


Food Effect and Formulation: How Soluble Fillers Affect the Disintegration and Dissolution of Tablets in Viscous Simulated Fed State Media
 , ,
, , 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Tablet Formulation
2.3. Composition of the Testing Media
2.4. Tablet Hardness Measurements
2.5. Disintegration Test
2.5.1. Compendial Disintegration Test Under Simulated Fasted-State Conditions
2.5.2. CNC Disintegration Test Under Simulated Fed-State Conditions
2.6. Dissolution Tests
2.6.1. Dissolution Testing in a Compendial Device Under Simulated Fasted- and Fed-State Conditions
2.6.2. Dissolution Testing in the CNC Device Under Simulated Fed-State Conditions
2.6.3. Analytics
2.7. Porosimetry
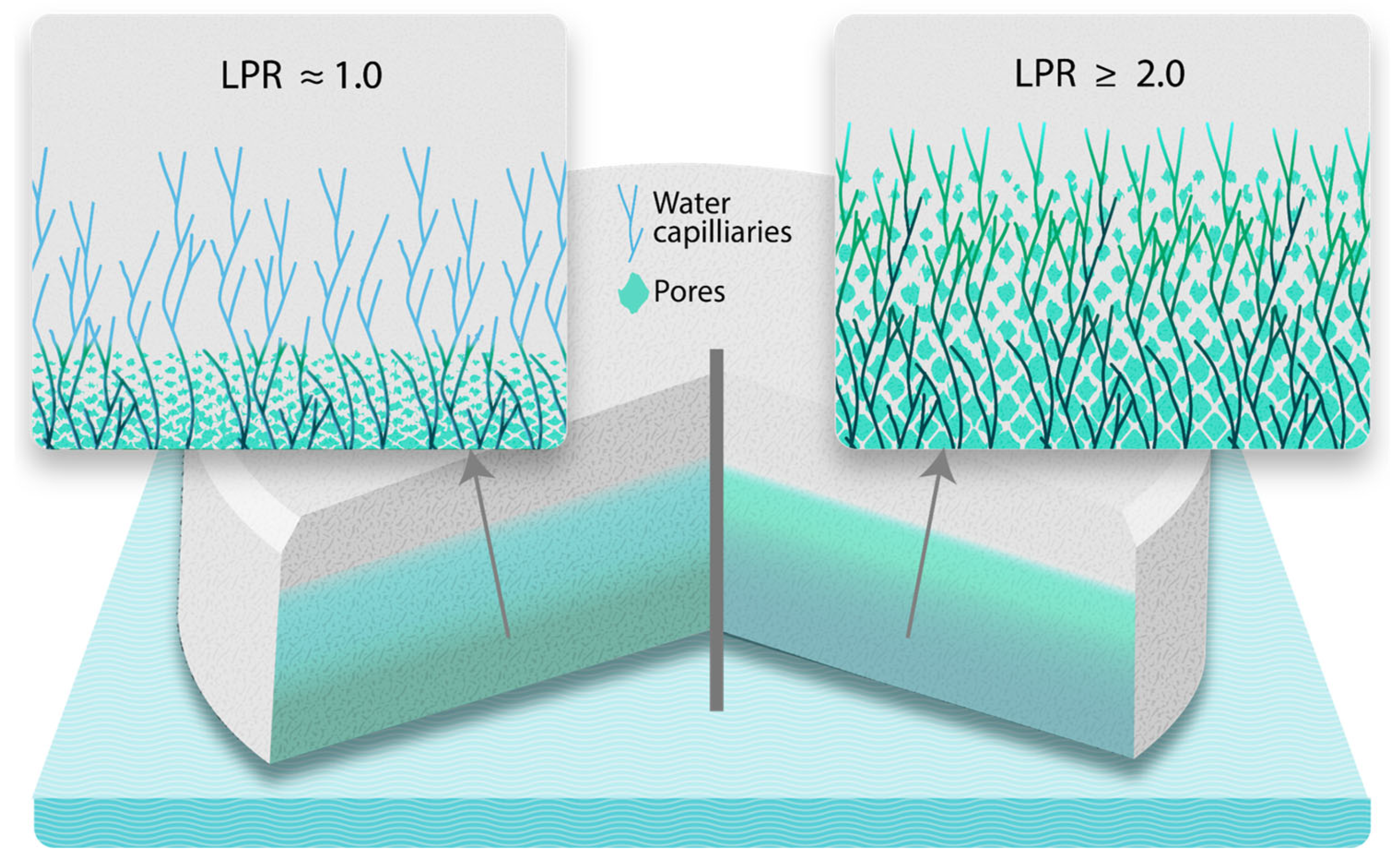
2.8. Working Principle—Liquid Penetration Ratio (LPR)
3. Results
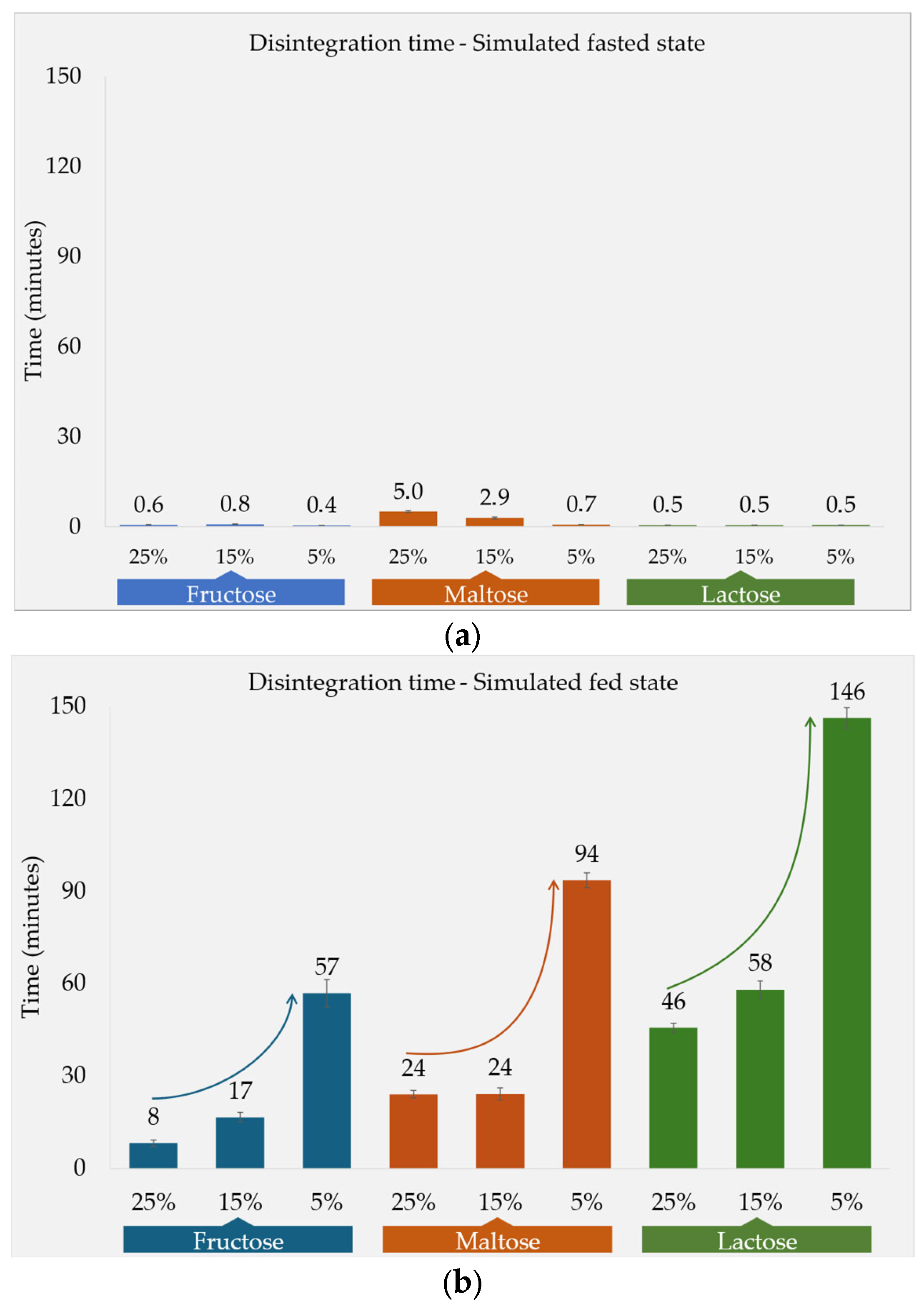
3.1. Disintegration Times and Tablet Properties
3.1.1. Fasted-State Conditions
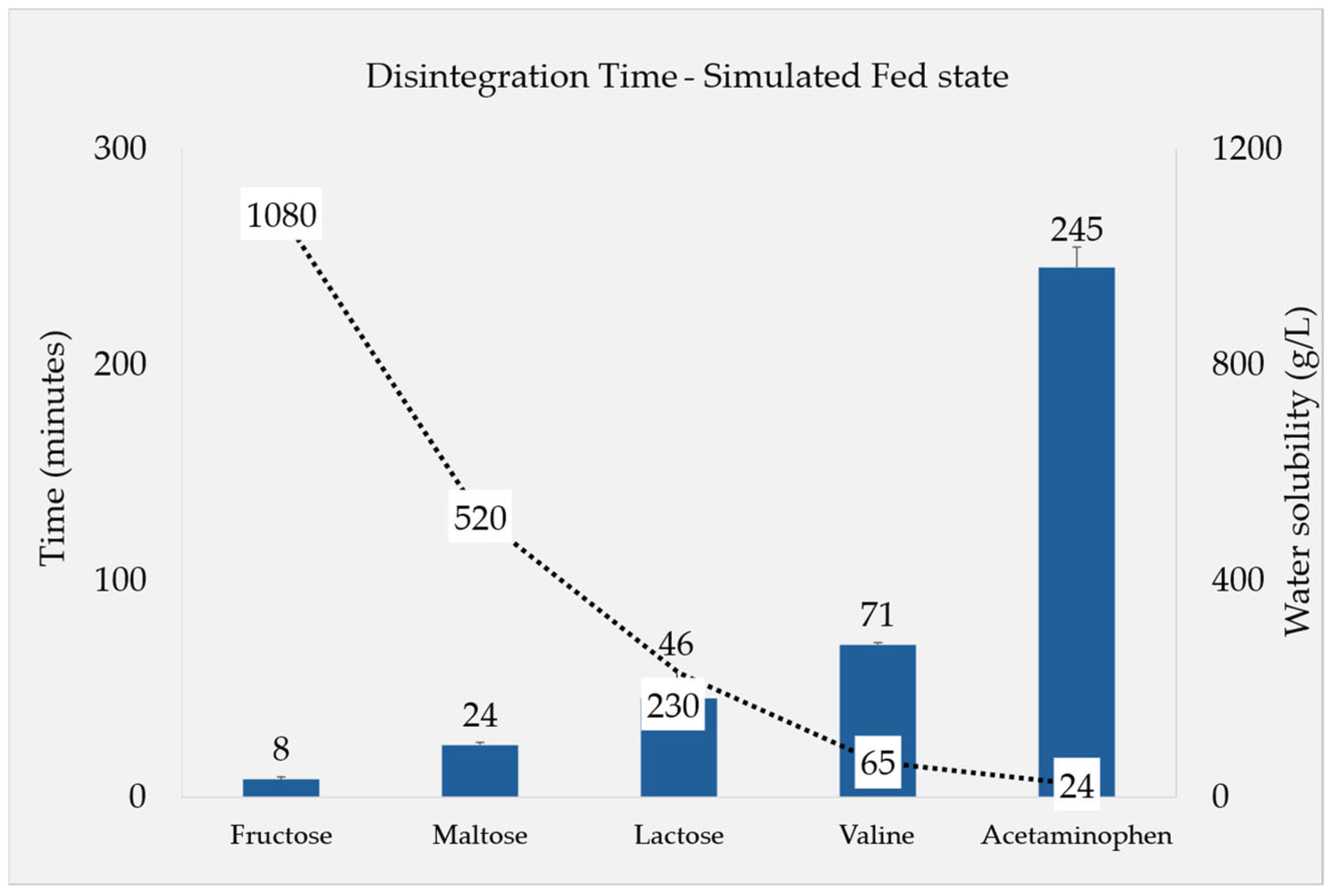
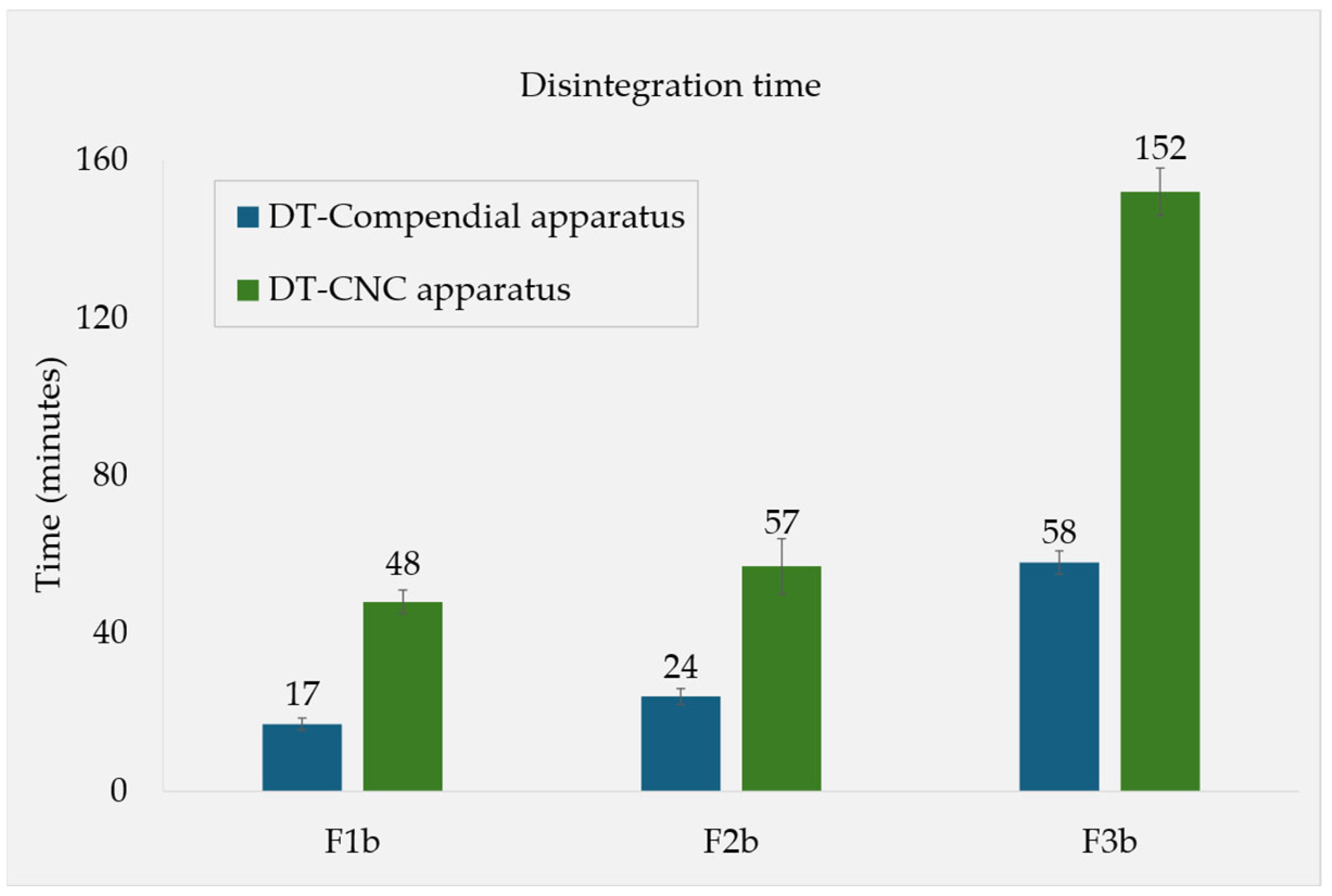
3.1.2. Fed-State Conditions
3.2. Determination of Porosity and Physical Characterization
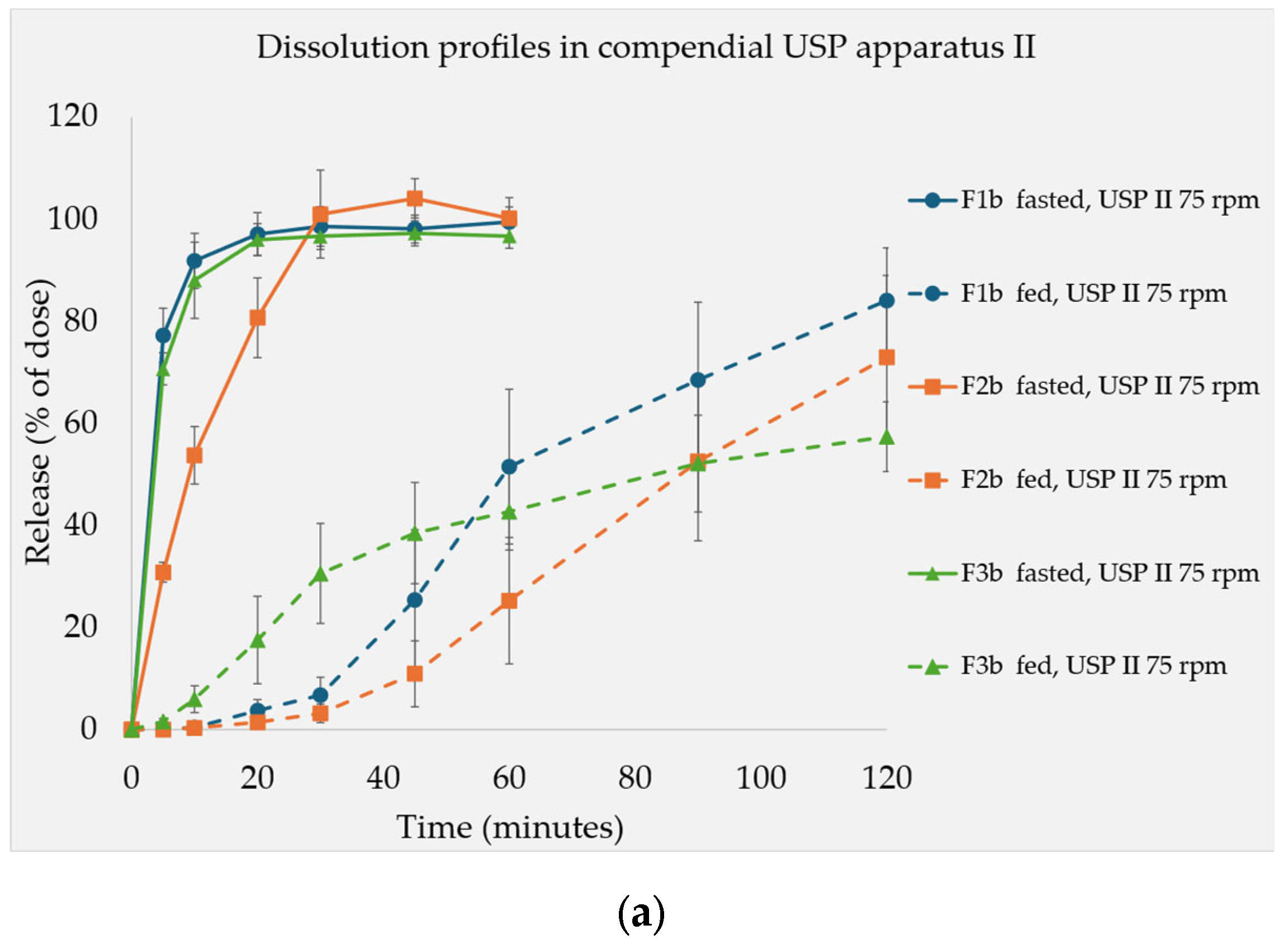
3.3. Dissolution Test Resutls
4. Discussion
4.1. Disintegration and Dissolution Under the Simulated Fasted State
4.2. Tablet Disintegration and LPR
4.3. Dissolution Tests Under Fed State
4.4. Physical Characterization
4.5. Filler Quantity
4.6. Practical Implications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| API | Active pharmaceutical ingredient |
| CNC | Computed numerical control |
| DCP | Dicalcium phosphate |
| DT | Disintegration time |
| HCl | Hydrochloric acid |
| HPMC | Hydroxypropyl methylcellulose |
| ICH | International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use |
| IR | Immediate release |
| LPR | Liquid penetration ratio |
| MCC | Microcrystalline cellulose |
| MPa | Megapascal |
| NaCl | Sodium chloride |
