1. Introduction
Traditionally, lipid nanoparticles and other nanoparticle delivery systems were commonly manufactured using complex techniques, often resulting in large particles requiring additional steps to reduce size, making these techniques hard to scale up and costly to maintain [
1]. As nanoparticle technology has advanced, so too have manufacturing methods. In an effort to create user-friendly, cost-effective and scalable nanoparticle manufacturing methods, microfluidic mixing was developed. Microfluidic mixing can briefly be described as the mixing of two or more fluids within a microscale environment [
2], with advantages including higher encapsulation efficiencies, the possibility to incorporate both lipophilic and hydrophilic drugs, and an overall decreased cost and time constraint [
3,
4]. Microfluidics results in nanoparticle formation in a single-step process involving the mixing of an aqueous phase (commonly including the moiety to be entrapped dissolved in buffer, e.g., mRNA in the case of LNPs) with a lipid phase (dissolved in solvent, e.g., ethanol). Indeed, microfluidics is now routinely involved in LNP manufacture, and Pratsinis et al. demonstrated using a Design of Experiments (DoE) approach that when compared to solvent injection methods, microfluidics resulted in small LNPs with a more homogenous population [
5]. Manufacturing using microfluidic devices can involve either active mixing (requiring external forces, e.g., vibration or electrical forces) or passive mixing (e.g., cross-flow or flow-focusing), which will additionally have variations in internal structures and diameters of mixers. This, in turn, can influence the critical quality attributes (CQAs) of the resulting LNP formulations [
6,
7,
8]. The degree of mixing can also be influenced by the channel length and contact surface area of the liquid/liquid interface [
8].
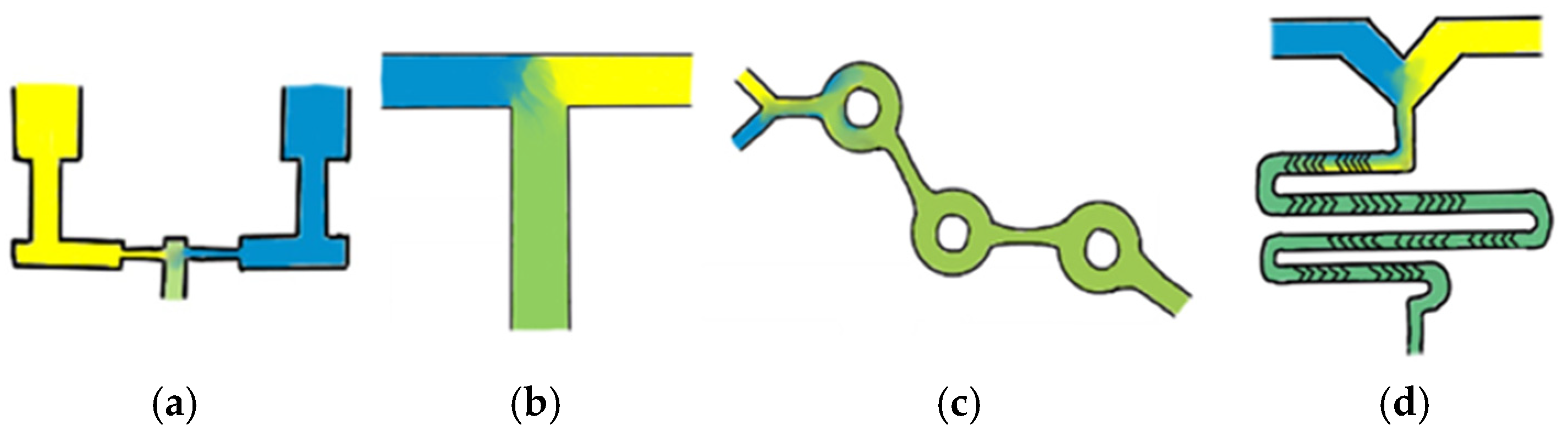
One of the earliest and easiest to use microfluidic mixer designs is a standard T-junction mixer, which typically involves the aqueous and lipid phases being injected directly into each other at high speeds, resulting in the formation of LNPs. This method often enables precise control over size; however, it requires increased waste volumes to allow for fluid mixing equilibration, making it less suitable for small-scale research. Additionally, the high forces generated in these mixers can disrupt droplet formation at the interface, potentially affecting LNP uniformity and stability [
8,
9]. Despite this, T-junction mixers have demonstrated significant clinical success, with Pfizer BioNTech manufacturing their COVID-19 vaccine using a proprietary T-mixer design [
10]. A second common design, known as impinging jet mixers, features a set-up in which fluids are constricted and directed toward each other at high speeds, resulting in nanoprecipitation. These mixers can be particularly useful when dealing with higher viscosity liquids. However, similar to T-mixers, these mixers are subject to high shear forces [
7]. Despite these challenges, impinging jet mixers help overcome the complexities of fluidic control through fewer fluid channels and a simpler design [
6]. As LNP systems have gained popularity, the demand for precise control over LNP CQAs, coupled with enhanced scalability, has increased. Mixers that excel in these parameters often harness chaotic flow, differing from traditional methods by incorporating groves or advections that disrupt laminar flow [
3,
7]. One example of a chaotic flow mixer is the staggered herringbone mixer (SHM), which contains two inlet channels flowing into a mixer with ‘v’ shaped advections, causing transverse flow and enhancing the mixing of the two fluids. Another example is the toroidal mixer (TM), which causes fluids to wrap around each other due to circular flow path structures [
7,
8,
11]. Despite their advantages, industry-leading chaotic mixers are often expensive, rely heavily on single-use plastics, and are not the only viable option for producing small, homogenous LNP populations. Simplified schematic representations of these microfluidic mixers are shown in
Figure 1; however, these mixer designs encompass only a small portion of the diverse range of available mixing methods.
Overall, different mixer designs can produce LNPs with varied CQAs, making it essential to understand how each mixer type influences nanoparticle characteristics to successfully select the most appropriate system. Beyond mixer design, the internal size of the mixer also plays a crucial role in LNP formation. For example, Maeki et al. conducted a study in which they designed two chaotic advection mixers with different channel depths and demonstrated that, for a liposomal formulation, a larger channel depth resulted in higher mixing efficiency [
12]. Additionally, passive and active mixers can be combined to refine process parameters further. For instance, Peng et al. coupled ultrasonic microreactors with either co-flow or confined impingement jet mixers to manufacture liposomal formulations, illustrating the potential of hybrid approaches [
13]. Furthermore, key process parameters, such as the speed at which the fluids are delivered (total flow rate; TFR) and the ratio at which the fluids are delivered (flow rate ratio; FRR), are vital in controlling the CQAs of LNPs. For example, McMillan et al. and Roces et al. both demonstrated that varying FRRs offered a precise method for controlling LNP size [
14,
15].
Here, we briefly highlighted a small portion of the extensive diversity of microfluidic mixer design. This study aims to determine whether low-cost, reusable microfluidic mixers can produce LNPs within a standard target CQA range with high payload entrapment and preclinical efficacy to allow for the low-cost and rapid screening of LNP formulations. To do this, two microfluidic mixing devices, a T-junction mixer (TM) and a confined impingement jet mixer (IJM), both equipped with automated syringe drivers, alongside a manual mixing process (pipette mixing (PM)), were used to manufacture LNPs. These LNPs were then subjected to orthogonal analysis techniques and preclinical in vitro and in vivo testing. A high throughput pipette mixing robot was also employed to assess PM as an LNP screening and optimization platform. Overall, this study aimed to establish if low-cost microfluidic mixers can reliably and reproducibly produce LNPs suitable for screening and testing of LNPs.
