
A Novel Human Biospecimen Repository for Clinical and Molecular Investigation of Thoracic Aortopathy


Abstract
:1. Introduction
2. Methods
2.1. Eligibility and Enrollment Processes
- Diagnosis of aortic disease including TAA or dissection, aortic tortuosity, or aortic hypoplasia/stenosis.
- Diagnosis of a syndrome or genetic abnormality that poses risk for the development of aortopathy.
- Diagnosis of aortic valve disease.
- Family members of eligible subjects.
- Control subjects having aortic tissue removed during a surgical procedure, such as during heart transplantation.
- Organ donors who have authorized the use of their specimens for research.
2.2. Clinical and Family Data Collection
2.2.1. Clinical Data
2.2.2. Family Data
2.3. Cardiac Imaging Data
2.3.1. CT Scans
2.3.2. Echocardiograms
2.4. Blood Sample Collection
Special Considerations
2.5. Tissue Sample Collection
2.5.1. Formalin Fixation
2.5.2. Glutaraldehyde Fixation
2.5.3. Primary Aortic Cell Culture
2.5.4. Tissue Specimen Freezing
2.5.5. Aortic Valve Tissue
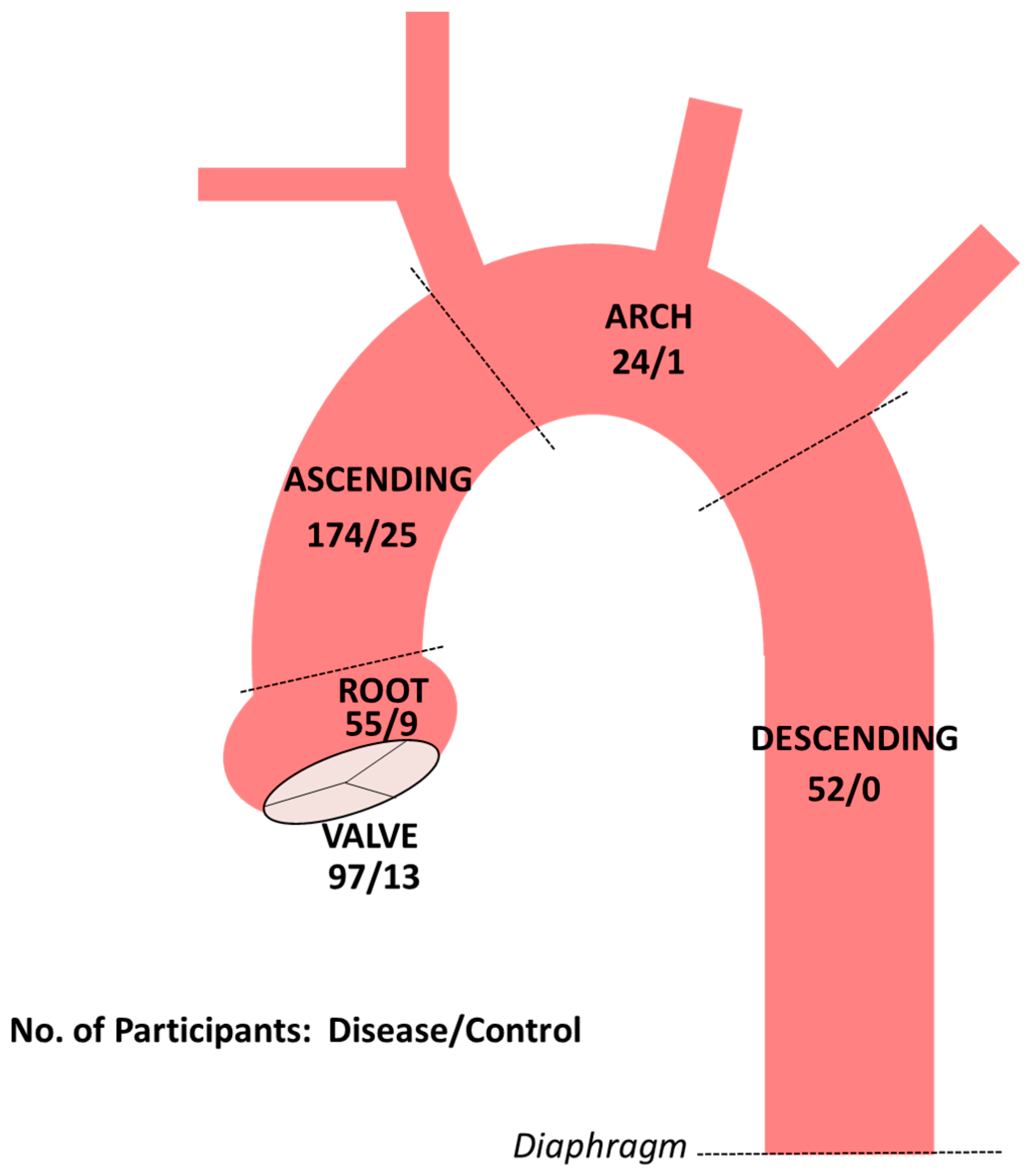
3. Results
4. Discussion
4.1. Incomplete Understanding of Genetic Causes of Aortopathy and Aortic Valve Disease
4.2. Undiscovered Mechanisms of Pathogenesis
4.3. The Clinical Challenge of Risk Classification
4.4. Aortic SMCs: Cellular Disease Model and Substrate for Experimentation
4.5. Opportunities to Build upon the Foundation of CHAR
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Isselbacher, E.M.; Cardenas, C.L.L.; Lindsay, M.E. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.-Q.; Guo, D.-C.; Prakash, S.K.; McDonald, M.-L.N.; Johnson, R.J.; Wang, M.; Regalado, E.S.; Russell, L.; Cao, J.-M.; Kwartler, C.; et al. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011, 7, e1002118. [Google Scholar] [CrossRef]