| NaOH | Sodium hydroxide |
| PVP | Polyvinyl pyrrolidone |
| RSD | Relative standard deviation |
| SD | Standard deviation |
| SSG | Sodium starch glycolate |
| US FDA | US Food and Drug Administration |
| USP | United States Pharmacopeia |
References
- Ghosh, S. Oral Solid Dosage Pharmaceutical Formulation Market Outlook: Oral Solid Dosage Pharmaceutical Formulation Market Forecast by Tablets, Capsules, and Other Dosage Forms from 2024 to 2034. Surging Demand for Innovative Oral Dosage Formulations Driven by Shift Towards Personalized Medicine and Customized Treatments. FMI Insights into Biodegradable Polymer Sustainability REP-GB-2782. 2017. Available online: https://www.futuremarketinsights.com/reports/oral-solid-dosage-pharmaceutical-formulation-market# (accessed on 12 April 2024).
- Horkovics-Kovats, S. Disintegration Rate and Properties of Active Pharmaceutical Ingredient Particles as Determined from the Dissolution Time Profile of a Pharmaceutical Formulation: An Inverse Problem. J. Pharm. Sci. 2014, 103, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Berardi, A.; Bisharat, L.; Quodbach, J.; Abdel Rahim, S.; Perinelli, D.R.; Cespi, M. Advancing the understanding of the tablet disintegration phenomenon—An update on recent studies. Int. J. Pharm. 2021, 598, 120390. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shi, C.; Fang, Y.; Zhao, H.; Mu, Y.; Zhao, L.; Shen, L. A comprehensive understanding of disintegrants and disintegration quantification techniques: From the perspective of tablet microstructure. J. Drug Deliv. Sci. Technol. 2023, 88, 104891. [Google Scholar] [CrossRef]
- Radwan, A.; Wagner, M.; Amidon, G.L.; Langguth, P. Bio-predictive tablet disintegration: Effect of water diffusivity, fluid flow, food composition and test conditions. Eur. J. Pharm. Sci. 2014, 57, 273–279. [Google Scholar] [CrossRef]
