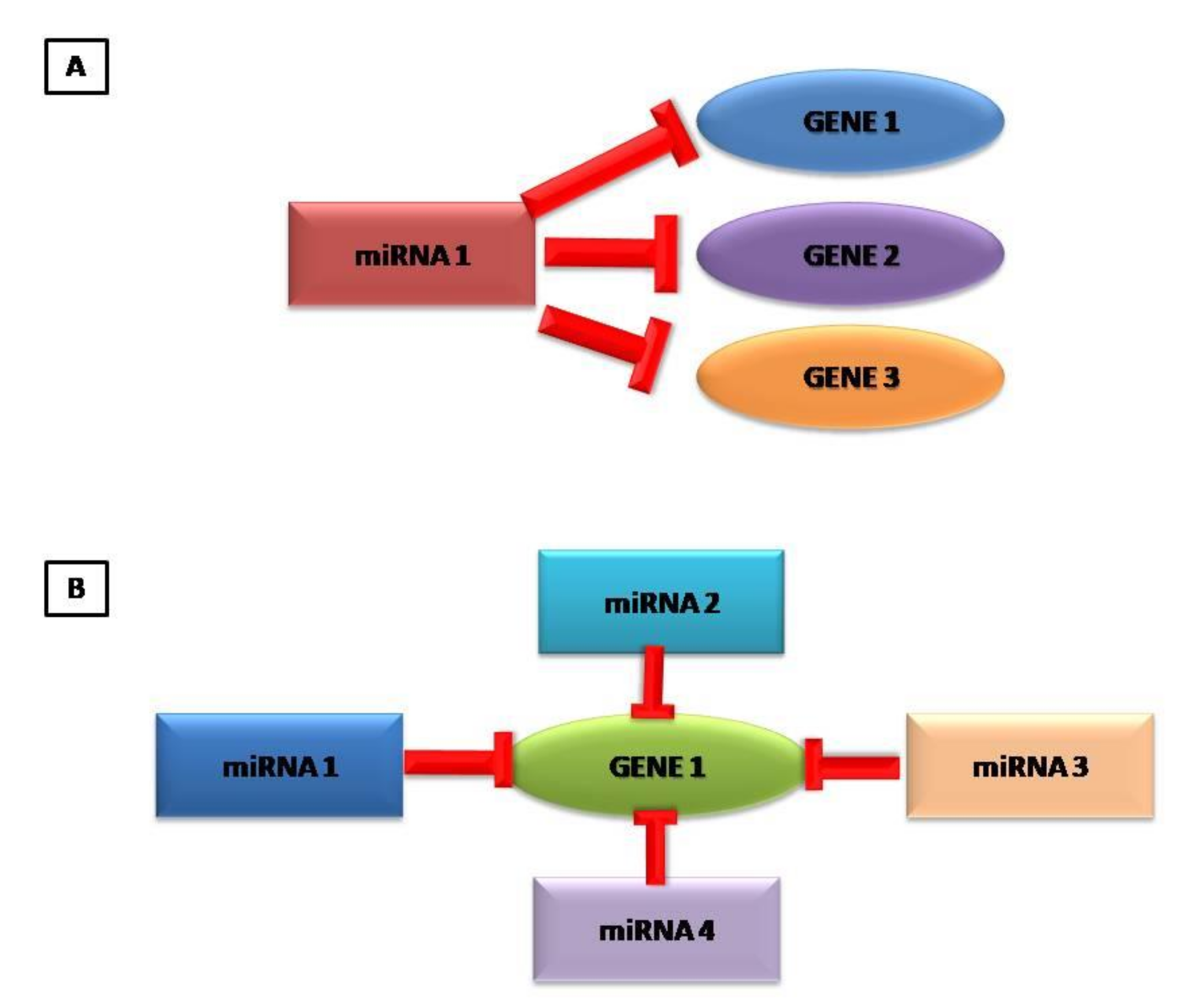
2. MiRNAs: What Are They?
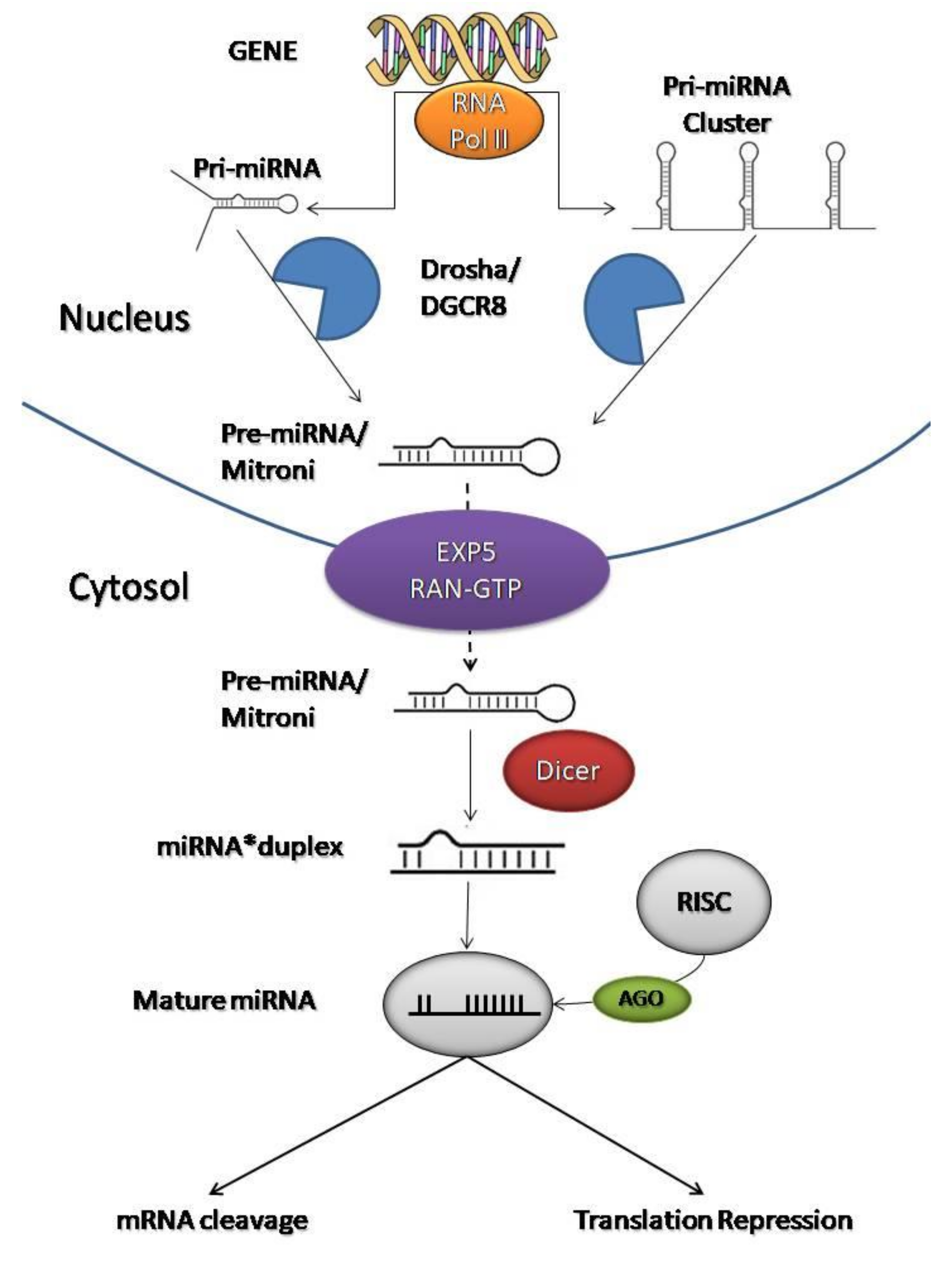
2.1. The miRNA Factory: How They Work
2.2. MiRNA as Biomarkers
3. MicroRNA and Cardiomyopathies
3.1. Hypertrophic Cardiomyopathy
MiRNA and Hypertrophic Cardiomyopathy
3.2. Dilated Cardiomyopathy
MiRNA and Dilated Cardiomyopathy
3.3. Arrhythmogenic Cardiomyopathy
MiRNA and Arrhythmogenic Cardiomyopathy
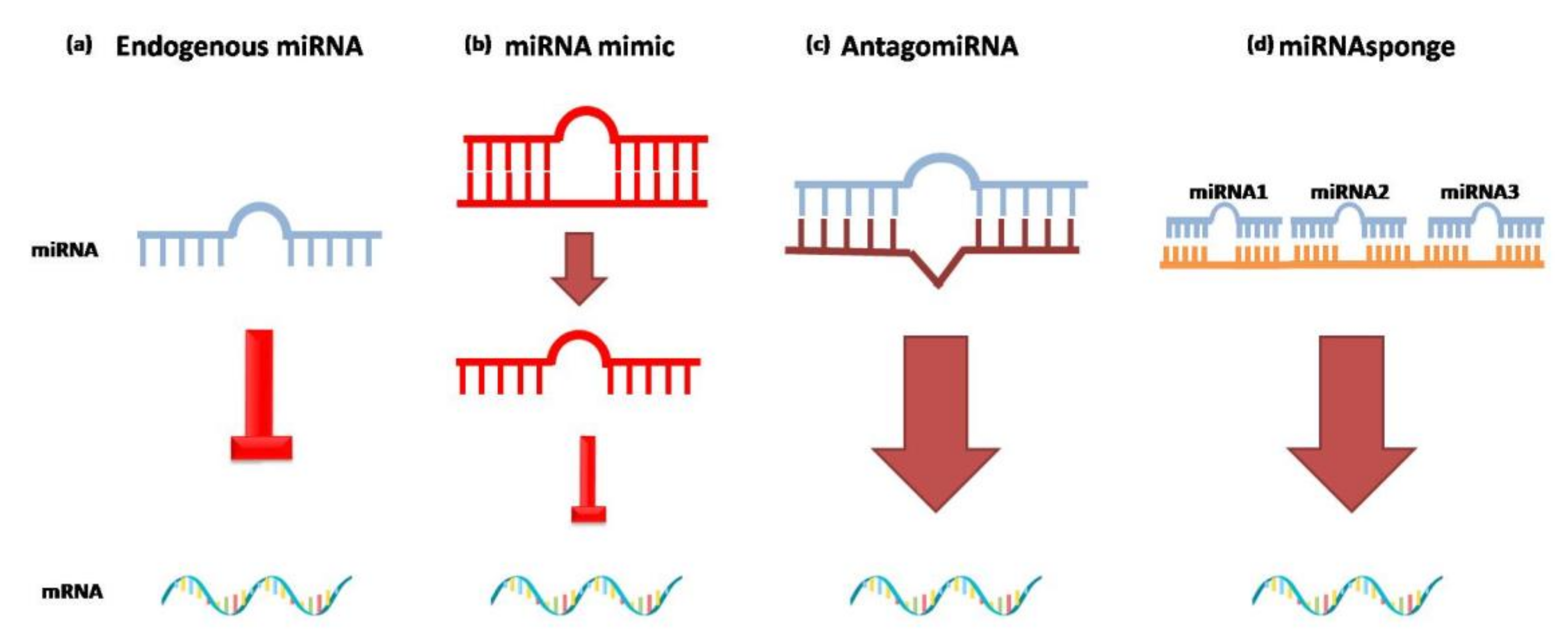
4. How to Train a miRNA: A Possible Therapy
5. Conclusions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circ. J. 2018, 137, e67–e492. [Google Scholar]
- Maillet, M.; van Berlo, J.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2012, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling--concepts and clinical implications: A consensus paper from an inter-national forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kamińska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.R.; Gay, M.S.; Zhang, L. Epigenetic mechanisms in heart development and disease. Drug Discov. Today. 2015, 20, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Hata, A. Functions of MicroRNAs in Cardiovascular Biology and Disease. Annu. Rev. Physiol. 2013, 75, 69–93. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, G.; Latronico, M.V.; Cavarretta, E. microRNAs in cardiovascular diseases: Current knowledge and the road ahead. J. Am. Coll Cardiol. 2014, 63, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Quiat, D.; Olson, E.N. MicroRNAs in cardiovascular disease: From pathogenesis to prevention and treatment. J. Clin. Investig. 2013, 123, 11–18. [Google Scholar] [CrossRef]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs Add a New Dimension to Cardiovascular Disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef]
- Bronze-Da-Rocha, E. MicroRNAs Expression Profiles in Cardiovascular Diseases. BioMed Res. Int. 2014, 2014, 1–23. [Google Scholar] [CrossRef]
- Klimczak, D.; Pączek, L.; Jażdżewski, K.; Kuch, M. MicroRNAs: Powerful regulators and potential diagnostic tools in cardio-vascular disease. Kardiol. Pol. 2015, 73, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating microRNAs: Novel biomarkers and extracellular communicators in cardi-ovascular disease? Circ. Res. 2012, 110, 483–495. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Alexander, R.P.; Fang, G.; Rozowsky, J.; Snyder, M.; Gerstein, M.B. Annotating non-coding regions of the genome. Nat. Rev. Genet. 2010, 11, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Carninci, P. Long non-coding RNA modifies chromatin: Epigenetic silencing by long non-coding RNAs. Bioessays. 2011, 33, 830–839. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature. 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Biophys. Acta. 2010, 1803, 1231–1243. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol Cell 2016, 64, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Anglicheau, D.; Muthukumar, T.; Suthanthiran, M. MicroRNAs: Small RNAs With Big Effects. Transplantation 2010, 90, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Fu, G.; Brkić, J.; Hayder, H.; Peng, C. MicroRNAs in Human Placental Development and Pregnancy Complications. Int. J. Mol. Sci. 2013, 14, 5519–5544. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs Modulate Hematopoietic Lineage Differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam Encodes a Developmentally Regulated microRNA that Controls Cell Proliferation and Regulates the Proapoptotic Gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef]
- Wilfred, B.R.; Wang, W.-X.; Nelson, P.T. Energizing miRNA research: A review of the role of miRNAs in lipid metabolism, with a prediction that miR-103/107 regulates human metabolic pathways. Mol. Genet. Metab. 2007, 91, 209–217. [Google Scholar] [CrossRef]
- Barwari, T.; Joshi, A.; Mayr, M. MicroRNAs in Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 68, 2577–2584. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Micro-array analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef]