2. Materials and Methods
2.1. Materials
The ionizable lipids Heptadecan-9-yl 8-((2-hydroxyethyl)[6-oxo-6-(undecyloxy)hexyl]amino) octanoate (SM-102) and (6Z, 9Z, 28Z, 31Z)-Heptatriaconta-6,9,28,31-tetraen-19-yl 4-(dimethylamino)butanoate (D-Lin-MC3-DMA) were purchased from Broadpharm (San Diego, CA, USA). 1,2-dimyristoylrac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2000), 1,2-distearoyl-sn-glycero-3-phosphocholine, 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), and 1,2-dioleoyl-3-dimethylammonium-propane (DODAP) were acquired from Avanti Polar Lipids (Alabaster, AL, USA). Cholesterol, polyadenylic acid (poly A), heat-inactivated fetal bovine serum (FBS) and HPLC-grade methanol were purchased from Merck Life Science UK Ltd. (Gillingham, UK). Ethanol was purchased from Suppelco (Bellefonte, PA, USA). Sodium citrate buffer, Tris-HCL buffer, Triethylamine acetate buffer, propidium iodide, Trypan Blue, and the Quanti-iT RiboGreen RNA Assay kit were purchased from Thermo Fisher Scientific (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). EZ CapTM Firefly Luciferase mRNA (5-moUTP) and EZ CapTM Cy5 Firefly Luciferase mRNA (5-moUTP) were purchased from APExBIO (Houston, TX, USA). CPI (Centre for Process Innovation, Sedgefield, UK) supplied Green Lantern mRNA. A Rabbit Reticulocyte Lysate kit, ONE-Glo™ Luciferase Assay System, and VivoGlo™ Luciferin were acquired from Promega Ltd. (Chilworth, UK). Dulbecco’s Minimal Essential Media (DMEM), Minimal Essential Media (MEM), sodium pyruvate, penicillin/streptomycin, and TrypLE were purchased from Gibco Technologies. Ultrapure water was supplied through an in-house system. HEK-293 cells were purchased from American Tissue Culture Collection (ATCC) (CRL-1573).
2.2. Microfluidic Mixer Set-Up
The T-junction mixer (TM) consisted of a 3-port manifold T-junction (Darwin Microfluidics); aqueous and lipid phase inlets flow directly into each other while the LNP outlet is positioned perpendicularly. The impingement jet mixer (IJM) consisted of a 5-port confined impinging jet mixer (CIJM) (Holland Applied Technologies, Burr Ridge, IL, USA); 3 of the 5 ports were used with the aqueous and lipid phase inlets flowing down, then directly into each other, with the LNP outlet positioned perpendicularly for sample collection. Both microfluidic mixers were combined with World Precision Instrument AL-1010 syringe pumps, which were automated using the SyringePumpPro software v1.7.0.0 to allow for precise control over mixing speeds and ratios. Pipette mixing (PM) was accomplished using Gilson P1000 and P200 pipettes (Dunstable, UK).
2.3. Manufacture of Lipid Nanoparticles (LNPs)
The aqueous phase consisted of poly A, Fluc mRNA, Green Lantern mRNA, or Cy5-Fluc mRNA diluted in 50 mM pH 5 citrate buffer to account for an N/P ratio of 6. The lipid phase consisted of DSPC, cholesterol, ionizable/cationic lipid (SM-102, D-Lin-MC3-DMA, DODAP, or DOTAP) and DMG-PEG2000 at a molar ratio of 10:38.5:50:1.5% dissolved in absolute ethanol (≥99.5%). LNPs were manufactured on the TM, IJM, or PM to achieve LNPs with a final payload concentration of 0.04 mg/mL and a final lipid concentration of 0.9 mg/mL. For TM and IJM, total flow rates (TFRs) were maintained at 15 mL/min, while the flow rate ratio (FRR) was kept constant at 3:1 (aqueous/lipid) for all mixers.
2.4. Purification of LNPs
Subsequent LNPs were purified using either dialysis or spin column chromatography. Dialysis purification consisted of LNPs being dialyzed against 10 mM pH 7.5 Tris buffer for 4 h using dialysis tubing with a molecular weight cut-off of 14 kDa. Spin column purification was carried out with LNPs being diluted 1:40 with 10 mM pH 7.5 Tris buffer, and centrifugal filtration was carried out using Amicon Ultra-15 columns at 2000× g 4 °C for approximately 10 min or until the desired volume was achieved.
2.5. LNP Characterization
The critical quality attributes (CQAs) of LNPs were monitored using Dynamic Light Scattering (DLS)/Electrophoretic Light Scattering (ELS). The z-average diameter (nm), polydispersity index, and zeta potential (mV) were measured using the Zetasizer Ultra (Malvern Panalytical, Ltd., Worcestershire, UK). The LNPs were diluted to a 0.1 mg/mL lipid concentration for analysis. Pre-purification measurements were diluted in citrate buffer (viscosity 1.28 cP and refractive index 1.47), post-purification measurements were diluted in Tris buffer (viscosity 1.0037 cP and refractive index 1.337), and zeta potential measurements were diluted in water (viscosity 0.8872 cP and refractive index 1.33).
Payload encapsulations were quantified using the Quanti-iTTM RiboGreenTM Assay Kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA. High (0–1000 ng) and low (0–50 ng) curves were prepared using the encapsulated payload to quantify the encapsulated material from Triton-treated and non-Triton-treated wells. Briefly, samples were diluted to a concentration of 3 μg/mL payload and mixed with either 50 μL of 2% Triton-TE or 1× TE buffer before being incubated for 15 min at 37 °C. Following on, a 200× and 500× RiboGreen solution was added to the Triton-treated and non-Triton-treated wells, respectively, to account for a 1:1 dilution. The fluorescence was then measured using the Glomax (Promega, Chilworth, UK) at excitation 475 nm and emission 500–550 nm.
2.6. Nanoparticle Tracking Analysis
Nanoparticle Tracking Analysis (NTA) was conducted using the NS300 equipped with a 488 nm laser block (Malvern Panalytical, Worcestershire, UK). Samples were diluted 1:5000 (PM) or 1:2500 (TM/IJM) in Tris buffer. Each sample was measured five times for 60 s at ambient temperature, with flow settings at a constant setting of 200, with a camera level of 9 and a screen gain of 6. Processing analysis was conducted using a detection threshold of 4, with the software version NTA 3.4 Build 3.4 003. The span parameter was calculated using ((D90-D10)/D50).
