
Human SMAD4 Genomic Variants Identified in Individuals with Heritable and Early-Onset Thoracic Aortic Disease

 and
and 
Abstract
:1. Introduction
2. Methods
2.1. Study Population
2.2. Whole Exome Sequencing
2.3. Mapping of Genomic Variants
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sánchez-Martínez, R.; Iriarte, A.; Mora-Luján, J.M.; Patier, J.L.; López-Wolf, D.; Ojeda, A. For the RiHHTa investigators of the rare diseases working group from the spanish society of internal medicine. Current HHT genetic overview in Spain and its phenotypic correlation: Data from RiHHTa registry. Orphanet J. Rare Dis. 2020, 15, 138. [Google Scholar] [CrossRef]
- Isselbacher, E.M.; Cardenas, C.L.L.; Lindsay, M.E. Hereditary Influence in thoracic aortic aneurysm and dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [Green Version]
- Isselbacher, E.M. Thoracic and abdominal aortic aneurysms. Circulation 2005, 111, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Philippakis, A.A.; Azzariti, D.R.; Beltran, S.; Brookes, A.J.; Brownstein, C.A.; Brudno, M.; Rehm, H.L. The matchmaker exchange: A platform for rare disease gene discovery. Hum. Mutat. 2015, 36, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.-Y.; Guo, D.; Regalado, E.S.; Shen, H.; Coselli, J.S.; Estrera, A.L.; Milewicz, D.M. SMAD4 rare variants in individuals and families with thoracic aortic aneurysms and dissections. Eur. J. Hum. Genet. 2019, 27, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Regalado, E.; Shendure, J.; Nickerson, D.A.; Guo, D. Successes and challenges of using whole exome sequencing to identify novel genes underlying an inherited predisposition for thoracic aortic aneurysms and acute aortic dissections. Trends Cardiovasc. Med. 2014, 24, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; MacArthur, D.G. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Talkowski, M.E. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Baburajendran, N.; Jauch, R.; Tan, C.Y.Z.; Narasimhan, K.; Kolatkar, P.R. Structural basis for the cooperative DNA recognition by Smad4 MH1 dimers. Nucleic Acids Res. 2011, 39, 8213–8222. [Google Scholar] [CrossRef] [Green Version]