- Parrott, A.; James, J.; Goldenberg, P.; Hinton, R.B.; Miller, E.; Shikany, A.; Aylsworth, A.S.; Kaiser-Rogers, K.; Ferns, S.; Lalani, S.R.; et al. Aortopathy in the 7q11.23 microduplication syndrome. Am. J. Med. Genet. A 2015, 167, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Creamer, T.J.; Bramel, E.E.; MacFarlane, E.G. Insights on the pathogenesis of Aneurysm through the study of hereditary aortopathies. Genes 2021, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2008, 42, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Van de Laar, I.M.; Van der Linde, D.; Oei, E.H.; Bos, P.K.; Bessems, J.H.; Bierma-Zeinstra, S.M.; Van Meer, B.L.; Pals, G.; Oldenburg, R.A.; Bekkers, J.A.; et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet. 2012, 49, 47–57. [Google Scholar] [CrossRef]
- MacCarrick, G.; Black, J.H., III; Bowdin, S.; El-Hamamsy, I.; Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Sponseller, P.D.; Loeys, B.; Dietz, H.C., III. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet. Med. 2014, 16, 576–587. [Google Scholar] [CrossRef]
- Meester, J.A.N.; Verstraeten, A.; Schepers, D.; Alaerts, M.; Van Laer, L.; Loeys, B.L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann. Cardiothorac. Surg. 2017, 6, 582–594. [Google Scholar] [CrossRef] [Green Version]
- Meraviglia, V.; Zanon, A.; Lavdas, A.; Schwienbacher, C.; Silipigni, R.; Di Segni, M.; Chen, H.-S.V.; Pramstaller, P.P.; Hicks, A.; Rossini, A. Generation of induced pluripotent stem cells from frozen buffy coats using non-integrating episomal plasmids. J. Vis. Exp. 2015, 100, e52885. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Coutinho, T.; Chu, M.W.; Ouzounian, M. Sex differences in thoracic aortic disease: A review of the literature and a call to action. J. Thorac. Cardiovasc. Surg. 2020, 160, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Andelfinger, G.U.; Arnaud, P.; Boileau, C.; Callewaert, B.L.; et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E., Jr.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, e266–e369. [Google Scholar]
- Landis, B.J.; Ware, S.M.; James, J.; Shikany, A.R.; Martin, L.J.; Hinton, R.B. Clinical stratification of pediatric patients with idiopathic thoracic aortic aneurysm. J. Pediatr. 2015, 167, 131–137.e5. [Google Scholar] [CrossRef]
- Bravo-Jaimes, K.; Prakash, S.K. Genetics in bicuspid aortic valve disease: Where are we? Prog. Cardiovasc. Dis. 2020, 63, 398–406. [Google Scholar] [CrossRef]