- Abdallah, D.B.; Charoo, N.A.; Elgorashi, A.S. Comparative binding and disintegrating property of Echinochloa colona starch (difra starch) against maize, sorghum, and cassava starch. Pharm. Biol. 2014, 52, 935–943. [Google Scholar] [CrossRef]
- Quodbach, J.; Kleinebudde, P. A critical review on tablet disintegration. Pharm. Dev. Technol. 2016, 21, 763–774. [Google Scholar] [CrossRef]
- Dvořák, J.; Tomas, J.; Lizoňová, D.; Schöngut, M.; Dammer, O.; Pekárek, T.; Beránek, J.; Štěpánek, F. Investigation of tablet disintegration pathways by the combined use of magnetic resonance imaging, texture analysis and static light scattering. Int. J. Pharm. 2020, 587, 119719. [Google Scholar] [CrossRef]
- Abdullahu, B.; Lajçi, A.; Shehu, V.; Krasniqi, S.; Islami, H. Study of Formulation of Pharmaceutical Forms of Paracetamol in Medical Practice. Med. Arch. 2010, 64, 196. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Saffari, M.; Dehghani, F.; Langrish, T. Incorporation of acetaminophen as an active pharmaceutical ingredient into porous lactose. Int. J. Pharm. 2016, 499, 217–227. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Radwan, A.; Amidon, G.L.; Langguth, P. Mechanistic investigation of food effect on disintegration and dissolution of BCS class III compound solid formulations: The importance of viscosity. Biopharm. Drug Dispos. 2012, 33, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Kindgen, S.; Rach, R.; Nawroth, T.; Abrahamsson, B.; Langguth, P. A Novel Disintegration Tester for Solid Dosage Forms Enabling Adjustable Hydrodynamics. J. Pharm. Sci. 2016, 105, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Picazo, A.; Colón-Useche, S.; Gonzalez-Alvarez, M.; Gonzalez-Alvarez, I.; Bermejo, M.; Langguth, P. Effect of thickener on disintegration, dissolution and permeability of common drug products for elderly patients. Eur. J. Pharm. Biopharm. 2020, 153, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Washburn, E.W. The Dynamics of Capillary Flow. Phys. Rev. 1921, 17, 273. [Google Scholar] [CrossRef]
