In Vitro Metabolism of DWP16001, a Novel Sodium-Glucose Cotransporter 2 Inhibitor, in Human and Animal Hepatocytes

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Metabolic Stability
2.3. Metabolite Profiling in Human and Animal Hepatocytes
2.4. Screening of CYP and UGT Enzymes Responsible for the Metabolism of DWP16001
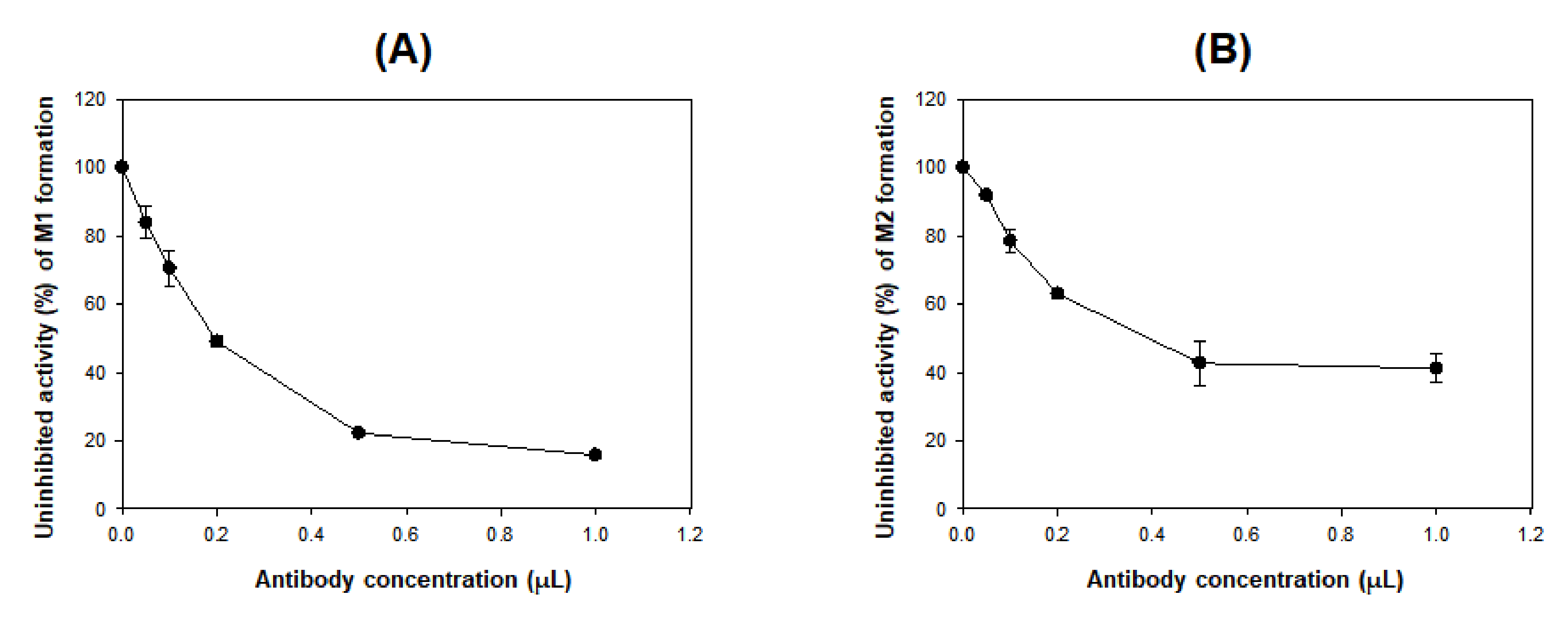
2.5. Immunoinhibition of DWP16001 Metabolism to M1 and M2 by anti-CYP3A4 Antibody
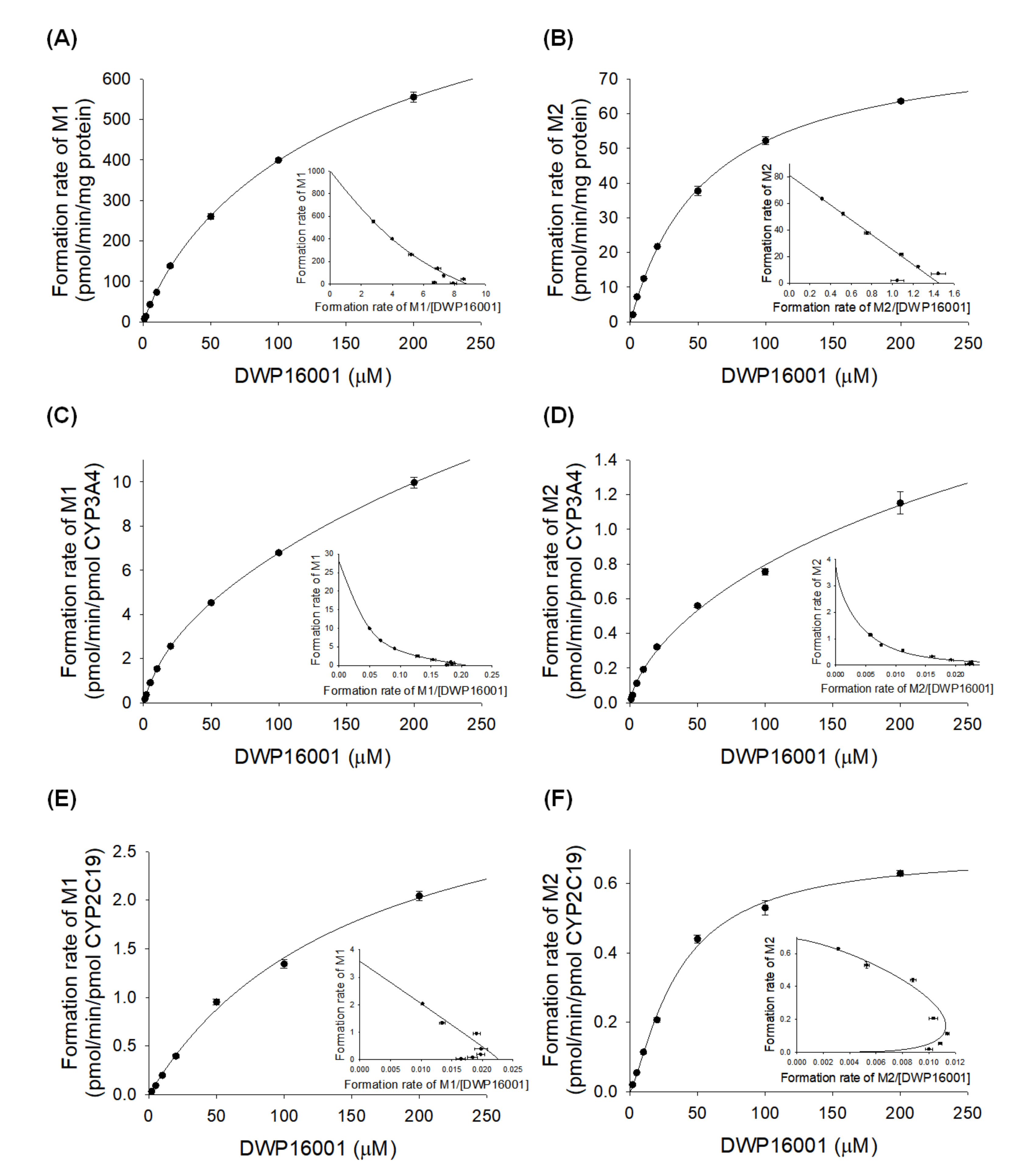
2.6. Enzyme Kinetics for the Formation of M1 and M2 from DWP16001 in Human Liver Microsomes and cDNA-Expressed CYP3A4 or CYP2C19 Supersomes
2.7. LC-MS Analysis
3. Results
3.1. Metabolic Stability of DWP16001 in Hepatocytes
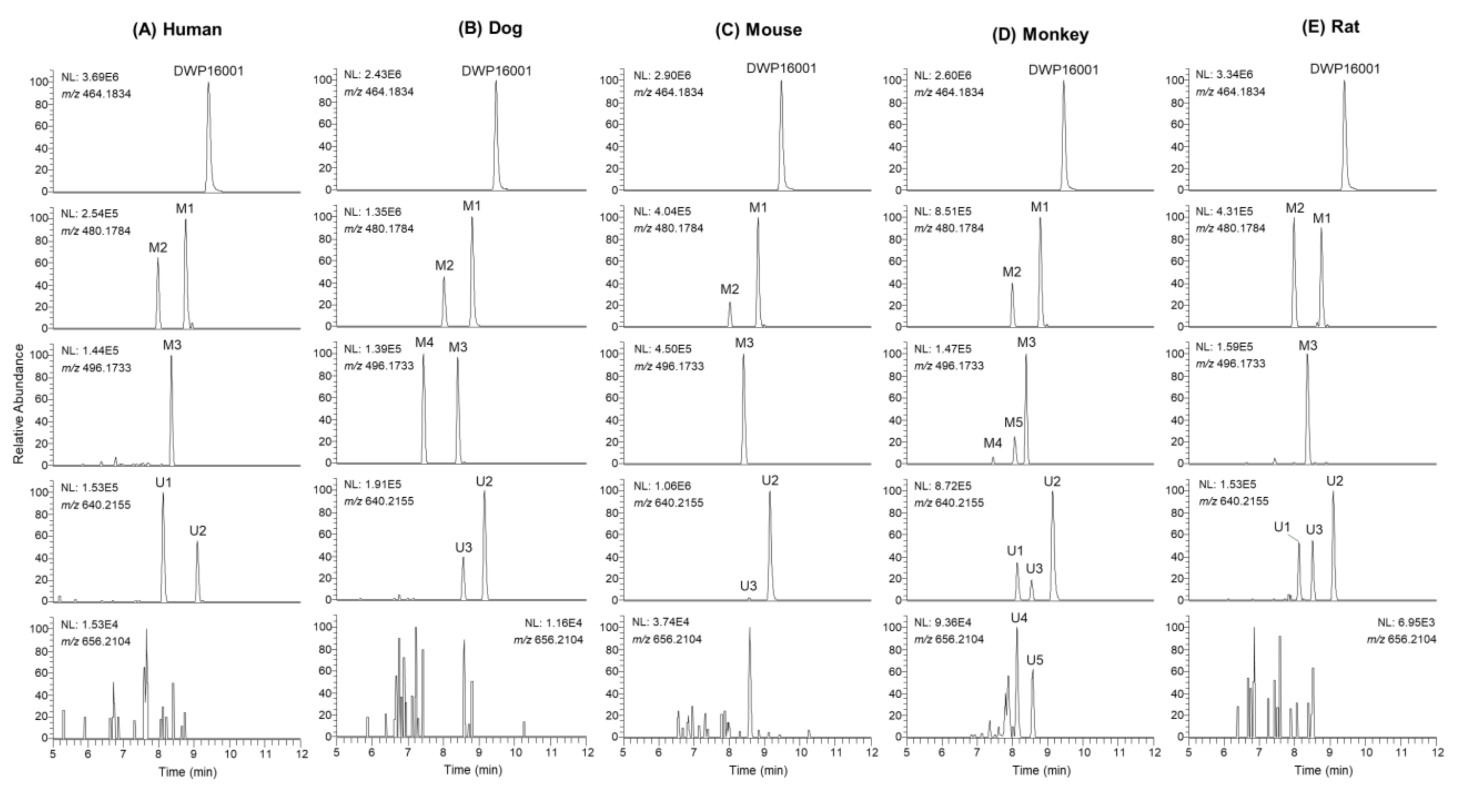
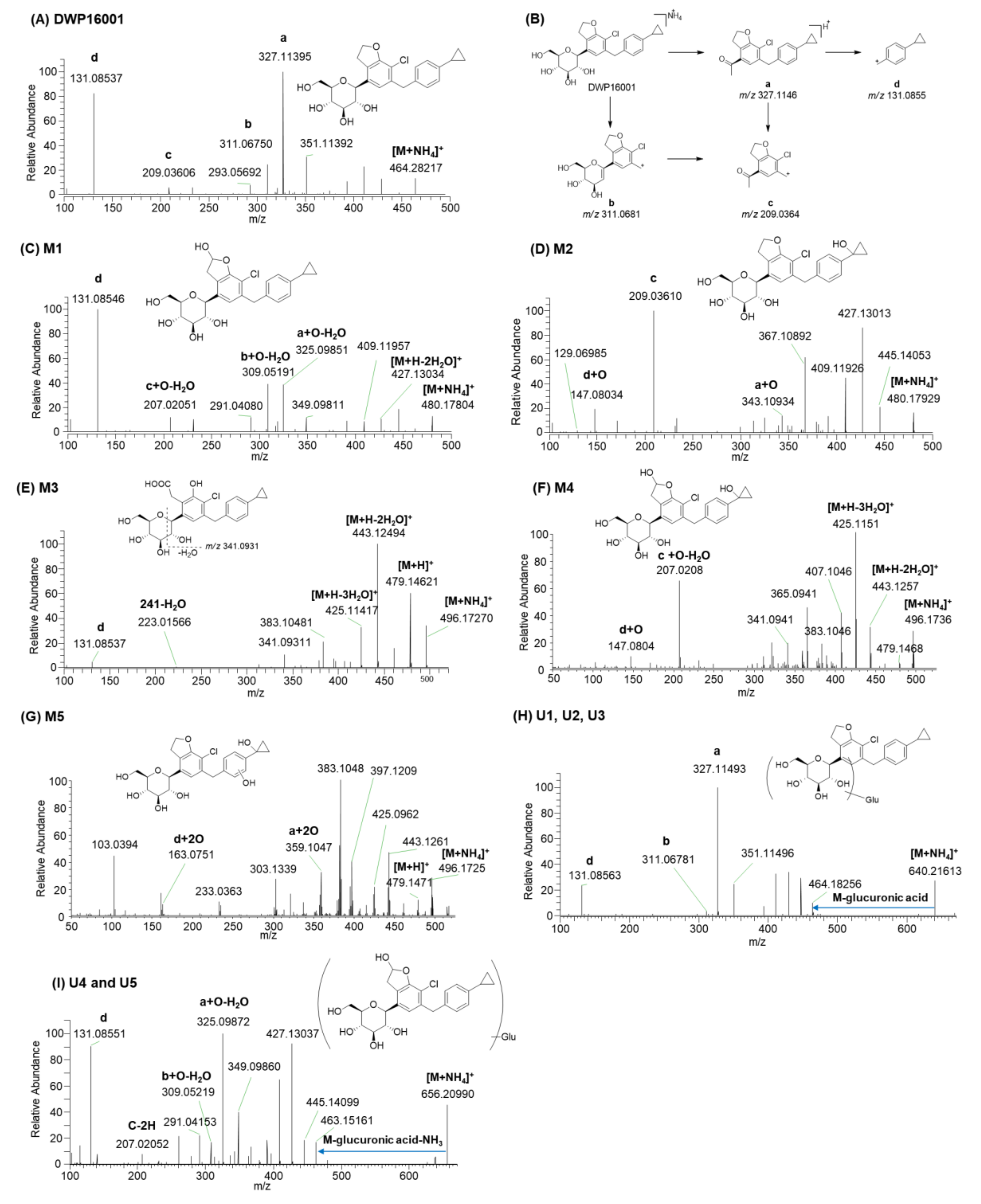
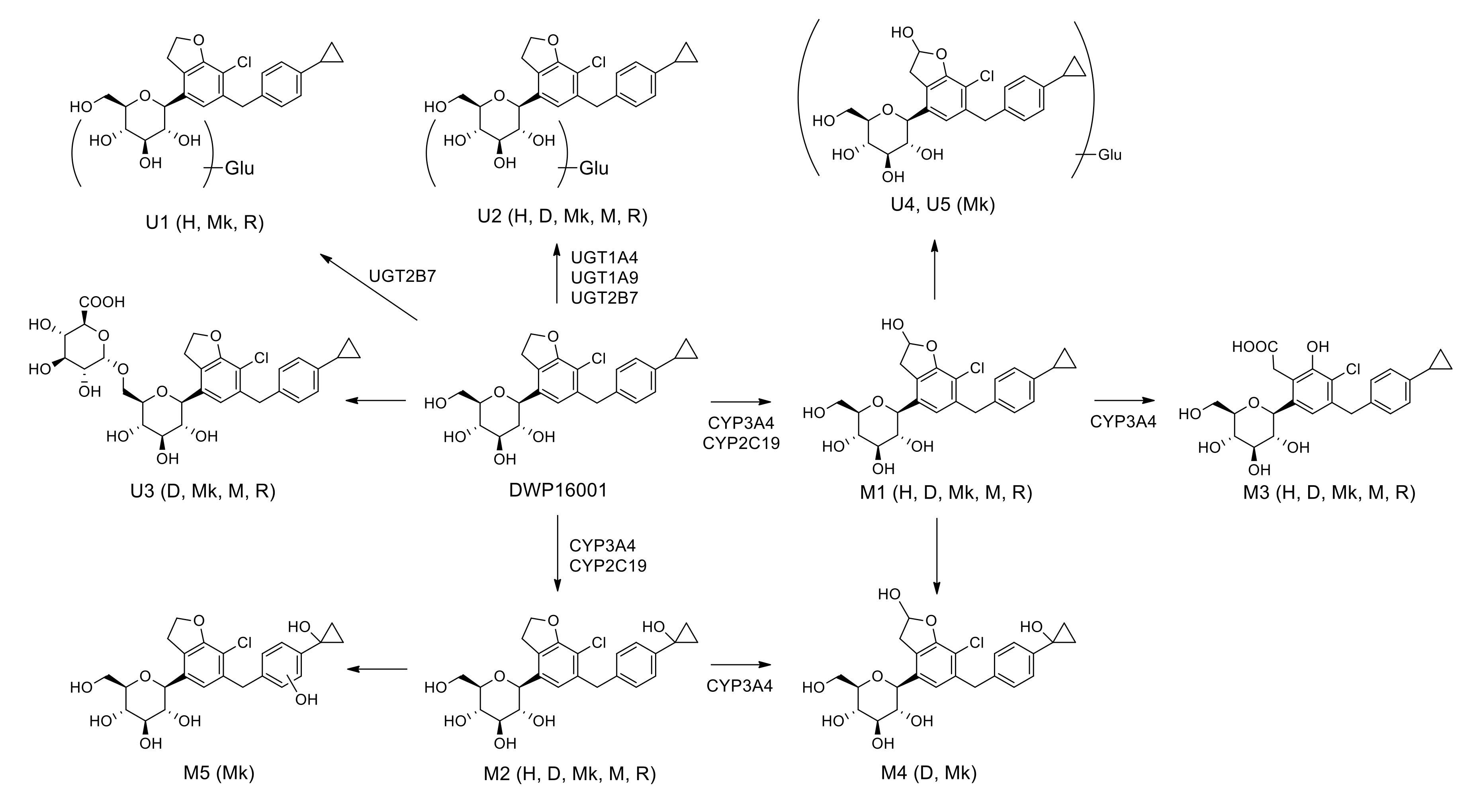
3.2. Metabolite Identification of DWP16001 in Hepatocytes
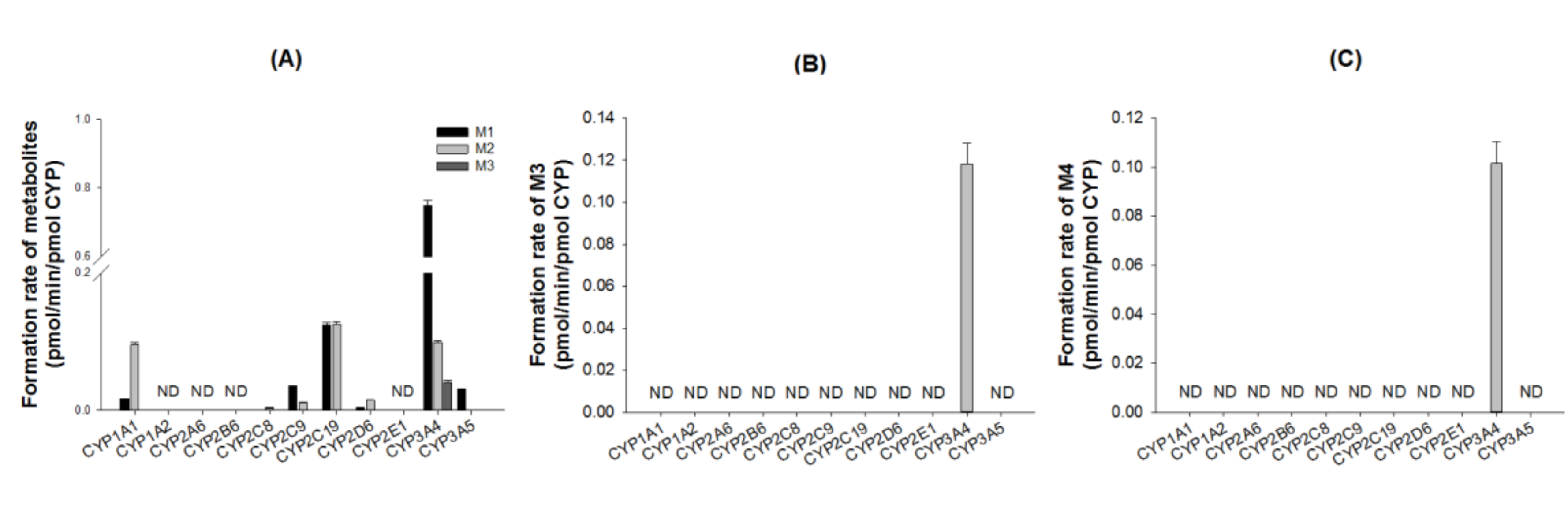
3.3. Characterization of Drug-Metabolizing Enzymes Responsible for DWP16001 Metabolism
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; IDF: Brussels, Belgium, 2019. [Google Scholar]