2.7. In Vitro Protein Expression of LNPs
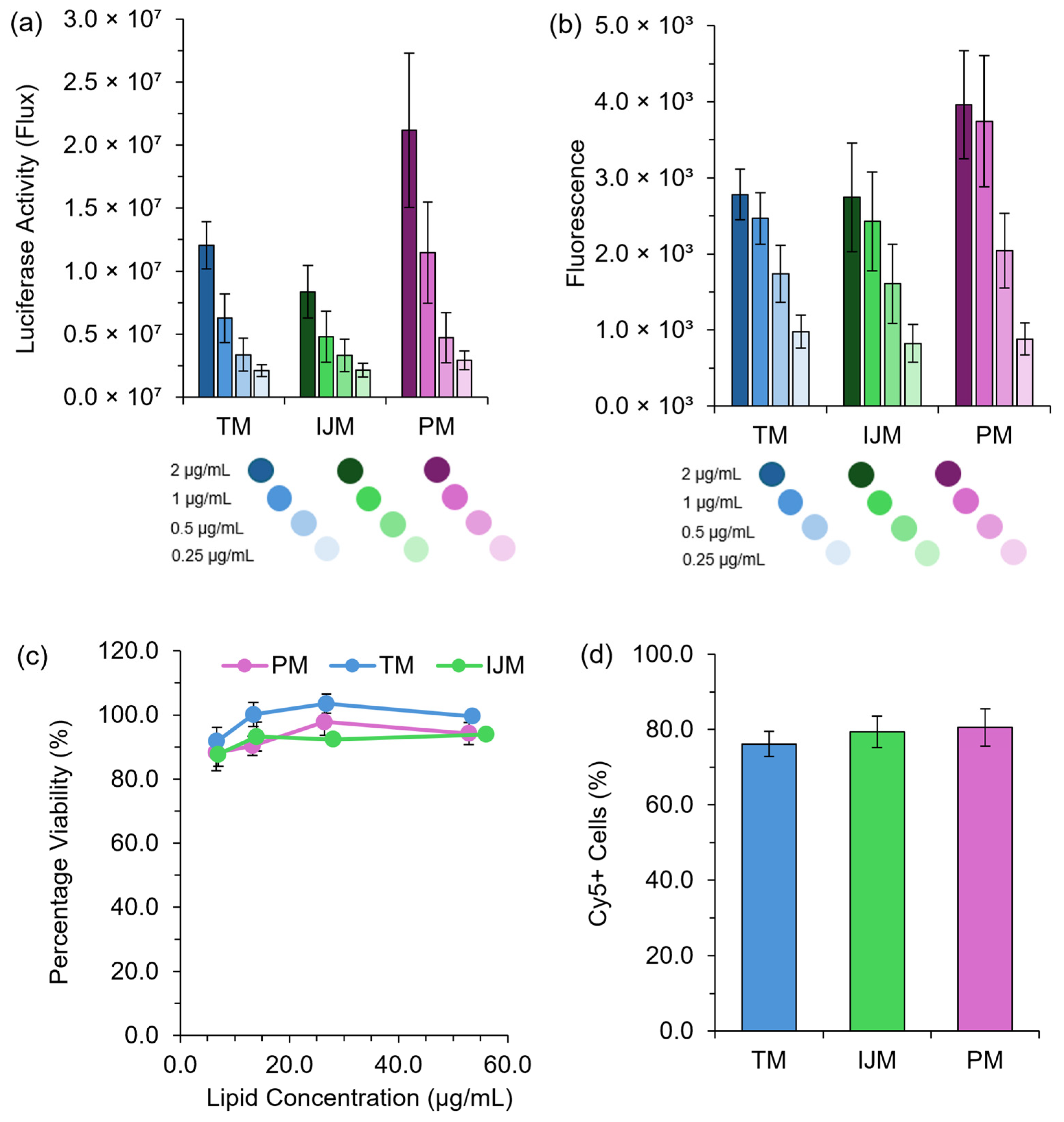
HEK-293 cells were seeded at 1.5 × 104 cells/well in either a 96-well white (Fluc) or black (Green Lantern) clear bottom plate in Minimal Essential Media (MEM) (supplemented with 10% FBS, 5% sodium pyruvate, and 5% penicillin streptomycin) and incubated at 37 °C (5% CO2) for 48 h. Following incubation, cells were treated with LNPs serially diluted to achieve mRNA concentrations of 2–0.25 μg/mL and left to incubate for a further 48 h at 37 °C (5% CO2). For Fluc-LNPs, to measure results, 100 μL of One-Glo luciferase was added to each well, and luminescence was measured using the Glomax (Promega, Chilworth, UK). For Green Lantern-LNPs, the fluorescence was measured using the Polarstar Omega Plate Reader (BMG LABTECH Ltd., Aylesbury, UK).
2.8. Measuring LNP Uptake Using Flow Cytometry
For LNP uptake, cells were plated in a 12-well plate at a concentration of 5 × 105 cells/well and incubated for 48 h at 37 °C (5% CO2). Then, 24 h pre-analysis, the dead cell control was treated with 0.1% Triton-MEM and 4 h pre-analysis, LNPs were added to the appropriate well at a mRNA concentration of 2 μg/mL. Sample preparation was carried out at room temperature, spent media was removed from wells and cells were washed with DBPS; TrypLE was added to all wells and cells were allowed to dissociate at 37 °C (5% CO2) for 2 min. After this, MEM was added to all wells, and samples were spun at 300× g for 5 min until a cell pellet was observed. The supernatant was removed, and the cell pellet was resuspended in DPBS, and where appropriate, 2 drops of propidium iodide were added. Samples remained at room temperature for 15 min before being transferred onto ice until analysis was complete. Flow cytometry measurements were done using the Attune NxT Flow Cytometer (Thermo Fisher) using the BL2 laser to monitor PI and the RL1 laser to monitor Cy5 fluorescence.
Flow cytometry analysis was gated based off of three control groups, unstained live cells (
Figure S1), stain live cells (
Figure S2) and stained dead cells (
Figure S3), once gating was appropriate for all three controls, analysis of samples was undertaken.