- Crestani, C.E.; Bernardo, A.; Costa, C.B.B.; Giulietti, M. Fructose Solubility in Mixed (Ethanol + Water) Solvent: Experimental Data and Comparison among Different Thermodynamic Models. J. Chem. Eng. Data 2013, 58, 3039–3045. [Google Scholar] [CrossRef]
- Gong, X.; Wang, C.; Zhang, L.; Qu, H. Solubility of Xylose, Mannose, Maltose Monohydrate, and Trehalose Dihydrate in Ethanol–Water Solutions. J. Chem. Eng. Data 2012, 57, 3264–3269. [Google Scholar] [CrossRef]
- Fox, P.F.; Uniacke-Lowe, T.; McSweeney, P.L.H.; O’Mahony, J.A. Dairy Chemistry and Biochemistry; Springer: Cham, Switzerland, 2015; ISBN 978-3-319-14892-2. [Google Scholar]
- Zhang, C.; Liu, B.; Wang, X.; Wang, H. Measurement and Correlation of the Solubilities of l-Valine in Water, Ethanol, N, N -Dimethylformamide, Acetone, and Isopropyl Alcohol between (293.15 and 343.15) K. J. Chem. Eng. Data 2014, 59, 2704–2708. [Google Scholar] [CrossRef]
- Kalantzi, L.; Reppas, C.; Dressman, J.B.; Amidon, G.L.; Junginger, H.E.; Midha, K.K.; Shah, V.P.; Stavchansky, S.A.; Barends, D.M. Biowaiver monographs for immediate release solid oral dosage forms: Acetaminophen (paracetamol). J. Pharm. Sci. 2006, 95, 4–14. [Google Scholar] [CrossRef]
- Zaheer, K.; Langguth, P. Formulation strategy towards minimizing viscosity mediated negative food effect on disintegration and dissolution of immediate release tablets. Drug Dev. Ind. Pharm. 2018, 44, 444–451. [Google Scholar] [CrossRef]
- Higuchi, M.; Nishida, S.; Yoshihashi, Y.; Tarada, K.; Sugano, K. Prediction of coning phenomena for irregular particles in paddle dissolution test. Eur. J. Pharm. Sci. 2015, 76, 213–216. [Google Scholar] [CrossRef]