- Tat, V.; Forest, C.P. The role of SGLT2 inhibitors in managing type 2 diabetes. JAAPA 2018, 31, 35–40. [Google Scholar] [CrossRef]
- Tahara, A.; Takasu, T.; Yokono, M.; Imamura, M.; Kurosaki, E. Characterization and comparison of sodium-glucose cotransporter 2 inhibitors: Part 2. Antidiabetic effects in type 2 diabetic mice. J. Pharmacol. Sci. 2016, 131, 198–208. [Google Scholar] [CrossRef]
- Ito, H.; Shinozaki, M.; Nishio, S.; Abe, M. SGLT2 inhibitors in the pipeline for the treatment of diabetes mellitus in Japan. Expert Opin. Pharmacother. 2016, 17, 2073–2084. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A. Update on developments with SGLT2 inhibitors in the management of type 2 diabetes. Drug Des. Dev. Ther. 2014, 8, 1335–1380. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2018, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Shaw, W.; Barrett, T.D.; Weidner-Wells, M.; Deng, H.; Matthews, D.R.; et al. Canagliflozin and renal outcomes in type 2 diabetes: Results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol. 2018, 6, 691–704. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Garcia-Ropero, A.; Mancini, D.; Pinney, S.P.; Contreras, J.P.; Fergus, I.; Abascal, V.; Moreno, P.; Atallah-Lajam, F.; Tamler, R.; et al. Rationale and design of the EMPA-TROPISM trial (ATRU-4): Are the “Cardiac Benefits” of empagliflozin independent of its hypoglycemic activity? Cardiovasc. Drugs Ther. 2019, 33, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ropero, A.; Badimon, J.J.; Santos-Gallego, C.G. The pharmacokinetics and pharmacodynamics of SGLT2 inhibitors for type 2 diabetes mellitus: The latest developments. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1287–1302. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-K.; Nam, S.J.; Ji, H.-Y.; Park, M.J.; Choi, J.-S.; Song, I.-S. Comparative pharmacokinetics and pharmacodynamics of a novel sodium-glucose cotransporter 2 inhibitor, DWP16001, with dapagliflozin and ipragliflozin. Pharmaceutics 2020, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.S.; Dalvie, D.K. Cytochrome P450 and non-cytochrome P450 oxidative metabolism: Contributions to the pharmacokinetics, safety and efficacy of xenobiotics. Drug Metab. Dispos. 2016, 44, 1229–1245. [Google Scholar] [CrossRef] [PubMed]