- Schubert, J.A.; Landis, B.J.; Shikany, A.R.; Hinton, R.B.; Ware, S.M. Clinically relevant variants identified in thoracic aortic aneurysm patients by research exome sequencing. Am. J. Med Genet. A 2016, 170, 1288–1294. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Beil, A.; Hornsby, W.; Uhlmann, W.R.; Aatre, R.; Arscott, P.; Wolford, B.; Eagle, K.A.; Yang, B.; McNamara, J.; Willer, C.; et al. Disclosure of clinically actionable genetic variants to thoracic aortic dissection biobank participants. BMC Med. Genom. 2021, 14, 66. [Google Scholar] [CrossRef] [PubMed]
- Habashi, J.P.; Judge, D.; Holm, T.M.; Cohn, R.D.; Loeys, B.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Tierney, E.S.S.; Levine, J.C.; Atz, A.A.; et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef] [Green Version]
- Bowman, M.A.H.; Eagle, K.A.; Milewicz, D.M. Update on clinical trials of losartan with and without β-blockers to block aneurysm growth in patients with Marfan syndrome: A review. JAMA Cardiol. 2019, 4, 702–707. [Google Scholar] [CrossRef]
- Lee, V.S.; Halabi, C.M.; Hoffman, E.P.; Carmichael, N.; Leshchiner, I.; Lian, C.G.; Bierhals, A.J.; Vuzman, D.; Mecham, R.P.; Frank, N.Y.; et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc. Natl. Acad. Sci. USA 2016, 113, 8759–8764. [Google Scholar] [CrossRef] [Green Version]
- Halushka, M.K.; Angelini, A.; Bartoloni, G.; Basso, C.; Batoroeva, L.; Bruneval, P.; Buja, L.M.; Butany, J.; D’Amati, G.; Fallon, J.T.; et al. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association for European Cardiovascular Pathology: II. Noninflammatory degenerative diseases-nomenclature and diagnostic criteria. Cardiovasc. Pathol. 2016, 25, 247–257. [Google Scholar] [CrossRef]
- MacFarlane, E.G.; Parker, S.J.; Shin, J.Y.; Ziegler, S.G.; Creamer, T.J.; Bagirzadeh, R.; Bedja, D.; Chen, Y.; Calderon, J.F.; Weissler, K.; et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J. Clin. Investig. 2019, 129, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Rateri, D.L.; Moorleghen, J.J.; Majesky, M.W.; Daugherty, A. Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta—Brief report. Arter. Thromb. Vasc. Biol. 2017, 37, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- Landis, B.J.; Schubert, J.A.; Lai, N.; Jegga, A.G.; Shikany, A.R.; Foroud, T.; Ware, S.M.; Hinton, R.B. Exome sequencing identifies candidate genetic modifiers of syndromic and familial thoracic aortic aneurysm severity. J. Cardiovasc. Transl. Res. 2017, 10, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.P.; Yang, Z.; Cordes, T.M.; Markham, L.W.; Landis, B.J. Characterization of the rate of aortic dilation in young patients with thoracic aortic aneurysm. Pediatr. Cardiol. 2021, 42, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Aubart, M.; Gazal, S.; Arnaud, P.; Benarroch, L.; Gross, M.-S.; Buratti, J.; Boland, A.; Meyer, V.; Zouali, H.; Hanna, N.; et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur. J. Hum. Genet. 2018, 26, 1759–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, A.; Bancone, C.; Cobellis, G.; Buonocore, M.; Santarpino, G.; Fischlein, T.J.M.; Cipollaro, M.; De Feo, M.; Della Corte, A. A possible early biomarker for bicuspid aortopathy: Circulating transforming growth factor β-1 to soluble endoglin ratio. Circ. Res. 2017, 120, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
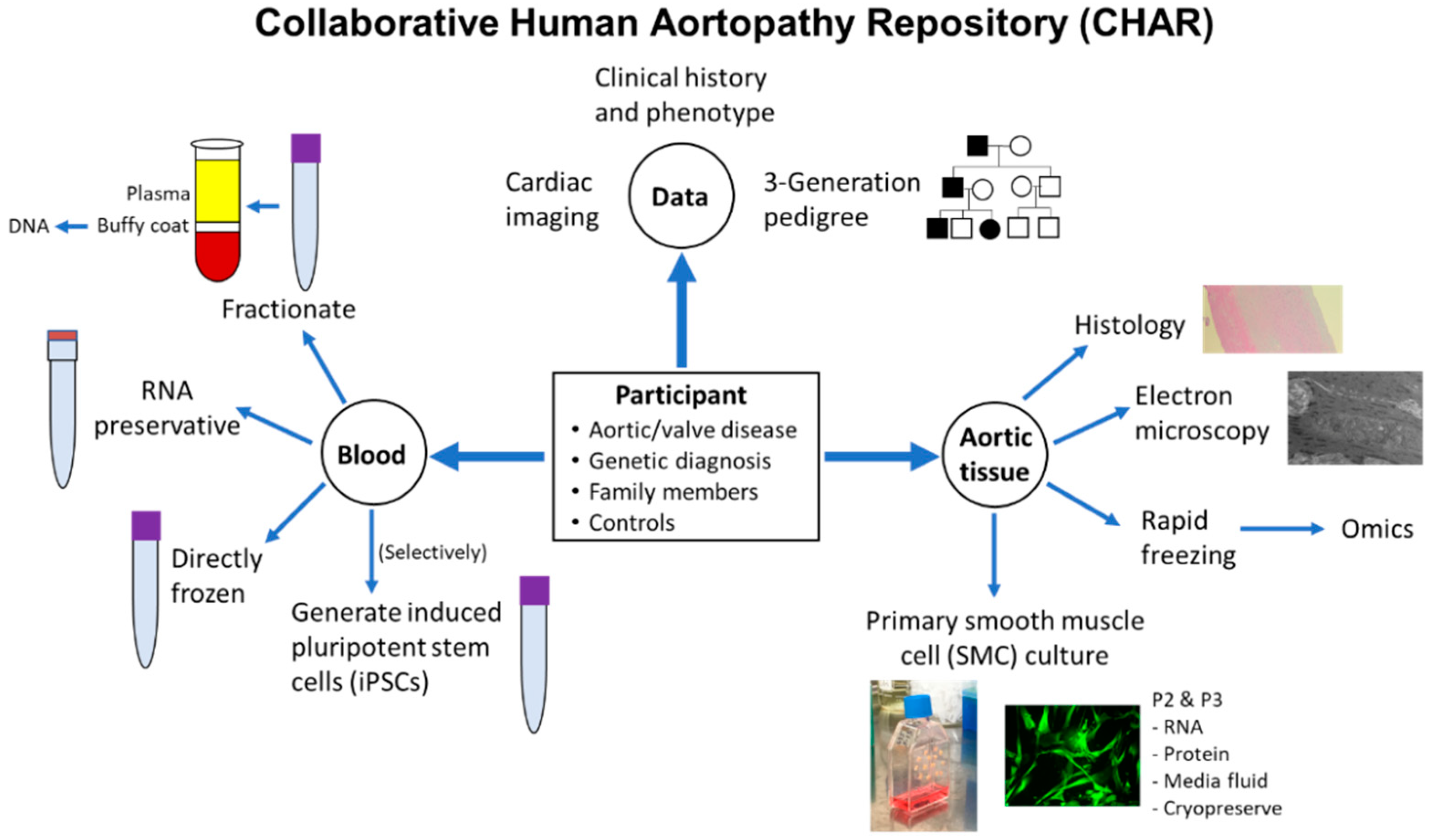

{kind=link}
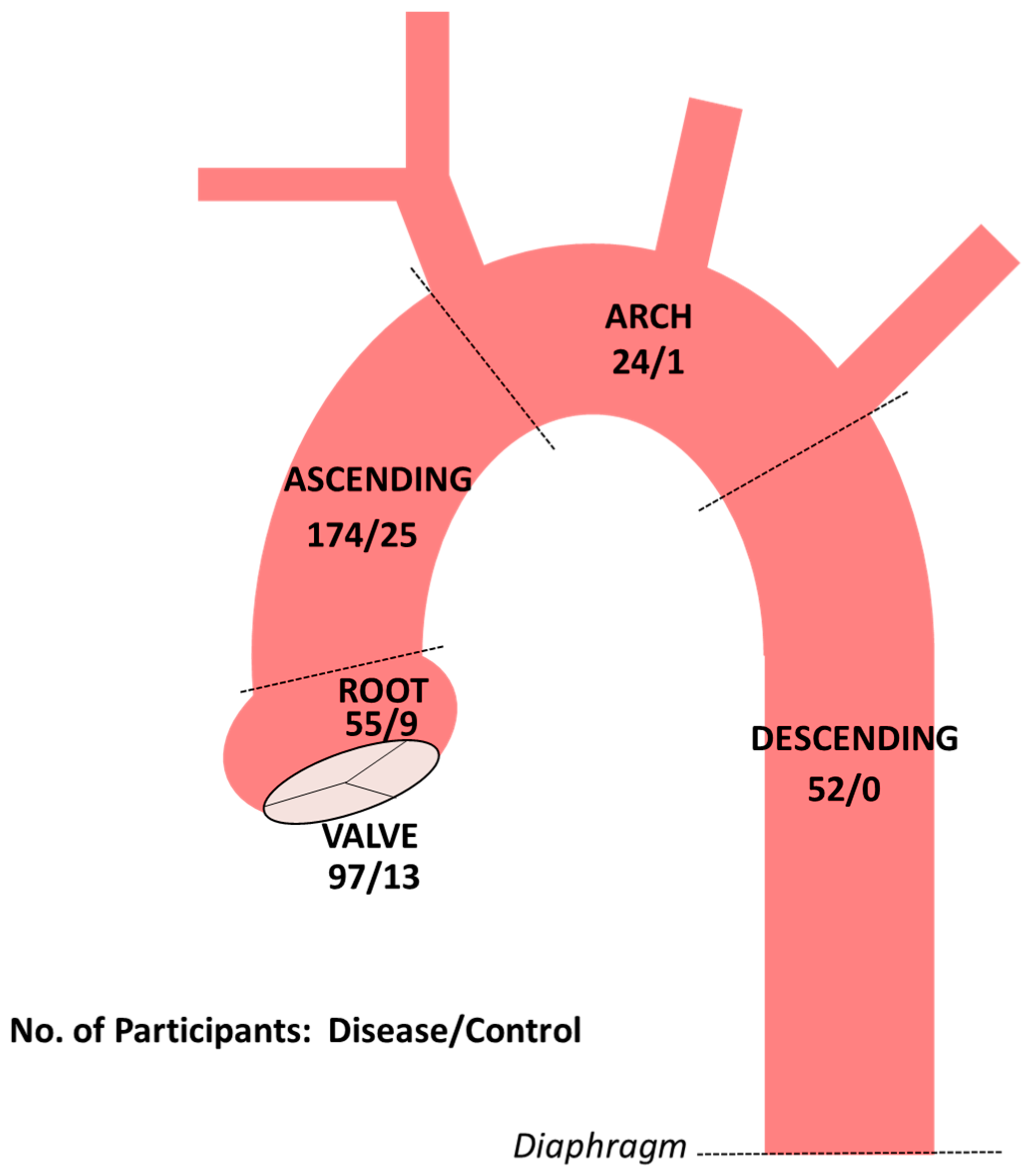{kind=link}
| Data Category | Components |
|---|---|
| Clinical data | Demographics Anthropometrics Medical history Surgical history Medication history Genetic evaluations Cardiac imaging performed |
| Family history | Three-generation pedigree (scripted) |
| Cardiac imaging | CT scans: Segmental aortic diameters; description of cardiovascular and non-cardiovascular findings Echocardiograms: Proximal aortic diameters; aortic valve function Cardiac MRI/MRA: Segmental aortic diameters |
| Biospecimen collection and processing | Blood Aortic/Valve Tissue |
| Category | Diagnosis or Event (Y/N) | Details |
|---|---|---|
| Aortic | Thoracic aortic aneurysm | Location |
| Thoracic aortic dissection | Date Stanford type (A vs. B) | |