- Al-Sharabi, M.; Markl, D.; Mudley, T.; Bawuah, P.; Karttunen, A.-P.; Ridgway, C.; Gane, P.; Ketolainen, J.; Peiponen, K.-E.; Rades, T.; et al. Simultaneous investigation of the liquid transport and swelling performance during tablet disintegration. Int. J. Pharm. 2020, 584, 119380. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, T.; Masuda, Y.; Yamamoto, T.; Yonemochi, E.; Terada, K. Formulation design of a novel fast-disintegrating tablet. Int. J. Pharm. 2005, 306, 83–90. [Google Scholar] [CrossRef]
- Desai, P.M.; Liew, C.V.; Heng, P.W.S. Assessment of disintegration of rapidly disintegrating tablets by a visiometric liquid jet-mediated disintegration apparatus. Int. J. Pharm. 2013, 442, 65–73. [Google Scholar] [CrossRef]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. (Eds.) Handbook of Pharmaceutical Excipients, 6th ed.; APhA (PhP) Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Lourdin, D.; Colonna, P.; Ring, S.G. Volumetric behaviour of maltose-water, maltose-glycerol and starch-sorbitol-water systems mixtures in relation to structural relaxation. Carbohydr. Res. 2003, 338, 2883–2887. [Google Scholar] [CrossRef]
- Parojcić, J.; Vasiljević, D.; Ibrić, S.; Djurić, Z. Tablet disintegration and drug dissolution in viscous media: Paracetamol IR tablets. Int. J. Pharm. 2008, 355, 93–99. [Google Scholar] [CrossRef]
- Vrbanac, H.; Trontelj, J.; Kalčič, Š.; Legen, I. Mechanistic study of model drug release from HPMC matrices in fed gastric media. J. Drug Deliv. Sci. Technol. 2020, 60, 102034. [Google Scholar] [CrossRef]
- Berardi, A.; Bauhuber, S.; Sawafta, O.; Warnke, G. Alginates as tablet disintegrants: Understanding disintegration mechanisms and defining ranges of applications. Int. J. Pharm. 2021, 601, 120512. [Google Scholar] [CrossRef]
- Geisslinger, G.; Menzel, S.; Gudermann, T.; Hinz, B.; Ruth, P.; Mutschler, E. Mutschler Arzneimittelwirkungen; Wissenschaftliche Verlagsgesellschaft Stuttgart: Stuttgart, Germany, 2019; ISBN 9783804740549. [Google Scholar]