- Cerny, M.A. Prevalence of non-cytochrome P450-mediated metabolism in Food and Drug Administration-approved oral and intravenous drugs: 2006–2015. Drug Metab. Dispos. 2016, 44, 1246–1252. [Google Scholar] [CrossRef]
- Hwang, D.; Kim, J.-H.; Shin, Y.; Choi, W.G.; Kim, S.; Ho, Y.-Y.; Lee, J.Y.; Kang, H.C.; Lee, H.S. Identification of catalposide metabolites in human liver and intestinal preparations and characterization of the relevant sulfotransferase, UDP-glucuronosyltransferase, and carboxylesterase enzymes. Pharmaceutics 2019, 11, 355. [Google Scholar] [CrossRef]
- Bohnert, T.; Gan, L.S. The role of drug metabolism in drug discovery. In Enzyme Inhibition in Drug Discovery and Development: The Good and the Bad; Chuang Lu, A.P.L., Ed.; Wiley: Hoboken, NJ, USA, 2010; pp. 91–176. [Google Scholar]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Atkinson, A.J., Jr.; Kushner, W. Clinical pharmacokinetics. Ann. Rev. Pharmacol. Toxicol. 1979, 19, 105–127. [Google Scholar] [CrossRef]
- Mamidi, R.N.V.S.; Cuyckens, F.; Chen, J.; Scheers, E.; Kalamaridis, D.; Lin, R.; Silva, J.; Sha, S.; Evans, D.C.; Kelley, M.F.; et al. Metabolism and excretion of canagliflozin in mice, rats, dogs, and humans. Drug Metab. Dispos. 2014, 42, 903–916. [Google Scholar] [CrossRef]
- Obermeier, M.; Yao, M.; Khanna, A.; Koplowitz, B.; Zhu, M.; Li, W.; Komoroski, B.; Kasichayanula, S.; Discenza, L.; Washburn, W.; et al. In vitro characterization and pharmacokinetics of dapagliflozin (BMS-512148), a potent sodium-glucose cotransporter Type II inhibitor, in animals and humans. Drug Metab. Dispos. 2010, 38, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Nucci, G.; Amin, N.; Sharma, R.; Mascitti, V.; Tugnait, M.; Vaz, A.D.; Callegari, E.; Kalgutkar, A.S. Pharmacokinetics, metabolism, and excretion of the antidiabetic agent ertugliflozin (PF-04971729) in healthy male subjects. Drug Metab. Dispos. 