- Eulalio, A.; Huntzinger, E.; Izaurralde, E. Getting to the root of miRNA-mediated gene silencing. Cell 2008, 132, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Paci, P.; Colombo, T.; Farina, L. Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC Syst. Biol. 2014, 8, 83. [Google Scholar] [CrossRef]
- Schneider, T.; Hung, L.H.; Schreiner, S.; Starke, S.; Eckhof, H.; Rossbach, O.; Reich, S.; Medenbach, J.; Bindereif, A. CircR-NA-protein complexes: IMP3 protein component defines subfamily of circRNPs. Sci. Rep. 2016, 6, 31313. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution—Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Liu, C.; Yang, P.; He, S.; Liao, Q.; Kang, S.; Zhao, Y. Clustered microRNAs’ coordination in regulating protein-protein interaction network. BMC Syst. Biol. 2009, 3, 65. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcrip-tion units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, X.; Ma, Z.; Huo, Y.; Xiao, Z.; Li, Y.; Wang, Y. Dynamic mechanisms for pre-miRNA binding and export by Ex-portin-5. RNA 2011, 17, 1511–1528. [Google Scholar] [CrossRef]
- Hammond, S.M. Dicing and slicing: The core machinery of the RNA interference pathway. FEBS Lett. 2005, 579, 5822–5829. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Marshall, W.S.; Olson, E.N. Toward microRNA-based therapeutics for heart disease: The sense in antisense. Circ. Res. 2008, 103, 919–928. [Google Scholar] [CrossRef]
- Hutvagner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, É.; Tuschl, T.; Zamore, P.D. A Cellular Function for the RNA-Interference Enzyme Dicer in the Maturation of the let-7 Small Temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.-H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 Negatively Regulates Expression of the Hypertrophy-Associated Calmodulin and Mef2a Genes. Mol. Cell. Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef]
- Diniz, G.P.; Lino, C.A.; Moreno, C.R.; Senger, N.; Barreto-Chaves, M.L.M. MicroRNA-1 overexpression blunts cardiomyocyte hypertrophy elicited by thyroid hormone. J. Cell. Physiol. 2017, 232, 3360–3368. [Google Scholar] [CrossRef]
- Meijer, H.A.; Smith, E.M.; Bushell, M. Regulation of miRNA strand selection: Follow the leader? Biochem. Soc. Trans. 2014, 42, 1135–1140. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2018, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Pratt, A.; MacRae, I.J. The RNA-induced Silencing Complex: A Versatile Gene-silencing Machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast–derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Fan, G.-C. Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am. J. Cardiovasc. Dis. 2011, 1, 138–149. [Google Scholar] [PubMed]
- Lee, L.C.; Zhihong, Z.; Hinson, A.; Guccione, J.M. Reduction in Left Ventricular Wall Stress and Improvement in Function in Failing Hearts using Algisyl-LVR. J. Vis. Exp. 2013, e50096. [Google Scholar] [CrossRef]
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. siRNAs can function as miRNAs. Genes Dev. 2003, 17, 438–442. [Google Scholar] [CrossRef]
- Ameres, S.L.; Zamore, P. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef]
- Latronico, M.V.G.; Condorelli, G. MicroRNAs and cardiac pathology. Nat. Rev. Cardiol. 2009, 6, 418–429. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; van Tintelen, J.P.; Fokstuen, S.; Elliott, P.; van Langen, I.M.; Meder, B.; Richard, P.; Syrris, P.; Caforio, A.L.; Adler, Y.; et al. The current role of next-generation DNA sequencing in routine care of patients with hereditary cardiovascular conditions: A viewpoint paper of the European Society of Cardiology working group on myocardial and pericardial diseases and members of the European Society of Human Genetics. Eur. Hear. J. 2015, 36, 1367–1370. [Google Scholar] [CrossRef]
- Mencía, A.; Modamio-Høybjør, S.; Redshaw, N.; Morín, M.; Mayo-Merino, F.; Olavarrieta, L.; Aguirre, L.A.; del Castillo, I.; Steel, K.P.; Dalmay, T.; et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet. 2009, 41, 609–613. [Google Scholar] [CrossRef]