2.9. In Vivo Protein Expression Studies
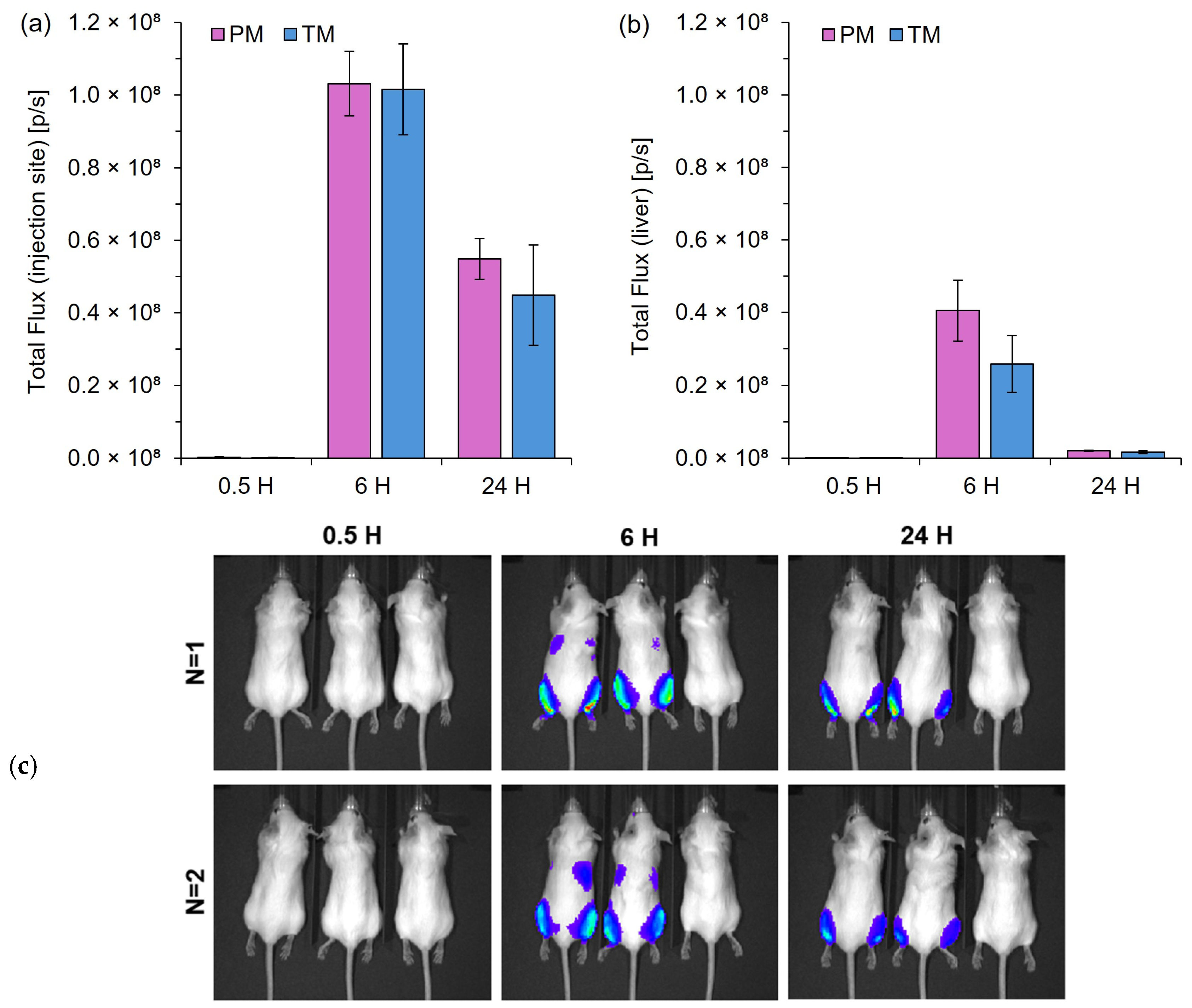
For comparing mixers, 14-week-old female BALB/c mice were intramuscularly injected with 50 μL/hind leg of LNPs, equating to 2 μg total Fluc mRNA. Then, 0.5 h post-injection, mice were subcutaneously injected with 100 μL of Vivo-Glo Luciferin, and after 10 min, bioluminescence signals were captured using an IVIS Spectrum imaging system (Revvity, Inc., Waltham, MA, USA). Bioluminescence signals were further captured at 0.5-, 6- and 24-h post-injection. For comparing formulations, 8-week-old male BALB/c mice were intramuscularly injected with 50 µL/hind leg of LNPs, equating to 2 µg total Fluc mRNA. Bioluminescence signals were captured as mentioned above, at 6- and 24-h post-injection. Bioluminescence signals were quantified using the Living Image 4.0 Software.
2.10. Cell-Free Protein Expression (CFPE)
A Rabbit Reticulocyte Lysate (RRL) system was used to analyze CFPE. Briefly, Fluc mRNA was extracted from LNPs by mixing 200 μL of sample with 750 μL ethanol and 25 μL sodium acetate at 3 M pH 5, before being centrifuged at 14,000 rpm for 20 min. Ethanol precipitation was carried out twice, and the resulting mRNA pellet was resuspended in 35 μL of RNAse-free water. Extracted mRNA was then used to complete reaction mixtures consisting of 35 μL rabbit reticulocyte lysate, 0.5 μL of both amino acid mixtures minus leucine or methionine, 1 μL RNasin® Ribonuclease Inhibitor, 2 μL of extracted mRNA, control mRNA (positive) or no mRNA (negative), and made up to a final volume of 50 μL with ultrapure water. The reaction mixtures were incubated at 30 °C for 1.5 h, and the reaction was terminated by placing them on ice. To measure protein expression, reaction mixtures were mixed with an equal volume of ONEGlo luciferase and luminescence was measured on the Glomax (Promega).
2.11. High Performance Liquid Chromatography–Evaporated Light Scattering Detector (HPLC-ELSD)
Lipid recovery was measured using the Shimadzu HPLC-ELSD III system. LNPs were injected at a volume of 10 μL at a flow rate of 0.5 mL/min, and lipids were separated using a Waters Column XBridge® C18 Column (internal diameter 3 mm, column length 150 mm, pore size 130 Å, and 3.5 μm particle size) with a detection temperature of 50 °C and a column oven temperature of 45 °C. Mobile phase A consisted of 0.01 M Triethylamine acetate and water, while mobile phase B consisted of 0.01 M Triethylamine acetate and ethanol. The run time was 15 min with a binary gradient, with mobile phase B increasing from 70 to 100% from 0 to 7 min with a drop back to 70/30% B/A gradient at 12.1 min. Samples were diluted 1:5 (v/v) with mobile phase B before analysis. Concentration curves were developed for each lipid, and all analysis was done using the Shimadzu post-run software v5.124 SP1.
2.12. Frit-Inlet Asymmetric Flow Field-Flow Fractionation (FI-AF4) Multiplexed with Multiangle Light Scattering, Dynamic Light Scattering, and UV Detection
The asymmetric flow field-flow fractionation module (AF2000, Postnova Analytics, Malvern, UK) was hyphenated with an inline multidetector (MD), multiangle light scattering (MALS-PN3621, Postnova Analytics), UV detector (PN3242, 260 nm, 0.5 sensitivity—Postnova Analytics), and DLS Zetasizer Nano ZS System (Malvern Panalytical, Worcestershire, UK). A frit-inlet channel (300 mm × 60 mm × 40 mm) used a 350 μm spacer, 10 kDa MWCO amphiphilic regenerated cellulose membrane and 10 mM pH 7.5 Tris as the eluent liquid for separation at 25 °C. The FI-AF4-MD methodology was used from Davidson et al. [
16] with a cross-flow (XF) initial hold of 15 min at 0.75 mL/min, followed by a 60 min exponential XF decay from 0.75 mL/min to 0 mL/min and a hold of 0 mL/min for 5 min; throughout separation, a detector flow (DF) of 0.3 mL/min was used.
PM, TM, and IJM LNP samples were diluted to 0.5 mg/mL lipid concentration in eluent liquid prior to analysis. A 20 μL sample was injected using a 100 μL injection loop. The sample recovery, radius of gyration (Rg) from the MALS detector, hydrodynamic radius (Rh) from the inline DLS, and shape factor were calculated as per Abdulrahman et al. [
17]. The collected MALS data were analyzed using the AF2000 software v2.2.01 (Postnova Analytics, Germany), which uses a spherical fit model across 52°–124° angles.
2.13. High-Throughput Formulation of LNPs Using Automated Platform
The aqueous phase consisted of Green Lantern mRNA diluted in 5 mM pH 4.5 citrate buffer. The mRNA concentration was kept at 0.05 mg/mL in all formulations. The lipid phase again consisted of DSPC, cholesterol, and DMG-PEG2000 and SM-102, D-Lin-MC3-DMA, DODAP, or DOTAP at a molar ratio of 10:38.5:50:1.5% dissolved in ethanol. The lipids and mRNA solutions were mixed in a robotic liquid handling system (JANUS G3 automated workstation). A robotic liquid handling system prepared the lipid mixture and mRNA dilutions for the high-throughput process. The mixing volume ratio of the aqueous/lipids in ethanol was maintained at 3:1, and the pipetting speed was set at 600 µL/s. LNP formulations were diluted 10-fold in Tris sucrose buffer to achieve a final concentration of 3.75 µg/mL mRNA.