- European Directorate for the Quality of Medicines & Healthcare. European Pharmacopoeia: Doxycycline Monohydrate; 01/2017:0820, 11.8th ed.; European Directorate for the Quality of Medicines & Healthcare: Strasborg, France, 2025. [Google Scholar]
- European Directorate for the Quality of Medicines & Healthcare. European Pharmacopoeia: Doxycycline Hyclate; 01/2017:0272, 11.8th ed.; European Directorate for the Quality of Medicines & Healthcare: Strasbourg, France, 2025. [Google Scholar]
- National Academies of Sciences, Engineering, and Medicine. Assessment of Long-Term Health Effects of Antimalarial Drugs When Used for Prophylaxis: Doxycline; The National Academies Press: Washington, DC, USA, 2020; ISBN 978-0-309-67210-8. [Google Scholar]
- Rote Liste® Service GmbH. Fachinformation (Specialist Information): Rifampicin: EREMFAT® 450 mg, Film tablets. Available online: https://www.fachinfo.de/fi/pdf/021100 (accessed on 14 April 2025).
- Rote Liste® Service GmbH. Fachinformation (Specialist Information): Pyrazinamide: PYRAFAT® 500 mg, Film tablets. Available online: https://www.fachinfo.de/fi/detail/001771/pyrafat-r-500-mg-filmtabletten (accessed on 14 April 2025).
- Rote Liste® Service GmbH. Fachinformation (Specialist Information): Isoniazide: ISOZID® 450 mg, Film tablets. Available online: https://www.fachinfo.de/fi/pdf/020692 (accessed on 14 April 2025).
- Hardin, T.C.; Graybill, J.R.; Fetchick, R.; Woestenborghs, R.; Rinaldi, M.G.; Kuhn, J.G. Pharmacokinetics of itraconazole following oral administration to normal volunteers. Antimicrob. Agents Chemother. 1988, 32, 1310–1313. [Google Scholar] [CrossRef]
- Ochoa, D.; Saiz-Rodríguez, M.; González-Rojano, E.; Román, M.; Sánchez-Rojas, S.; Wojnicz, A.; Ruiz-Nuño, A.; García-Arieta, A.; Abad-Santos, F. High-Fat Breakfast Increases Bioavailability of Albendazole Compared to Low-Fat Breakfast: Single-Dose Study in Healthy Subjects. Front. Pharmacol. 2021, 12, 664465. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | Percentage Content of Soluble Fillers and DCP Used, 30% of Total Formulation | |||||