- Ghai, V.; Wang, K. Recent progress toward the use of circulating microRNAs as clinical biomarkers. Arch. Toxicol. 2016, 90, 2959–2978. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids—the mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.-L.T.; Schmittgen, T.D.; et al. Detection of microRNA Expression in Human Peripheral Blood Microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuan, Y.; Cho, J.-H.; McClarty, S.; Baxter, D.; Galas, D.J. Comparing the MicroRNA Spectrum between Serum and Plasma. PLoS ONE 2012, 7, e41561. [Google Scholar] [CrossRef]
- Moldovan, L.; Batte, K.E.; Trgovcich, J.; Wisler, J.; Marsh, C.B.; Piper, M. Methodological challenges in utilizing miRNAs as circulating biomarkers. J. Cell Mol. Med. 2014, 18, 371–390. [Google Scholar] [CrossRef]
- Haider, B.A.; Baras, A.S.; McCall, M.N.; Hertel, J.A.; Cornish, T.C.; Halushka, M.K. A critical evaluation of microRNA bi-omarkers in non-neoplastic disease. PLoS ONE 2014, 9, e89565. [Google Scholar]
- Kim, Y.-K. Extracellular microRNAs as Biomarkers in Human Disease. Chonnam Med J. 2015, 51, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 9, e89565. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, D.; McKenna, W.J. Genetics of inherited cardiomyopathy. Eur. Hear. J. 2011, 33, 296–304. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [PubMed]
- Palacín, M.; Reguero, J.R.; Martín, M.; Molina, B.D.; Morís, C.; Alvarez, V.; Coto, E. Profile of MicroRNAs Differentially Produced in Hearts from Patients with Hypertrophic Cardiomyopathy and Sarcomeric Mutations. Clin. Chem. 2011, 57, 1614–1616. [Google Scholar] [CrossRef]
- Palacín, M.; Coto, E.; Reguero, J.R.; Morís, C.; Alvarez, V. Profile of microRNAs in the plasma of hypertrophic cardiomyopa-thy patients compared to healthy controls. Int. J. Cardiol. 2013, 167, 3075–3076. [Google Scholar]
- Roncarati, R.; Viviani Anselmi, C.; Losi, M.A.; Papa, L.; Cavarretta, E.; Da Costa Martins, P.; Contaldi, C.; Saccani Jotti, G.; Franzone, A.; Galastri, L.; et al. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 920–927. [Google Scholar]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178, 6p following 178. [Google Scholar] [CrossRef]
- Derda, A.A.; Thum, S.; Lorenzen, J.M.; Bavendiek, U.; Heineke, J.; Keyser, B.; Stuhrmann, M.; Givens, R.C.; Kennel, P.J.; Schulze, P.C.; et al. Blood-based microRNA signatures differentiate various forms of cardiac hypertrophy. Int. J. Cardiol. 2015, 196, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Latronico, M.V.G.; Costinean, S.; Lavitrano, M.L.; Peschle, C.; Condorelli, G. Regulation of Cell Size and Contractile Function by AKT in Cardiomyocytes. Ann. N. Y. Acad. Sci. 2004, 1015, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Kuster, D.W.; Mulders, J.; Cate, F.T.; Michels, M.; dos Remedios, C.G.; Martins, P.A.D.C.; van der Velden, J.; Oudejans, C.B. MicroRNA transcriptome profiling in cardiac tissue of hypertrophic cardiomyopathy patients with MYBPC3 mutations. J. Mol. Cell. Cardiol. 2013, 65, 59–66. [Google Scholar] [CrossRef]
- Ming, S.; Shui-Yun, W.; Wei, Q.; Jian-Hui, L.; Ru-Tai, H.; Lei, S.; Mei, J.; Hui, W.; Ji-Zheng, W. miR-139-5p inhibits isopro-terenol-induced cardiac hypertrophy by targetting c-Jun. Biosci. Rep. 2018, 38, BSR20171430. [Google Scholar] [CrossRef]
- Sun, D.; Li, C.; Liu, J.; Wang, Z.; Liu, Y.; Luo, C.; Chen, Y.; Wen, S. Expression Profile of microRNAs in Hypertrophic Cardi-omyopathy and Effects of microRNA-20 in Inducing Cardiomyocyte Hypertrophy Through Regulating Gene MFN2. DNA Cell Biol. 2019, 38, 796–807. [Google Scholar] [CrossRef]
- Wang, H.; Chen, F.; Tong, J.; Li, Y.; Cai, J.; Wang, Y.; Li, P.; Hao, Y.; Tian, W.; Lv, Y.; et al. Circulating microRNAs as novel biomarkers for dilated cardiomyopathy. Cardiol. J. 2017, 24, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Raso, A.; Dirkx, E.; Philippen, L.E.; Fernandez-Celis, A.; De Majo, F.; Pinto, V.; Sansonetti, M.; Juni, R.; el Azzouzi, H.; Calore, M.; et al. Therapeutic Delivery of miR-148a Suppresses Ventricular Dilation in Heart Failure. Mol. Ther. 2018, 27, 584–599. [Google Scholar] [CrossRef]