2.14. Physicochemical Characterization of LNPs by Automation
Critical quality attributes (CQAs) of LNPs were monitored using Dynamic Light Scattering (DLS). The z-average diameter (nm) and polydispersity index of LNPs were measured in a 96-well plate using a DynaPro plate reader II (Wyatt Technology UK Ltd., Wilmslow, UK). LNPs were diluted 10-fold in Tris buffer using a CyBio FeliX liquid handler (Analytik Jena UK Ltd., London, UK) for size measurement. Solvent parameters for sucrose 2% were applied for analysis, including a viscosity of 1.055 cP and refractive index of 1.336 (25 °C).
The payload encapsulation efficiency (EE%) was quantified using the Quanti-iTTM RiboGreenTM RNA Assay Kit (InvitrogenTM) following the manufacturer’s instructions and adapted to be performed on a CyBio FeliX liquid handler (Analytik Jena), integrated with a PreciseFlex 750 robotic arm, STX44 incubator (LiCONiC, Montabaur, Germany), labware hotel, barcode reader, and Synergy H1 microplate reader (BioTek, Winooski, VT, USA). A high (0–1000 ng) standard curve was prepared using the encapsulated payload to quantify RNA from Triton-treated and non-Triton-treated wells. Briefly, samples were diluted to a concentration of 1 µg/mL mRNA and mixed with 50 µL of either 2% Triton-TE or 1× TE buffer before incubation at 37 °C for 15 min. Following incubation, a 200× RiboGreen solution was added to all wells for a final 1:1 dilution. Fluorescence was then measured at an excitation of 485 nm and an emission of 535 nm.
2.15. High-Throughput In Vitro Assay
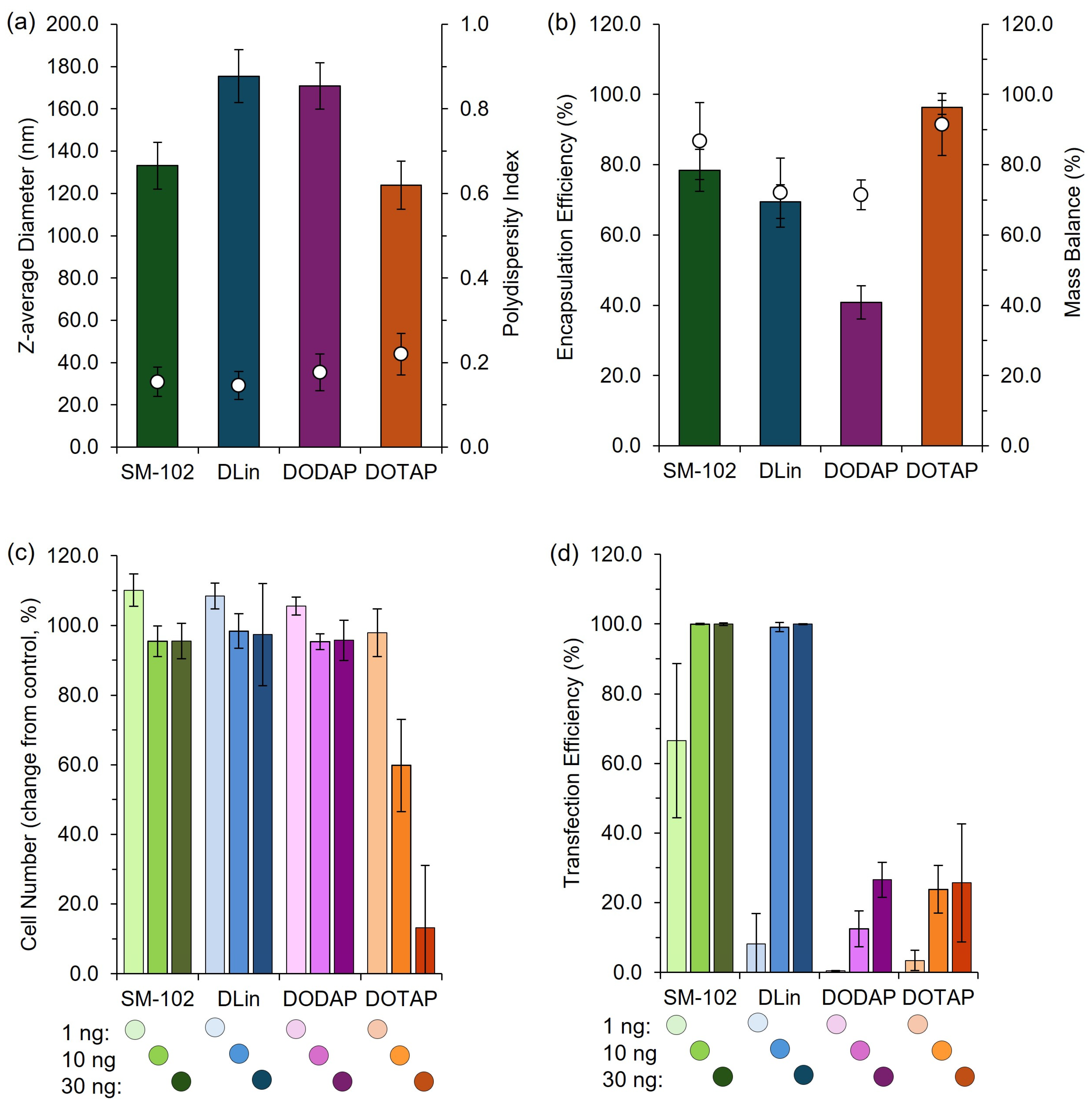
A high-throughput cell-based in vitro assay for interrogation of LNP efficacy was developed by the IDDC consortium to support formulation screening. LNPs encapsulating mGreenLantern mRNA were dosed using an Echo liquid handler at 1 ng, 10 ng, or 30 ng mRNA per well in a 384-well plate (Greiner µCLEAR®, Nürtingen, Germany). HeLa cells, which were previously modified by lentiviral transduction to express nuclear-localized mKate2, were seeded on top of LNPs at 2 × 103 cells per well in FluoroBrite DMEM (Gibco, Waltham, MA, USA) containing 10% fetal bovine serum (Sigma, St. Louis, MO, USA), GlutaMAX™ Supplement (Gibco) and 1% penicillin–streptomycin (Sigma), and incubated at 37 °C (5% CO2) for 42 h. Following incubation, cells were fixed with 4% paraformaldehyde and imaged using an Opera Phenix high-content screening system equipped with a 10 × 0.3 NA air objective and 488 nm and 561 nm laser lines. Images were analyzed in Harmony. Automated segmentation pipelines were constructed to identify nuclei and cytosolic mGreenLantern expression with single-cell resolution. The cell number was defined as the number of detected nuclei per well, and transfection efficiency as the proportion of these cells positive for mGreenLantern expression. Data were normalized to internal controls before plotting in Microsoft Excel (Version 16.84, Microsoft Corporation, Redmond, WA, USA).