2013, 41, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Miyata, A.; Hasegawa, M.; Hachiuma, K.; Mori, H.; Horiuchi, N.; Mizuno-Yasuhira, A.; Chino, Y.; Jingu, S.; Sakai, S.; Samukawa, Y.; et al. Metabolite profiling and enzyme reaction phenotyping of luseogliflozin, a sodium–glucose cotransporter 2 inhibitor, in humans. Xenobiotica 2017, 47, 332–345. [Google Scholar] [CrossRef]
- Chen, L.-Z.; Jungnik, A.; Mao, Y.; Philip, E.; Sharp, D.; Unseld, A.; Seman, L.; Woerle, H.-J.; Macha, S. Biotransformation and mass balance of the SGLT2 inhibitor empagliflozin in healthy volunteers. Xenobiotica 2015, 45, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Kasichayanula, S.; Liu, X.; LaCreta, F.; Griffen, S.C.; Boulton, D.W. Clinical Pharmacokinetics and Pharmacodynamics of Dapagliflozin, a selective inhibitor of sodium-glucose co-transporter Type 2. Clin. Pharmacokinet. 2014, 53, 17–27. [Google Scholar] [CrossRef]
- Meng, W.; Ellsworth, B.A.; Nirschl, A.A.; McCann, P.J.; Patel, M.; Girotra, R.N.; Wu, G.; Sher, P.M.; Morrison, E.P.; Biller, S.A. Discovery of dapagliflozin: A potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2008, 51, 1145–1149. [Google Scholar] [CrossRef]