- Yu, D.-C.; Li, Q.-G.; Ding, X.-W.; Ding, Y.-T. Circulating MicroRNAs: Potential Biomarkers for Cancer. Int. J. Mol. Sci. 2011, 12, 2055–2063. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of ca-nonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular car-diomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, S.; Dong, T.; Yang, J.; Xie, Y.; Wu, Y.; Kang, K.; Hu, S.; Gou, D.; Wei, Y. Profiling of differentially expressed microRNAs in arrhythmogenic right ventricular cardiomyopathy. Sci. Rep. 2016, 6, 28101. [Google Scholar] [CrossRef]
- Sommariva, E.; D’Alessandra, Y.; Farina, F.M.; Casella, M.; Cattaneo, F.; Catto, V.; Chiesa, M.; Stadiotti, I.; Brambilla, S.; Del-lo Russo, A.; et al. MiR-320a as a Potential Novel Circulating Bi-omarker of Arrhythmogenic CardioMyopathy. Sci. Rep. 2017, 7, 4802. [Google Scholar] [CrossRef]
- Yamada, S.; Hsiao, Y.-W.; Chang, S.-L.; Lin, Y.-J.; Lo, L.-W.; Chung, F.-P.; Chiang, S.-J.; Hu, Y.-F.; Tuan, T.-C.; Chao, T.-F.; et al. Circulating microRNAs in arrhythmogenic right ventricular cardiomyopathy with ventricular arrhythmia. Europace 2017, 20, f37–f45. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.-W.; Ren, X.-P.; Chen, J.; Liu, H.; Yang, J.; Medvedovic, M.; Hu, Z.; Fan, G.-C. MicroRNA-494 Targeting Both Proapoptotic and Antiapoptotic Proteins Protects Against Ischemia/Reperfusion-Induced Cardiac Injury. Circulation 2010, 122, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circ. J. 2006, 113, 1807–1816. [Google Scholar]
- Maron, B.J.; Haas, T.S.; Ahluwalia, A.; Murphy, C.J.; Garberich, R.F. Demographics and Epidemiology of Sudden Deaths in Young Competitive Athletes: From the United States National Registry. Am. J. Med. 2016, 129, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Movahed, M.R.; Strootman, D.; Bates, S.; Sattur, S. Prevalence of suspected hypertrophic cardiomyopathy or left ventricular hypertrophy based on race and gender in teenagers using screening echocardiography. Cardiovasc. Ultrasound 2010, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef]
- Kumar, K.R.; Mandleywala, S.N.; Link, M.S. Atrial and Ventricular Arrhythmias in Hypertrophic Cardiomyopathy. Card. Electrophysiol. Clin. 2015, 7, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Artham, S.M.; Lavie, C.J.; Milani, R.V.; Patel, D.A.; Verma, A.; Ventura, H.O. Clinical Impact of Left Ventricular Hypertrophy and Implications for Regression. Prog. Cardiovasc. Dis. 2009, 52, 153–167. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.; Weeks, K.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y. Genetics and Clinical Destiny: Improving Care in Hypertrophic Cardiomyopathy. Circulation 2010, 122, 2430–2440. [Google Scholar] [CrossRef]
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Haas, T.S.; Goodman, J.S. Hypertrophic Cardiomyopathy: One Gene … but Many Phenotypes. Am. J. Cardiol. 2014, 113, 1772–1773. [Google Scholar] [CrossRef]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Spirito, P.; Autore, C.; Formisano, F.; Assenza, G.E.; Biagini, E.; Haas, T.S.; Bongioanni, S.; Semsarian, C.; Devoto, E.; Musumeci, B.; et al. Risk of Sudden Death and Outcome in Patients With Hypertrophic Cardiomyopathy With Benign Presentation and Without Risk Factors. Am. J. Cardiol. 2014, 113, 1550–1555. [Google Scholar] [CrossRef]
- Lopes, L.R.; Rahman, M.S.; Elliott, P.M. A systematic review and meta-analysis of genotype-phenotype associations in pa-tients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013, 99, 1800–1811. [Google Scholar] [CrossRef]
- Keren, A.; Syrris, P.; McKenna, W.J. Hypertrophic cardiomyopathy: The genetic determinants of clinical disease expression. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 158–168. [Google Scholar] [CrossRef]
- Bos, J.M.; Towbin, J.A.; Ackerman, M.J. Diagnostic, prognostic, and therapeutic implications of genetic testing for hyper-trophic cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 201–211. [Google Scholar] [CrossRef]
- Ingles, J.; Sarina, T.; Yeates, L.; Hunt, L.; Macciocca, I.; McCormack, L.; Winship, I.; McGaughran, J.; Atherton, J.; Semsarian, C. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet. Med. 2013, 15, 972–977. [Google Scholar] [CrossRef]