2.16. Cryogenic Transmission Electron Microscopy
Fluc-LNP samples were prepared as normal and then concentrated to 0.08 mg/mL mRNA. Glow discharge was carried out on Lacey carbon film, 300 mesh copper grids (Agar Scientific, Rotherham, UK) for 30 s at 30 mA using the Emitech K100X (Quorum Technologies Ltd., Laughton, UK). A total of 3 μL of the LNP samples were blotted on either Whatman 597 paper using the Thermo Fisher Vitroblot Mk IV (25 °C, 95% humidity, blot time 3 s, and blot force 13 or 12) or Whatmann 1 paper using the Leica EM GP2 plunge-freezer before being plunge frozen in liquid ethane. Samples were cooled in liquid nitrogen and held in a Gatan Elsa cryo-specimen holder until being transferred to the JEOL F2-Cryo (200 kV) with Direct Electron DE-20 camera for image capture. The acquisition software was SerialEM version 4.1, with images being processed on Relion 5.0.0 and Image J.JS v0.5.8.
2.17. Ethics Statement
Animal experiments and experimental procedures were carried out in accordance with UK Home Office regulations and the University of Strathclyde Animal Welfare and Ethical Review Board regulations under project license number PPL PP1650440. BALB/c mice were bred and maintained in the Biological Procedures Unit at the University of Strathclyde, Glasgow, and experiment design and reporting adhered to the ARRIVE guidelines.
2.18. Statistical Analysis
All results represent the mean ± standard deviation of three independent replicates. Statistical significance was determined using an ANOVA with Tukey’s post hoc test using Minitab software (v19.2). For in vivo studies, 2–3 mice were used for each condition to give 4–6 regions of interest for the analysis of the injection site and 2–3 regions of interest for the analysis of livers, with results representing the mean ± standard error of the mean (SEM). No data points were excluded.
4. Discussion
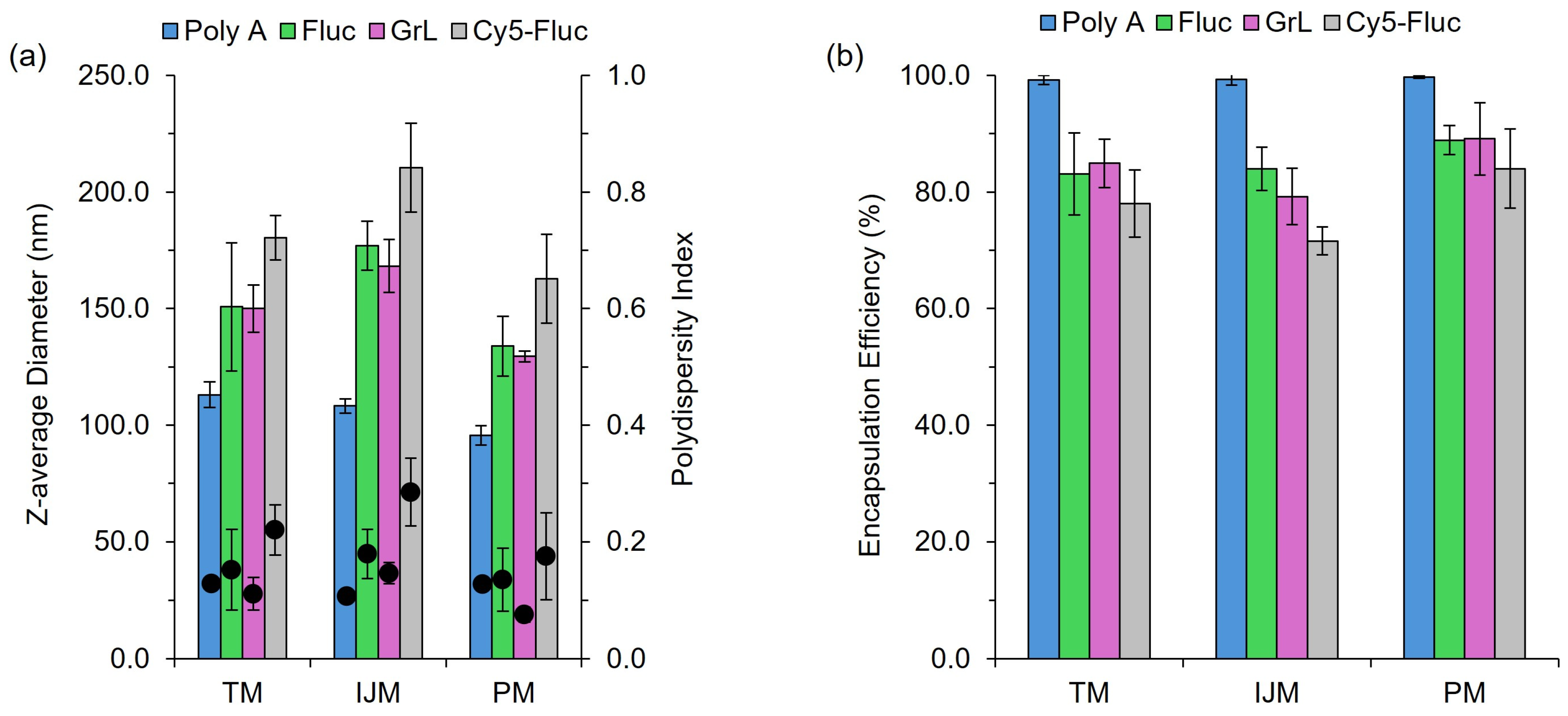
Our research demonstrates that low-cost microfluidic mixers can reliably produce LNPs, highlighting their potential as effective tools for affordable small-scale production and high-throughput screening applications. We evaluated three mixing methods: TM, IJM, and PM. While PM may lack precise control over CPPs and is not scalable, it proved effective for small-scale LNP manufacture. Characterization studies showed that CQAs were influenced more by payload (
Figure 2) than the mixing method (
Figure 3,
Figure 4,
Figure 5,
Figure 6,
Figure 7 and
Figure 8); for example, poly A yielded smaller LNPs with higher encapsulation efficiencies, whereas mRNA constructs increased particle size. This is likely because poly A is a small, linear molecule compared to mRNA, which can exhibit enhanced secondary structures that may lead to a less tightly packed LNP structure. Cy5-Fluc mRNA also increased the LNP size, possibly due to interactions with the fluorescent tag and lipid components.
We also observed a decrease in encapsulation for LNPs containing Cy5-Fluc (
Figure 2), possibly due to the Cy5-tag interfering with the RiboGreen assay. Specifically, Cy5 has an excitation/emission spectrum of approximately 650/670 nm, while Ribogreen operates at approximately 500/525 nm; this spectral overlap suggests that Cy5 has the potential to fluorescently quench the RiboGreen signal, thereby affecting the fluorescence readout. To mitigate this effect, Cy5-Fluc mRNA was used during the assay to build the calibration curves. While not fully quantified in this study, this potential interference should be considered when interpreting encapsulation data involving fluorescently labelled RNA. Fluorescent tags, particularly cyanine dyes, are widely used in LNP and liposomal research and generally do not significantly affect particle characteristics [
20]. For example, Ma et al. investigated in vivo mRNA delivery using SM-102-LNPs tagged with Cy5 and DiR, reporting sizes around 100 nm [
21]. However, their LNPs were produced at a higher flow rate and purified via dialysis. In contrast, we used spin column purification in our studies, introducing a rapid pH change and high centrifugal forces that may contribute to these differences. Schober et al. also examined the impact of payload size in DLin- and ALC-0315-LNPs, finding no significant effect on size [
22]. Therefore, the low-cost mixers tested in our studies may be more sensitive to nucleic acid payload, which may also explain differences in encapsulation efficiency.