- BabuRajendran, N.; Palasingam, P.; Narasimhan, K.; Sun, W.; Prabhakar, S.; Jauch, R.; Kolatkar, P.R. Structure of Smad1 MH1/DNA complex reveals distinctive rearrangements of BMP and TGF-beta effectors. Nucleic Acids Res. 2010, 38, 3477–3488. [Google Scholar] [CrossRef]
- Chai, N.; Li, W.-X.; Wang, J.; Wang, Z.-X.; Yang, S.-M.; Wu, J.-W. Structural basis for the Smad5 MH1 domain to recognize different DNA sequences. Nucleic Acids Res. 2015, 43, 9051–9064. [Google Scholar] [CrossRef] [Green Version]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of thoracic and abdominal aortic diseases. Circ. Res. 2019, 124, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Renard, M.; Francis, C.; Ghosh, R.; Scott, A.F.; Witmer, P.D.; Adès, L.C.; Backer, J.D. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J. Am. Coll. Cardiol. 2018, 72, 605–615. [Google Scholar] [CrossRef]
- Boileau, C.; Guo, D.-C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Milewicz, D.M. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.-C.; Pannu, H.; Tran-Fadulu, V.; Papke, C.L.; Yu, R.K.; Avidan, N.; Milewicz, D.M. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007, 39, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- LeMaire, S.A.; McDonald, M.-L.N.; Guo, D.-C.; Russell, L.; Miller, C.C.; Johnson, R.J.; Milewicz, D.M. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat. Genet. 2011, 43, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Milewicz, D.M.; Trybus, K.M.; Guo, D.-C.; Sweeney, H.L.; Regalado, E.; Kamm, K.; Stull, J.T. Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Milewicz, D.M.; Prakash, S.K.; Ramirez, F. Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: Insights from predisposing genes and mouse models. Annu. Rev. Med. 2017, 68, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Chu, G.C.; Dunn, N.R.; Anderson, D.C.; Oxburgh, L.; Robertson, E.J. Differential requirements for Smad4 in TGFbeta-dependent patterning of the early mouse embryo. Development 2004, 131, 3501–3512. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Liu, B.; Yao, H.; Li, F.; Weng, T.; Yang, G.; Yang, X. Essential role of endothelial smad4 in vascular remodeling and integrity. Mol. Cell. Biol. 2007, 27, 7683–7692. [Google Scholar] [CrossRef] [Green Version]
- Takaku, K.; Miyoshi, H.; Matsunaga, A.; Oshima, M.; Sasaki, N.; Taketo, M.M. Gastric and duodenal polyps in Smad4 (Dpc4) knockout mice. Cancer Res. 1999, 59, 6113–6117. [Google Scholar] [PubMed]
- Crist, A.M.; Lee, A.R.; Patel, N.R.; Westhoff, D.E.; Meadows, S.M. Vascular deficiency of Smad4 causes arteriovenous malformations: A mouse model of hereditary hemorrhagic telangiectasia. Angiogenesis 2018, 21, 363–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crist, A.M.; Zhou, X.; Garai, J.; Lee, A.R.; Thoele, J.; Ullmer, C.; Meadows, S.M. Angiopoietin-2 Inhibition rescues arteriovenous malformation in a Smad4 hereditary hemorrhagic telangiectasia mouse model. Circulation 2019, 139, 2049–2063. [Google Scholar] [CrossRef]
- Gallione, C.; Aylsworth, A.S.; Beis, J.; Berk, T.; Bernhardt, B.; Clark, R.D.; Marchuk, D.A. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP–HHT syndrome. Am. J. Med. Genet. Part A 2010, 152, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, C.; Mahaut, C.; Abhyankar, A.; Le Goff, W.; Serre, V.; Afenjar, A.; Cormier-Daire, V. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat. Genet. 2012, 44, 85–88. [Google Scholar] [CrossRef]
- Wu, L. Functional characteristics of a novel SMAD4 mutation from thoracic aortic aneurysms (TAA). Gene 2017, 628, 129–133. [Google Scholar] [CrossRef] [PubMed]
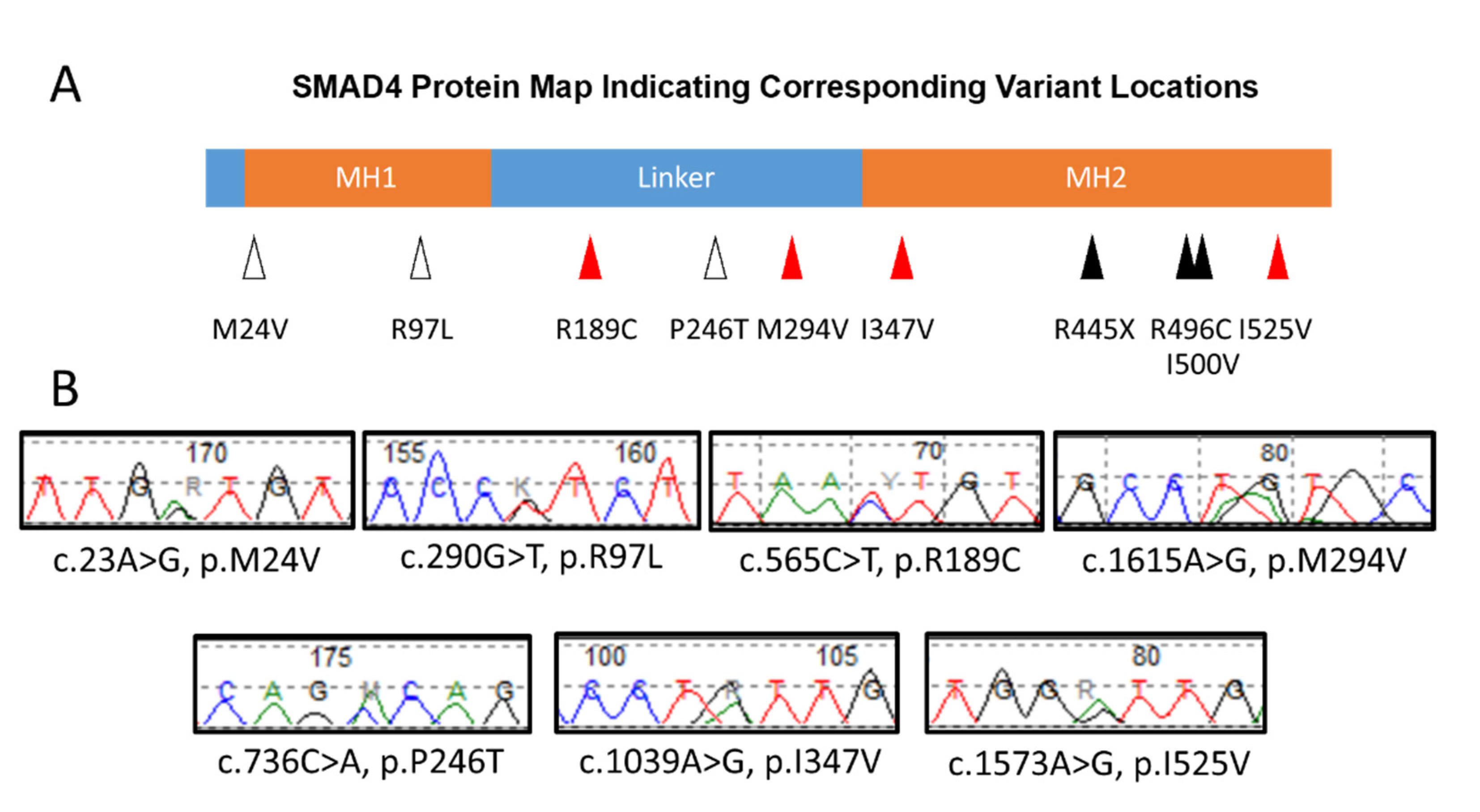

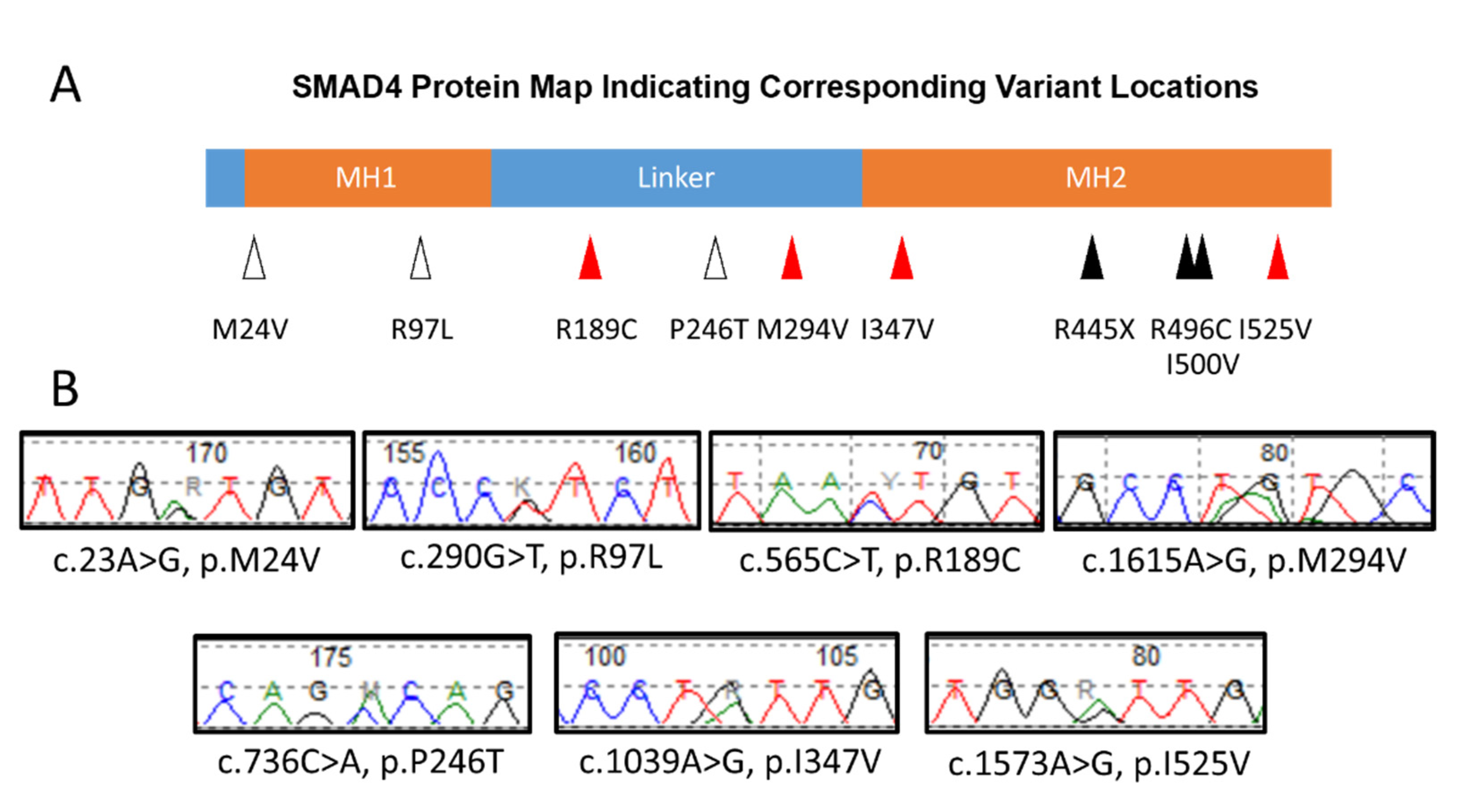{kind=link}
| Gene. | Aa alt | REVEL | CADD | SIFT4G | Clin Var | gnomAD v2.1.1 | ||
|---|---|---|---|---|---|---|---|---|
| Clinical Significance | MAF | Allele Count | Homozygote Count | |||||