| Thoracic aortic rupture | Date | |
| Abdominal aortic aneurysm | ||
| Abdominal aortic dissection | Date | |
| Abdominal aortic rupture | Date | |
| Arterial | Brain aneurysm | Specify artery |
| Aneurysm of other artery | Specify artery Diameter | |
| Dissection of other artery | Specify artery Date | |
| Rupture of other artery | Specify artery Date | |
| Valvar | Aortic valve disease | BAV (Y/N) Aortic stenosis (Y/N) Aortic regurgitation (Y/N) |
| Mitral valve prolapse | ||
| Cardiovascular risk factors | Hypertension | Age of diagnosis Medical therapy (Y/N) |
| High cholesterol | Age of diagnosis Medical therapy (Y/N) | |
| Diabetes | Type 1 vs. type 2 Age of diagnosis Medical therapy | |
| Cardiovascular disease | Stroke | Ischemic versus hemorrhagic Date |
| Coronary artery disease | Myocardial infarction (Y/N) | |
| Venous thrombosis | Deep venous thrombosis | Specify vein Date |
| Pulmonary embolism | Date | |
| Heart rhythm | Arrhythmia | Type Date |
| Syncope | Total number of lifetime episodes | |
| Cardiac arrest | Date | |
| Congenital | Congenital heart disease (excluding BAV) | Specify |
| Other | Specify | |
| Procedures | Aortic replacement | Date Hospital Segments that were replaced Dissection present (Y/N) |
| Aortic repair | Date Hospital Segments that were repaired Dissection present (Y/N) | |
| Endovascular repair | Date Hospital Segments that were repaired | |
| Aortic valve replacement | Date Hospital Simultaneous aortic replacement (Y/N) | |
| Mitral valve replacement | Date Hospital | |
| Mitral valve repair | Date Hospital | |
| Coronary artery bypass graft | Date Hospital | |
| Coronary artery stent or balloon angioplasty | Date Hospital | |
| Pacemaker or implantable cardioverter/defibrillator | Date Hospital | |
| Other | Specify |
| Category | Diagnosis (Y/N) | Details |
|---|---|---|
| Skeletal | Pectus excavatum | Surgery required (Y/N) |
| Pectus carinatum | Surgery required (Y/N) | |
| Joint hyperflexibility | ||
| Scoliosis | Age of diagnosis X-ray evaluation (Y/N) Surgery required (Y/N) | |
| Club foot | Right/left/bilateral Surgery required (Y/N) | |