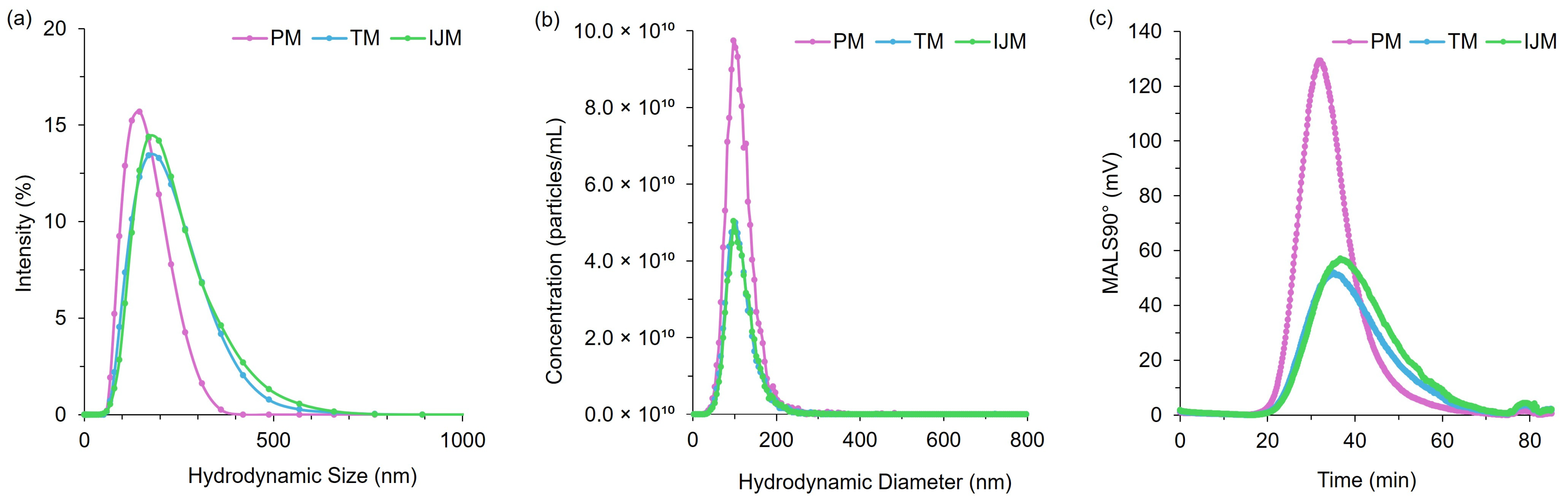
It is generally recognized that LNP size is a critical factor in determining biodistribution and transfection efficiency, and previous studies have shown that various mixing methods can significantly influence particle size. To investigate this, we used orthogonal characterization techniques to investigate the physicochemical properties of the LNPs produced by different mixers. DLS measurements showed that PM LNPs had smaller hydrodynamic diameters and lower PDI than TM and IJM mixers (
Table 2 and
Figure 6). While NTA showed minimal differences in mean particle size across the mixers, PM LNPs displayed a higher particle concentration and narrower size span, indicating a more uniform population (
Table 3 and
Figure 6). AF4-MALS analysis supported these findings, with PM LNPs exhibiting lower Rg values and narrower elution profiles, indicating a more compact structure (
Table 4 and
Figure 6). Shape factor analysis (
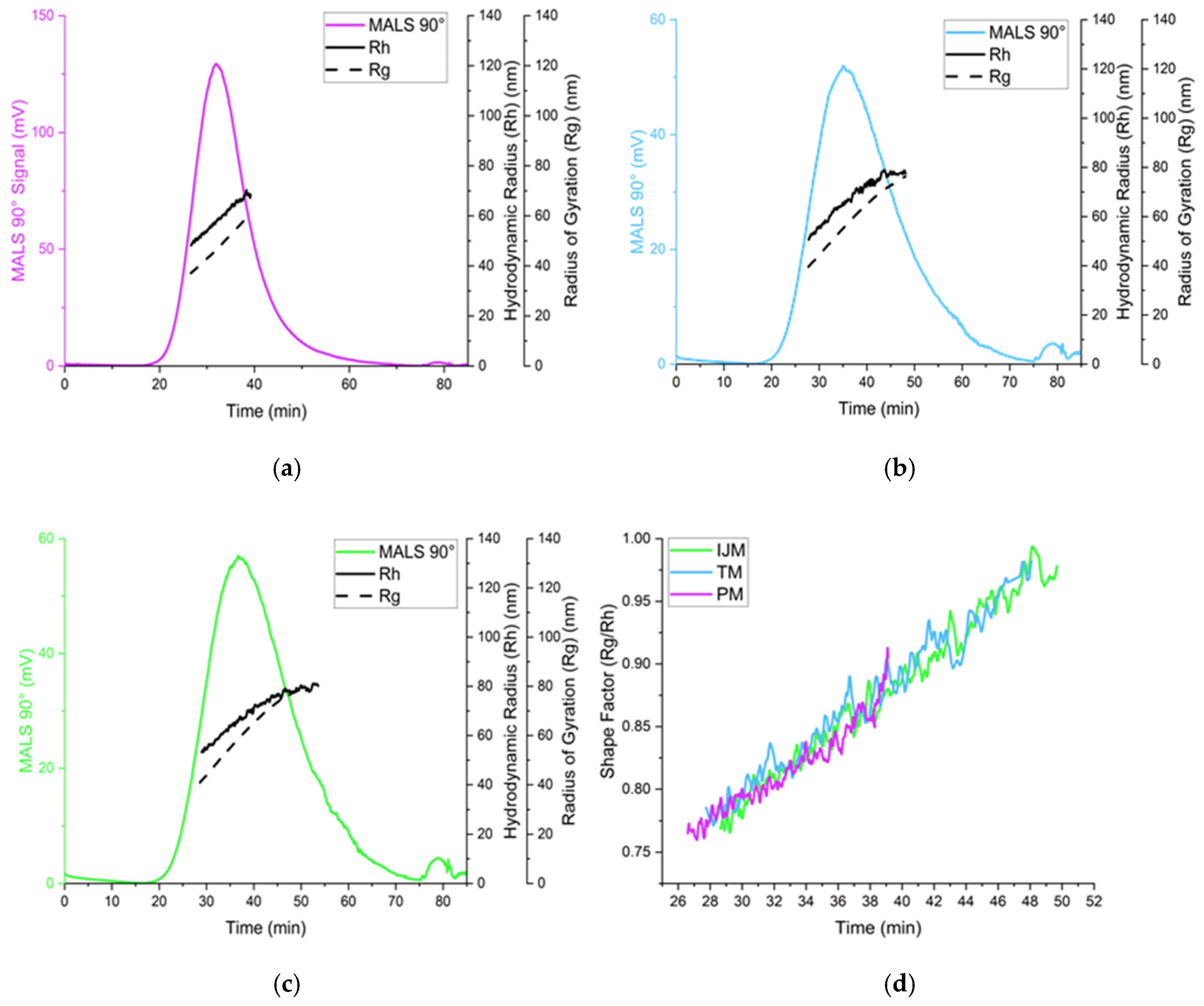
Table 4 and
Figure 7) showed all LNPs were generally spherical (Rg/Rh ~0.775), but TM and IJM particles displayed shape factor that increases over time, suggesting some elongation or fusion events. These differences were not visually evident from cryo-TEM images (
Figure 8), which showed similar external morphology and bleb features across all samples. This highlights the importance of employing complementary characterization techniques to fully understand LNP behavior, as cryo-TEM alone does not easily show population-level differences.