| SMAD4 | M24V | 0.579 | 26.3 | D | Uncertain significance | 3.19 × 10−5 | 1 | 0 |
| SMAD4 | R97L | 0.925 | 32 | D | Conflicting interpretations | |||
| SMAD4 | R189C | 0.24 | 23.5 | T | Conflicting interpretations | 3.64 × 10−4 | 103 | 1 |
| SMAD4 | P246T | 0.214 | 22.5 | D | Uncertain significance | 7.95 × 10−6 | 2 | 0 |
| SMAD4 | M294V | 0.35 | 22.2 | T | Conflicting interpretations | 1.74 × 10−4 | 49 | 0 |
| SMAD4 | I347V | 0.387 | 20.4 | T | Uncertain significance | 7.95 × 10−6 | 2 | 0 |
| SMAD4 | R445X | Pathogenic | 3.98 × 10−6 | 1 | 0 | |||
| SMAD4 | R496C | Pathogenic/Likely pathogenic | 7.95 × 10−6 | 2 | 0 | |||
| SMAD4 | I500V | Pathogenic | 3.98 × 10−6 | 1 | 0 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhave, S.A.; Guo, D.; Angelov, S.N.; Bamshad, M.J.; Nickerson, D.A.; Milewicz, D.M.; Wallingford, M.C. Human SMAD4 Genomic Variants Identified in Individuals with Heritable and Early-Onset Thoracic Aortic Disease. Cardiogenetics 2021, 11, 132-138. https://doi.org/10.3390/cardiogenetics11030015
Bhave SA, Guo D, Angelov SN, Bamshad MJ, Nickerson DA, Milewicz DM, Wallingford MC. Human SMAD4 Genomic Variants Identified in Individuals with Heritable and Early-Onset Thoracic Aortic Disease. Cardiogenetics. 2021; 11(3):132-138. https://doi.org/10.3390/cardiogenetics11030015
Chicago/Turabian StyleBhave, Shreyas A., Dongchuan Guo, Stoyan N. Angelov, Michael J. Bamshad, Deborah A. Nickerson, Dianna M. Milewicz, and Mary C. Wallingford. 2021. "Human SMAD4 Genomic Variants Identified in Individuals with Heritable and Early-Onset Thoracic Aortic Disease" Cardiogenetics 11, no. 3: 132-138. https://doi.org/10.3390/cardiogenetics11030015
APA StyleBhave, S. A., Guo, D., Angelov, S. N., Bamshad, M. J., Nickerson, D. A., Milewicz, D. M., & Wallingford, M. C. (2021). Human SMAD4 Genomic Variants Identified in Individuals with Heritable and Early-Onset Thoracic Aortic Disease. Cardiogenetics, 11(3), 132-138. https://doi.org/10.3390/cardiogenetics11030015