|---|---|---|---|---|---|---|
| Fructose | Maltose | Lactose | Valine | Acetaminophen | DCP | |
| F1a | 25 | -- | -- | -- | -- | 5 |
| F1b | 15 | -- | -- | -- | -- | 15 |
| F1c | 5 | -- | -- | -- | -- | 25 |
| F2a | -- | 25 | -- | -- | -- | 5 |
| F2b | -- | 15 | -- | -- | -- | 15 |
| F2c | -- | 5 | -- | -- | -- | 25 |
| F3a | -- | -- | 25 | -- | -- | 5 |
| F3b | -- | -- | 15 | -- | -- | 15 |
| F3c | -- | -- | 5 | -- | -- | 25 |
| F4 | -- | -- | -- | 25 | -- | 5 |
| F5 | -- | -- | -- | -- | 25 | 5 |
| Water solubility g/L | 1080 [16] | 520 [17] | 230 [18] | 65 [19] | 24 [20] | Considered insoluble |
| Parameter | Value |
|---|---|
| Absorption wavelength | 242 nm |
| Linear range | 1–14 µg/mL |
| Calibration curve | |
| R2 | 1.000 |
| Slope | 0.0634 |
| Intercept | −0.0001 |
| Limit of detection | 0.09 µg/mL |
| Limit of quantification | 0.26 µg/mL |
| Intraday precision (triplicate) | |
| 4.0 µg/mL | ±0.68% relative standard deviation (RSD) |
| 8.0 µg/mL | ±0.23% RSD |
| 12.0 µg/mL | ±0.53% RSD |
| Interday precision | |
| (8.0 µg/mL at three consecutive days) | ±0.34% RSD |
| Formulation Code | DT—Fasted State, Compendial App., Mean ± SD (n = 6) (Minutes) | DT—Fed State, Compendial App., Mean ± SD (n = 6) (Minutes) | DT—Fed State, CNC App., Mean ± SD (n = 3) (Minutes) | Tablet Weight, Mean ± SD (n = 3)(mg) | Tablet Hardness, Mean ± SD (n = 3)(N) |
|---|---|---|---|---|---|
| F1a | 0.61 ± 0.11 | 8 ± 01 | * | 650 ± 05 | 64 ± 04 |
| F1b | 0.78 ± 0.09 | 17 ± 02 | 48 ± 03 | 651 ± 05 | 76 ± 02 |
| F1c | 0.43 ± 0.03 | 57 ± 04 | * | 648 ± 03 | 92 ± 05 |
| F2a | 5.02 ± 0.31 | 24 ± 01 | * | 653 ± 03 | 113 ± 02 |
| F2b | 2.87 ± 0.39 | 24 ± 02 | 57 ± 07 | 649 ± 05 | 87 ± 05 |
| F2c | 0.71 ± 0.05 | 94 ± 02 | * | 649 ± 02 | 78 ± 02 |
| F3a | 0.52 ± 0.03 | 46 ± 01 | * | 653 ± 01 | 91 ± 06 |