Our findings are generally consistent with the existing literature regarding the impact of mixer choice. For instance, Misra et al. produced SM-102-LNPs using a confined impingement jet system, reporting sizes around 135 nm with high encapsulation efficiency (>95%), which is smaller than the IJM-generated LNPs in this study [
23]. However, these LNPs were manufactured under different conditions and included higher PEG content (2 mol%), which has been shown to reduce LNP size [
14,
24]. Similarly, Subraveti et al. produced Dlin-MC3-DMA-LNPs using a confined impingement jet mixer, achieving sizes below 100 nm at physiological pH; however, their study also used dialysis purification and had a different lipid composition, which may have contributed to smaller particle sizes [
25]. These studies suggest that IJM-generated LNPs can be tuned to smaller sizes by carefully optimizing CPPs, such as the flow rate ratio, lipid composition, and purification method.
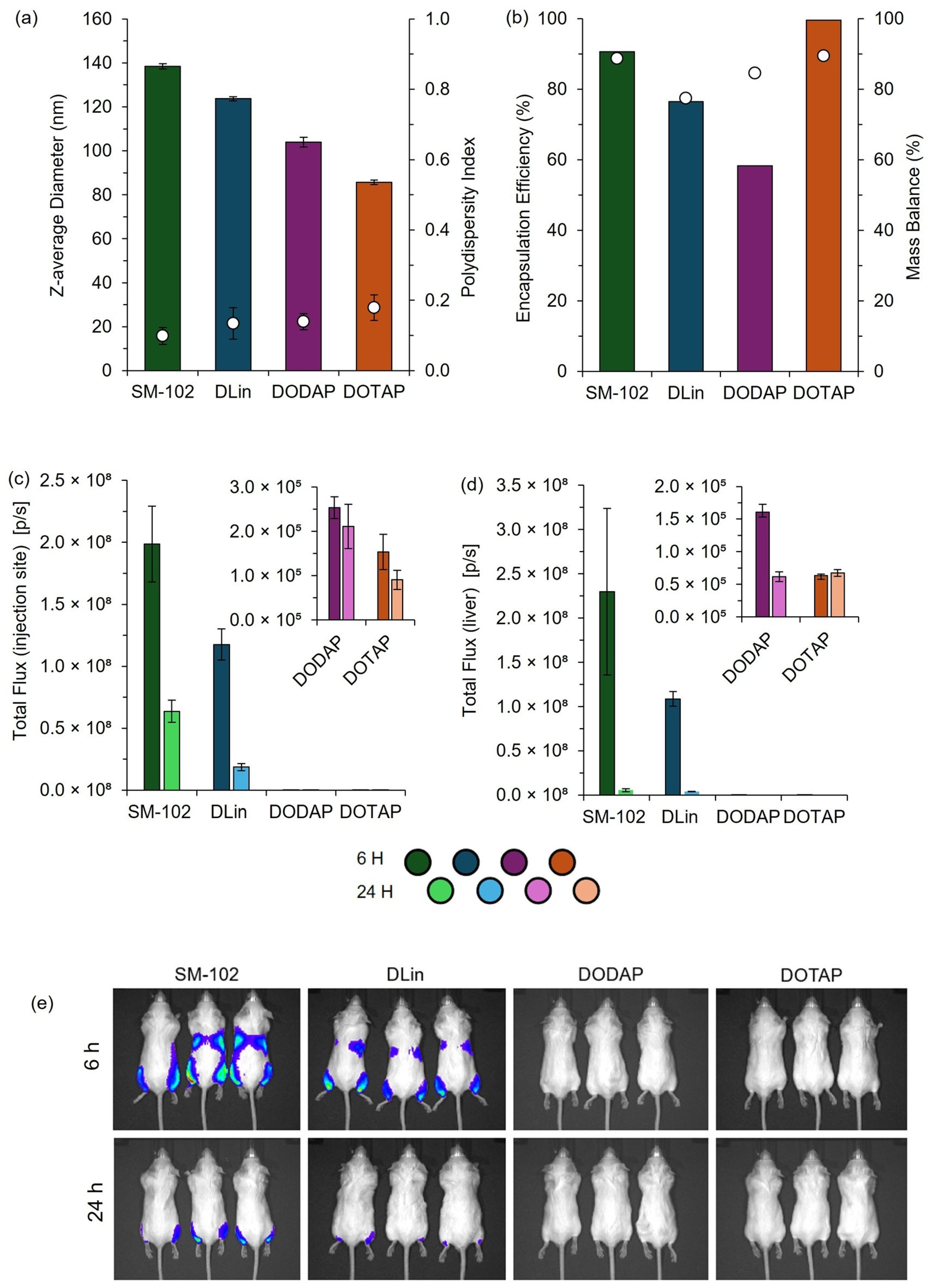
Further studies have investigated the impact of size on LNP biodistribution and expression efficiency. Di et al. reported that LNPs ≥ 180 nm exhibited reduced liver expression, while McMillan et al. demonstrated that larger LNPs correlated with significantly decreased in vivo expression [
14,
26]. Given that TM and PM produced more comparable sizes, they were prioritized for in vivo testing, aligning with previous reports that T-mixed LNPs typically range from 120 to 130 nm [
27]. Our results showed no significant difference in vivo expression between the TM and PM LNPs; however, studies comparing T-junction and chaotic mixers suggest that chaotic mixing methods may improve LNP efficiency. For example, Jürgens et al. found that staggered herringbone mixers (SHMs) outperformed T-mixing for mRNA LNPs [
27]. This effect could be attributed to chaotic mixers typically producing smaller LNPs, which could increase uptake as size-dependent effects on uptake are generally more pronounced at sizes below 100 nm [
28].
There are various reports on the performance of pipette-mixed LNPs. For example, Wang et al. found that pipette-mixed MC3-LNPs had larger sizes (~162 nm) and lower mRNA expression than microfluidic-generated LNPs [
29]. In contrast, Strelkova et al. demonstrated that manually mixed LNPs could outperform some microfluidic-generated formulations when process parameters were varied [
30]. These results, combined with the results we present, confirm that PM is a useful, low-cost option.
To further validate the use of pipette mixing as part of a larger high-throughput pipette mixing system, both high-throughput and manual pipette mixing were employed to generate clinically relevant cationic and ionizable LNPs for in vitro and in vivo testing. The observed expression profiles aligned with previous reports, with expression increasing from DOTAP > DODAP > DLin > SM-102, consistent with the known efficiency of ionizable lipids (DLin and SM-102) versus cationic lipids (DOTAP and DODAP) in nucleic acid delivery [
19]. While PM LNPs exhibited larger sizes, they maintained comparable expression profiles to published data, supporting PM as an effective tool for rapid LNP screening.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}