| Joint pain | Specify joint | |
| Arthritis | Specify joint Surgery required (Y/N) | |
| Orthotics required before age 30 | ||
| Bone fracture | Specify bone Age when occurred | |
| Osteoporosis | ||
| Ocular | Lens dislocation or subluxation | Right/left/bilateral Age of diagnosis Surgery required (Y/N) |
| Vision correction (glasses or contact lens) | Age first prescribed Prescription strength Reason (e.g., near- or far-sighted) | |
| Retinal detachment | Right/left/bilateral Age of diagnosis | |
| Glaucoma | Right/left/bilateral Age of diagnosis | |
| Cataract | Right/left/bilateral Age of diagnosis | |
| Macular degeneration | Right/left/bilateral Age of diagnosis | |
| Craniofacial | Orthodontia | |
| Palate expander | ||
| Tooth extraction for dental crowding | ||
| Cleft palate | ||
| Skin | Hyperextensible | |
| Wide atrophic scar | ||
| Striae | Specify body location(s) | |
| Easy bruising | ||
| Abdominal | Hernia | Location Surgery required (Y/N) Recurrence after surgery (Y/N) |
| Eosinophilic esophagitis | Age of diagnosis | |
| Bowel rupture | Age of diagnosis | |
| Microvascular | Frequent nose bleeds | |
| Immune | Kawasaki disease | Age of diagnosis Coronary artery dilation (Y/N) |
| Vasculitis | Specify diagnosis Age of diagnosis | |
| Development | Physical therapy in childhood | |
| Learning disability | ||
| ADHD | ||
| Speech delay | ||
| Neurologic | Seizure | Age Medical therapy (Y/N) |
| Chronic headache | Migraine type (Y/N) Age when started | |
| Pulmonary | Pneumothorax | Age Number of occurrences Spontaneous versus post-operative |
| Emphysema | Age of diagnosis | |
| Asthma | Age of diagnosis Severity | |
| Other medical diagnoses | Specify | |
| Other prior surgeries | Specify | |
| Obstetrical | Pregnancy | Number Number of births Number of spontaneous fetal losses |
| Uterine rupture | ||
| Lifestyle/social | Smoke cigarettes | Number of years Number of packs per day |
| Other tobacco products | Specify type | |
| Regular heavy weightlifting for exercise | Number of years | |
| Regular heavy weightlifting for non-exercise purpose (e.g., occupational) | Number of years |