| F3b | 0.53 ± 0.03 | 58 ± 03 | 152 ± 06 | 650 ± 02 | 88 ± 02 |
| F3c | 0.53 ± 0.03 | 146 ± 03 | * | 650 ± 02 | 91 ± 04 |
| F4 | 1.46 ± 0.07 | 71 ± 01 | * | 649 ± 01 | 76 ± 02 |
| F5 | 0.31 ± 0.03 | 245 ± 09 | * | 649 ± 01 | 41 ± 03 |
| Formulation | F1b | F2b | F3b |
|---|---|---|---|
| Bulk density (g/cm3) at 0 MPa | 1.2697 | 1.2630 | 1.2919 |
| Bulk density (g/cm3) at 400 MPa | 1.4390 | 1.3934 | 1.4069 |
| Average pore diameter (nm) | 33.34 | 41.06 | 35.32 |
| Porosity (%) | 11.67 | 9.36 | 8.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umer, M.F.; Stahl, V.; Al-Gousous, J.; Nawroth, T.; Sun, W.-J.; Wu, F.; Jiang, W.; Gao, Z.; Langguth, P. Food Effect and Formulation: How Soluble Fillers Affect the Disintegration and Dissolution of Tablets in Viscous Simulated Fed State Media. Pharmaceutics 2025, 17, 567. https://doi.org/10.3390/pharmaceutics17050567
Umer MF, Stahl V, Al-Gousous J, Nawroth T, Sun W-J, Wu F, Jiang W, Gao Z, Langguth P. Food Effect and Formulation: How Soluble Fillers Affect the Disintegration and Dissolution of Tablets in Viscous Simulated Fed State Media. Pharmaceutics. 2025; 17(5):567. https://doi.org/10.3390/pharmaceutics17050567
Chicago/Turabian StyleUmer, Muhammad Farooq, Valentin Stahl, Jozef Al-Gousous, Thomas Nawroth, Wei-Jhe Sun, Fang Wu, Wenlei Jiang, Zongming Gao, and Peter Langguth. 2025. "Food Effect and Formulation: How Soluble Fillers Affect the Disintegration and Dissolution of Tablets in Viscous Simulated Fed State Media" Pharmaceutics 17, no. 5: 567. https://doi.org/10.3390/pharmaceutics17050567
APA StyleUmer, M. F., Stahl, V., Al-Gousous, J., Nawroth, T., Sun, W.-J., Wu, F., Jiang, W., Gao, Z., & Langguth, P. (2025). Food Effect and Formulation: How Soluble Fillers Affect the Disintegration and Dissolution of Tablets in Viscous Simulated Fed State Media. Pharmaceutics, 17(5), 567. https://doi.org/10.3390/pharmaceutics17050567