- Hoffmann, B.; Schmidt-Traub, H.; Perrot, A.; Osterziel, K.J.; Gessner, R. First mutation in cardiac troponin C, L29Q, in a pa-tient with hypertrophic cardiomyopathy. Hum. Mutat. 2001, 17, 524. [Google Scholar] [CrossRef]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef]
- Osio, A.; Tan, L.; Chen, S.N.; Lombardi, R.; Nagueh, S.F.; Shete, S.; Roberts, R.; Willerson, J.T.; Marian, A.J. Myozenin 2 Is a Novel Gene for Human Hypertrophic Cardiomyopathy. Circ. Res. 2007, 100, 766–768. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.L.; Bagnall, R.D.; Ingles, J.; Yeates, L.; Kennerson, M.; Donald, J.A.; Jormakka, M.; Lind, J.M.; Semsarian, C. Mutations in Alpha-Actinin-2 Cause Hypertrophic Cardiomyopathy: A Genome-Wide Analysis. J. Am. Coll. Cardiol. 2010, 55, 1127–1135. [Google Scholar] [CrossRef]
- Arimura, T.; Bos, J.M.; Sato, A.; Kubo, T.; Okamoto, H.; Nishi, H.; Harada, H.; Koga, Y.; Moulik, M.; Doi, Y.L.; et al. Cardiac Ankyrin Repeat Protein Gene (ANKRD1) Mutations in Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Mas, R.; Gutiérrez-Fernández, A.; Gómez, J.; Coto, E.; Astudillo, A.; Puente, D.A.; Reguero, J.R.; Álvarez, V.; Morís, C.; León, D. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat. Commun. 2014, 29, 5326. [Google Scholar] [CrossRef] [PubMed]
- Román, I.S.; Navarro, M.; Martínez, F.; Albert, L.; Polo, L.; Guardiola, J.; García-Molina, E.; Esparza, C.M.; Lopez-Ayala, J.M.; Molina, M.S.; et al. Unclassifiable arrhythmic cardiomyopathy associated with Emery-Dreifuss caused by a mutation in FHL1. Clin. Genet. 2016, 90, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Fernandes, A.R. Genetics of hypertrophic cardiomyopathy: Advances and pitfalls in molecular diagnosis and therapy. Appl. Clin. Genet. 2014, 7, 195–208. [Google Scholar] [CrossRef][Green Version]
- Van Putten, M.; Young, C.; Berg, S.V.D.; Pronk, A.; Hulsker, M.; Karnaoukh, T.G.; Vermue, R.; van Dijk, K.W.; de Kimpe, S.; Aartsma-Rus, A. Preclinical Studies on Intestinal Administration of Antisense Oligonucleotides as a Model for Oral Delivery for Treatment of Duchenne Muscular Dystrophy. Mol. Ther. Nucleic. Acids 2014, 3, e211. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; McKenna, W.J.; Watkins, H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopa-thy. Circ. Res. 2011, 109, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Su, M.; Wang, S.; Zou, Y.; Wang, X.; Wang, Y.; Cui, H.; Zhao, P.; Hui, R.; Wang, J. MiR-451 is decreased in hyper-trophic cardiomyopathy and regulates autophagy by targeting TSC1. J. Cell. Mol. Med. 2014, 18, 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Yang, Z.; Abdellatif, M.; Sayed, D. GTPase Activating Protein (Sh3 Domain) Binding Protein 1 Regulates the Pro-cessing of MicroRNA-1 during Cardiac Hypertrophy. PLoS ONE 2015, 10, e0145112. [Google Scholar]
- Xu, X.-D.; Song, X.-W.; Li, Q.; Wang, G.-K.; Jing, Q.; Qin, Y.-W. Attenuation of MicroRNA-22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J. Cell. Physiol. 2011, 227, 1391–1398. [Google Scholar] [CrossRef]
- Guan, X.; Wang, L.; Liu, Z.; Guo, X.; Jiang, Y.; Lu, Y.; Peng, Y.; Liu, T.; Yang, B.; Shan, H.; et al. miR-106a pro-motes cardiac hypertrophy by targeting mitofusin 2. J. Mol. Cell. Cardiol. 2016, 99, 207–217. [Google Scholar] [CrossRef]
- Cam, F.S.; Güray, M. Hypertrophic cardiomyopathy: Pathological features and molecular pathogenesis. Anadolu Kardiyol. Dergisi/The Anatol. J. Cardiol. 2004, 4, 327–330. [Google Scholar]
- Li, A.-L.; Lv, J.-B.; Gao, L. MiR-181a mediates Ang II-induced myocardial hypertrophy by mediating autophagy. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5462–5470. [Google Scholar]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. A Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving concepts in dilated cardiomyopathy. Eur. J. Hear. Fail. 2017, 20, 228–239. [Google Scholar] [CrossRef]