| Characteristic | Children and Adolescents (Age < 21 Years), n = 156 | Adults, n = 678 |
|---|---|---|
| Age at enrollment (years), mean ± SD | 10 ± 6 | 56 ± 14 |
| Sex, n (%) | ||
| Male | 99 (63) | 481 (71) |
| Female | 57 (37) | 197 (29) |
| Race, n (%) | ||
| White | 145 (93) | 630 (93) |
| Black or African American | 6 (4) | 39 (6) |
| Asian | 4 (2.4) | 7 (1) |
| Not available | 1 (0.6) | 2 (0.3) |
| Ethnicity, n (%) | ||
| Non-Hispanic | 152 (97) | 669 (99) |
| Hispanic | 4 (3) | 7 (1) |
| Not available | 0 | 2 (0.3) |
| Syndrome diagnosis, n (%) | ||
| Marfan, n (%) | 47 (30) | 45 (7) |
| Loeys–Dietz, n (%) | 12 (8) | 7 (1) |
| Vascular-type Ehlers–Danlos, n (%) | 7 (4.5) | 5 (0.7) |
| Turner, n (%) | 4 (3) | 2 (0.3) |
| Thoracic aortic disease, n (%) | ||
| Thoracic aortic aneurysm | 110 (71) | 577 (85) |
| Thoracic aortic dissection | 0 | 151 (22) |
| Thoracic aortic rupture | 0 | 5 (0.7) |
| Coarctation of the aorta | 2 (1.3) | 5 (0.7) |
| Bicuspid aortic valve, n (%) | 53 (34) | 170 (25) |
| Ventricular septal defect, n (%) | 5 (3) | 9 (1.3) |
| Atrial septal defect, n (%) | 2 (1.3) | 10 (1.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vujakovich, C.E.; Landis, B.J. A Novel Human Biospecimen Repository for Clinical and Molecular Investigation of Thoracic Aortopathy. Cardiogenetics 2021, 11, 148-163. https://doi.org/10.3390/cardiogenetics11030017
Vujakovich CE, Landis BJ. A Novel Human Biospecimen Repository for Clinical and Molecular Investigation of Thoracic Aortopathy. Cardiogenetics. 2021; 11(3):148-163. https://doi.org/10.3390/cardiogenetics11030017
Chicago/Turabian StyleVujakovich, Courtney E., and Benjamin J. Landis. 2021. "A Novel Human Biospecimen Repository for Clinical and Molecular Investigation of Thoracic Aortopathy" Cardiogenetics 11, no. 3: 148-163. https://doi.org/10.3390/cardiogenetics11030017
APA StyleVujakovich, C. E., & Landis, B. J. (2021). A Novel Human Biospecimen Repository for Clinical and Molecular Investigation of Thoracic Aortopathy. Cardiogenetics, 11(3), 148-163. https://doi.org/10.3390/cardiogenetics11030017