- Porcari, A.; De Angelis, G.; Romani, S.; Paldino, A.; Artico, J.; Cannatà, A.; Gentile, P.; Pinamonti, B.; Merlo, M.; Sinagra, G. Current diagnostic strategies for dilated cardiomyopathy: A comparison of imaging techniques. Expert Rev. Cardiovasc. Ther. 2018, 17, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Kramer, C.M. Role of Cardiac Magnetic Resonance intheDiagnosis and Prognosis ofNonischemicCardiomyopa-thy. JACC Cardiovasc. Imaging 2017, 10 Pt A, 1180–1193. [Google Scholar] [CrossRef]
- Dellefave, L.; McNally, E.M. The genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 2010, 25, 198–204. [Google Scholar] [CrossRef]
- Cho, K.W.; Lee, J.; Kim, A.Y. Genetic Variations Leading to Familial Dilated Cardiomyopathy. Mol. Cells 2016, 39, 722–727. [Google Scholar] [CrossRef]
- Naga Prasad, S.V.; Karnik, S.S. MicroRNAs--regulators of signaling networks in dilated cardiomyopathy. J. Cardiovasc. Transl. Res. 2010, 3, 225–234. [Google Scholar] [CrossRef]
- Miyamoto, S.D.; Karimpour-Fard, A.; Peterson, V.; Auerbach, S.R.; Stenmark, K.R.; Stauffer, B.; Sucharov, C.C. Circulating microRNA as a biomarker for recovery in pediatric dilated cardiomyopathy. J. Heart Lung Transplant. 2015, 34, 724–733. [Google Scholar] [CrossRef]
- Tao, L.; Yang, L.; Huang, X.; Hua, F.; Yang, X. Reconstruction and Analysis of the lncRNA-miRNA-mRNA Network Based on Competitive Endogenous RNA Reveal Functional lncRNAs in Dilated Cardiomyopathy. Front Genet. 2019, 10, 1149. [Google Scholar] [CrossRef]
- Chen, J.-F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Meyners, W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol. 2004, 97, 499–501. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Danieli, G.A.; Buja, G.; Daliento, L.; Fasoli, G.; Scognamiglio, R.; Corrado, D.; Thlene, G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23–q24. Hum. Mol. Genet. 1994, 3, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Pavei, A.; Michieli, P.; Schiavon, M.; Thiene, G. Trends in Sudden Cardiovascular Death in Young Competitive Athletes After Implementation of a Preparticipation Screening Program. JAMA 2006, 296, 1593–1601. [Google Scholar] [CrossRef]
- Thiene, G.; Corrado, D.; Basso, C. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet J. Rare Dis. 2007, 2, 45. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; McKenna, W.J. Reconciling the Protean Manifestations of Arrhythmogenic Cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2010, 3, 566–570. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young peo-ple. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Saguner, A.M.; Brunckhorst, C.; Duru, F. Arrhythmogenic ventricular cardiomyopathy: A paradigm shift from right to biventricular disease. World J. Cardiol. 2014, 6. [Google Scholar] [CrossRef]
- Hoorntje, E.T.; Te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmo-genic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ven-tricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Akdis, D.; Saguner, A.M.; Shah, K.; Wei, C.; Medeiros-Domingo, A.; von Eckardstein, A.; Lüscher, T.F.; Brunckhorst, C.; Chen, H.S.V.; Duru, F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: From a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur. Heart J. 2017, 38, 1498–1508. [Google Scholar] [CrossRef]
- Te Riele, A.S.J.M.; James, C.A.; Sawant, A.C.; Bhonsale, A.; Groeneweg, J.A.; Mast, T.P.; Murray, B.; Tichnell, C.; Dooijes, D.; van Tintelen, J.P.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in the Pediatric Population: Clinical Characterization and Comparison with Adult-Onset Disease. JACC Clin. Electrophysiol. 2015, 1, 551–560. [Google Scholar] [CrossRef]
- Gutiérrez, S.L.C.; Kamel, I.R.; Zimmerman, S.L. Current Concepts on Diagnosis and Prognosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. J. Thorac. Imaging 2016, 31, 324–335. [Google Scholar] [CrossRef]
- Pilichou, K.; Thiene, G.; Bauce, B.; Rigato, I.; Lazzarini, E.; Migliore, F.; Perazzolo Marra, M.; Rizzo, S.; Zorzi, A.; Daliento, L.; et al. Arrhythmogenic cardiomyopathy. Orphanet J. Rare Dis. 2016, 11, 33. [Google Scholar] [CrossRef]
- McNally, E.; MacLeod, H.; Dellefave-Castillo, L. Arrhythmogenic Right Ventricular Cardiomyopathy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1131/ (accessed on 17 October 2021).
- Protonotarios, N.; Tsatsopoulou, A. Naxos disease and Carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 2004, 13, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Vite, A.; Radice, G.L. N-Cadherin/Catenin Complex as a Master Regulator of Intercalated Disc Function. Cell Commun. Adhes. 2014, 21, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Vermij, S.H.; Abriel, H.; Van Veen, T.A.B. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef]
- Coonar, A.S.; Protonotarios, N.; Tsatsopoulou, A.; Needham, E.W.; Houlston, R.; Cliff, S.; Otter, M.I.; Murday, V.A.; Mattu, R.K.; McKenna, W.J. Gene for Arrhythmogenic Right Ventricular Cardiomyopathy with Diffuse Nonepidermolytic Palmoplantar Keratoderma and Woolly Hair (Naxos Disease) Maps to 17q21. Circulation 1998, 97, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Huerta, L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J. Am. Acad. Dermatol. 1998, 39, 418–421. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes di-lated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [PubMed]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in ar-rhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Heuser, A.; Plovie, E.R.; Ellinor, P.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant Desmocollin-2 Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Haan, A.D.D.; Tan, B.Y.; Zikusoka, M.N.; Lladó, L.I.; Jain, R.; Daly, A.; Tichnell, C.; James, C.; Amat-Alarcon, N.; Abraham, T.; et al. Comprehensive Desmosome Mutation Analysis in North Americans with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 428–435. [Google Scholar] [CrossRef]
- Fressart, V.; Duthoit, G.; Donal, E.; Probst, V.; Deharo, J.-C.; Chevalier, P.; Klug, D.; Dubourg, O.; Delacretaz, E.; Cosnay, P.; et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. Europace 2010, 12, 861–868. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed Modification of the Task Force Criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef]
- Bs, K.L.T.; Bs, D.J.T.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicatesCDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein αT-catenin are associated with ar-rhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Cao, Q.; Zhou, Q.; Xie, J.; Shen, Y.; Wan, R.; Yu, J.; Yan, S.; Marian, A.J.; Hong, K. Arrhythmogenic cardiomyopa-thy in a patient with a rare loss-of-function KCNQ1 mutation. J. Am. Heart Assoc. 2015, 4, e001526. [Google Scholar] [CrossRef]
- Forleo, C.; Carmosino, M.; Resta, N.; Rampazzo, A.; Valecce, R.; Sorrentino, S.; Iacoviello, M.; Pisani, F.; Procino, G.; Gerbino, A.; et al. Clinical and Functional Characterization of a Novel Mutation in Lamin A/C Gene in a Multigenerational Family with Arrhythmogenic Cardiac Laminopathy. PLoS ONE 2015, 10, e0121723. [Google Scholar] [CrossRef]
- Pilichou, K.; Remme, C.A.; Basso, C.; Campian, M.E.; Rizzo, S.; Barnett, P.; Scicluna, B.P.; Bauce, B.; van den Hoff, M.J.; de Bakker, J.M.; et al. Myocyte necrosis un-derlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 2009, 206, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.-F.; Haneline, L.S.; Field, L.J.; et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pilichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Multidiscipli-nary Study of Right Ventricular Dysplasia Investigators. Compound and digenic heterozygosity contributes to arrhythmo-genic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef]
- König, E.; Volpato, C.B.; Motta, B.M.; Blankenburg, H.; Picard, A.; Pramstaller, P.; Casella, M.; Rauhe, W.; Pompilio, G.; Meraviglia, V.; et al. Exploring digenic inheritance in arrhythmogenic cardiomyopa-thy. BMC Med. Genet. 2017, 18, 145. [Google Scholar] [CrossRef]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Kirschner, M.B.; van Zandwijk, N. Circulating microRNAs: Association with disease and potential use as bi-omarkers. Crit. Rev. Oncol. Hematol. 2011, 80, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A. The Hippo Pathway Is Activated and Is a Causal Mechanism for Adipogenesis in Arrhythmogenic Cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2016, 16, 167–179. [Google Scholar] [CrossRef]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Investig. 2017, 127, 761–771. [Google Scholar] [CrossRef]
- Dangwal, S.; Thum, T. microRNA Therapeutics in Cardiovascular Disease Models. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 185–203. [Google Scholar] [CrossRef]
- Van Rooij, E.; Olson, E.N. MicroRNA therapeutics for cardiovascular disease: Opportunities and obstacles. Nat. Rev. Drug Discov. 2012, 11, 860–872. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Webber, M.; Anderson, D.G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Control. Release 2016, 240, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Gurda, B.L.; Lataillade, A.D.G.D.; Bell, P.; Zhu, Y.; Yu, H.; Wang, P.; Bagel, J.; Vite, C.H.; Sikora, T.; Hinderer, C.; et al. Evaluation of AAV-mediated Gene Therapy for Central Nervous System Disease in Canine Mucopolysaccharidosis VII. Mol. Ther. 2016, 24, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, J.; Ramanujam, D.P.; Sassi, Y.; Ahles, A.; Jentzsch, C.; Werfel, S.; Leierseder, S.; Loyer, X.; Giacca, M.; Zentilin, L.; et al. MiR-378 Controls Cardiac Hypertrophy by Combined Repression of Mitogen-Activated Protein Kinase Pathway Factors. Circulation 2013, 127, 2097–2106. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Crippa, S.; Montecchiani, C.; Camps, J.; Cornaglia, A.I.; Boldrin, L.; Morgan, J.; Calligaro, A.; Casasco, A.; Orlacchio, A.; et al. Long-Term miR-669a Therapy Alleviates Chronic Dilated Cardiomyopathy in Dystrophic Mice. J. Am. Heart Assoc. 2013, 2, e000284. [Google Scholar] [CrossRef]
- Kwekkeboom, R.F.; Sluijter, J.P.; van Middelaar, B.J.; Metz, C.H.; Brans, M.A.; Kamp, O.; Paulus, W.J.; Musters, R.J. In-creased local delivery of antagomir therapeutics to the rodent myocardium using ultrasound and microbubbles. J. Control Release 2016, 222, 18–31. [Google Scholar] [CrossRef] [PubMed]
- De Cock, I.; Zagato, E.; Braeckmans, K.; Luan, Y.; de Jong, N.; De Smedt, S.; Lentacker, I. Ultrasound and microbubble mediated drug delivery: Acoustic pressure as determinant for uptake via membrane pores or endocytosis. J. Control. Release 2015, 197, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Kajander, S.; Joutsiniemi, E.; Saraste, M.; Pietilä, M.; Ukkonen, H.; Saraste, A.; Sipilä, H.T.; Teräs, M.; Mäki, M.; Airaksinen, J.; et al. Cardiac positron emission tomography/computed tomography imaging accurately detects anatomi-cally and functionally significant coronary artery disease. Circulation 2010, 122, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Hassinen, I.; Kivelä, A.; Hedman, A.; Saraste, A.; Knuuti, J.; Hartikainen, J.; Ylä-Herttuala, S. Intramyocardial Gene Therapy Directed to Hibernating Heart Muscle Using a Combination of Electromechanical Mapping and Positron Emission Tomogra-phy. Hum. Gene Ther. 2016, 27, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Philippen, L.E.; Dirkx, E.; Wit, J.B.; Burggraaf, K.; de Windt, L.J.; da Costa Martins, P.A. Antisense MicroRNA Therapeutics in Cardiovascular Disease: Quo Vadis? Mol. Ther. 2015, 23, 1810–1818. [Google Scholar] [CrossRef]
- Anand, S.; Majeti, B.K.; Acevedo, L.M.; Murphy, E.A.; Mukthavaram, R.; Scheppke, L.; Huang, M.; Shields, D.J.; Lindquist, J.N.; Lapinski, P.E.; et al. MicroRNA-132-mediated loss of p120RasGAP activates the endo-thelium to facilitate pathological angiogenesis. Nat. Med. 2010, 16, 909–914. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ncRNA Class | Function | References |
|---|---|---|
| Micro RNA (miRNA) | Fundamental role in gene regulation at the post-transcriptional level. They act either by cleaving the target mRNA or by inhibiting translation and therefore protein synthesis | [21] |
| long non-coding RNAs (lncRNAs) | They have various inhibitory functions by acting on transcription regulatory proteins (histone modifying enzymes and chromatin remodeling factors), mRNA and miRNA | [34,35] |
| circular RNAs (circRNAs) | Their function is not yet well known, however some experiments have shown that some of them can bind to specific proteins or to miRNAs blocking their functions. | [36,37] |
| Small nucleolar RNA (snoRNA) | Essential to drive nucleotide modifications and processing. | [38] |
| MiRNA | Regulation | Cardiomyopathy | Reference | Gene Target |
|---|---|---|---|---|
| miR-590-5p | Upregulated | Hypertrophic | [80] | TRIM, JPH1, POM121C |
| miR-92a | Upregulated | Hypertrophic | [80] | STAT, SUMO2, TBC1D1 |
| miR-483-5p | Upregulated | Hypertrophic | [81] | APOL, DLL4, FHL2 |
| miR-29a | Upregulated | Hypertrophic | [82] | PRTEN, AKT, NFAT, GSK3B, Elastin |
| miR-133 | Downregulated | Hypertrophic | [83] | CTGF, SERCA2a, NFATC4, MYH, SERCA |
| miR-155 | Downregulated | Hypertrophic | [84] | SOCS1, MEF2A, JARID2 AT1R |
| miR-1 | Downregulated | Hypertrophic | [85] | BCL1, CBX6, CCND1, CREB |
| miR-204 | Upregulated | Hypertrophic | [86] | ATXN1, CAPRIN1, CREB, OGT … |
| miR-139-5p | Downregulated | Hypertrophic | [87] | c-JUN, SRSF, Est-1, MEIS1, ZFX |
| miR-20 | Upregulated | Hypertrophic | [88] | STAT3, ATF2, DVL3 |
| miR-3135b | Upregulated | Dilated | [89] | FLNC, PRX, RBL1 |
| miR-3908 | Upregulated | Dilated | [89] | ADD2, FRMD4B, PDE11A |
| miR-5571-5p | Upregulated | Dilated | [89] | PPP2R2B, BMP7, TCF21, PSEN1 |
| miR-148a | Downregulated | Dilated | [90] | gp130, AKT,ITPR2 |
| miR-185 | Upregulated | Dilated | [91] | TFPI, Ctgf, ARHGEF, CAMK2 |
| miR-1251 | Upregulated | Arrhythmogenic | [92,93] | TMEM, ANK1, PROX1 |
| miR-21-3p, miR-21-5p | Upregulated | Arrhythmogenic | [92,93] | PITX2, CADM1, PVRL3, SLMAP |
| miR-212-3p | Upregulated | Arrhythmogenic | [92,93] | PLXNA2, PRDM16, TCF, PKP4 |
| miR-34a-5p | Upregulated | Arrhythmogenic | [92,93] | HCN, JPH, PKP2 |
| miR-135b | Upregulated | Arrhythmogenic | [92,93] | ERBB, FOXO1,TMEM, SCN5A |
| miR-138-5p | Downregulated | Arrhythmogenic | [92,93] | WNT9A, BMPR, AKAP11 |
| miR-193-3p | Downregulated | Arrhythmogenic | [92,93] | ALOX5, SOX2, L-MYC, KLF4 |
| miR-302 | Downregulated | Arrhythmogenic | [92,93] | FMR1, CAMTA1 |
| miR-491-3p | Downregulated | Arrhythmogenic | [92,93] | WNT, BMPR2, TGFBR2 |
| miR-575 | Downregulated | Arrhythmogenic | [92,93] | EPB41L5, HCN1, HCN4 |
| miR-4254 | Downregulated | Arrhythmogenic | [92,93] | CDR1AS, COL4A, HSPB7 |
| miR-4643 | Downregulated | Arrhythmogenic | [92,93] | RBM20, RAC1, VCAM1, PTPRC |
| miR-320a | Downregulated | Arrhythmogenic | [94] | CDH2, CTNNA3, DSC2 |
| miR-144-3p | Upregulated | Arrhythmogenic | [95] | CTNNA3, AREG, PROS1 |
| miR-145-5p | Upregulated | Arrhythmogenic | [95] | CDH2, DAG1, CITED2, TLL1, PAK7 |
| miR-185-5p | Upregulated | Arrhythmogenic | [95] | DLG2, NOX5,PRRT2 |
| miR-494 | Upregulated | Arrhythmogenic | [95,96] | PTEN, ROCK1, CaMKIIδ, FGFR2, LIF |
| miRNA Therapeutic Approach | Advantage | Limitation |
|---|---|---|
| miRNA mimics | Promote the expression of miRNAs | Low efficiency in the heart and vascular system; Can cause miRNA to over-act, potentially causing serious side effects; Easily degraded by nucleases; The chemistry of the construct is toxic; |
| miRNA inhibitors | Block the activity of miRNAs Directly bind the target of the miRNA sequence | Low efficiency in the heart and vascular system; Low target binding affinity; Unwanted genetic changes or off-target effects; Easily degraded by nucleases Difficult to create and to keep stable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amodio, F.; Caiazza, M.; Fimiani, F.; Calabrò, P.; Limongelli, G. MicroRNAs: From Junk RNA to Life Regulators and Their Role in Cardiovascular Disease. Cardiogenetics 2021, 11, 230-254. https://doi.org/10.3390/cardiogenetics11040023
Amodio F, Caiazza M, Fimiani F, Calabrò P, Limongelli G. MicroRNAs: From Junk RNA to Life Regulators and Their Role in Cardiovascular Disease. Cardiogenetics. 2021; 11(4):230-254. https://doi.org/10.3390/cardiogenetics11040023
Chicago/Turabian StyleAmodio, Federica, Martina Caiazza, Fabio Fimiani, Paolo Calabrò, and Giuseppe Limongelli. 2021. "MicroRNAs: From Junk RNA to Life Regulators and Their Role in Cardiovascular Disease" Cardiogenetics 11, no. 4: 230-254. https://doi.org/10.3390/cardiogenetics11040023
APA StyleAmodio, F., Caiazza, M., Fimiani, F., Calabrò, P., & Limongelli, G. (2021). MicroRNAs: From Junk RNA to Life Regulators and Their Role in Cardiovascular Disease. Cardiogenetics, 11(4), 230-254. https://doi.org/10.3390/cardiogenetics11040023