- Dong, S.T.; Niu, H.M.; Wu, Y.; Jiang, J.L.; Li, Y.; Jiang, K.Y.; Wang, X.; Zhang, M.F.; Han, M.F.; Meng, S.N. Plasma pharmacokinetic determination of canagliflozin and its metabolites in a type 2 diabetic rat model by UPLC-MS/MS. Molecules 2018, 23, 1229. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Tugnait, M.; Zhu, T.; Kimoto, E.; Miao, Z.; Mascitti, V.; Yang, X.; Tan, B.; Walsky, R.L.; Chupka, J.; et al. Preclinical species and human disposition of PF-04971729, a selective inhibitor of the sodium-dependent glucose cotransporter 2 and clinical candidate for the treatment of type 2 diabetes mellitus. Drug Metab. Dispos. 2011, 39, 1609–1619. [Google Scholar] [CrossRef]
- Zhang, W.; Li, X.; Ding, H.; Lu, Y.; Stilwell, G.E.; Halvorsen, Y.D.; Welihinda, A. Metabolism and disposition of the SGLT2 inhibitor bexagliflozin in rats, monkeys and humans. Xenobiotica 2020, 50, 559–569. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Liu, X.; Griffen, S.C.; Lacreta, F.P.; Boulton, D.W. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diabetes Obes Metab. 2013, 15, 280–283. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Liu, X.; Shyu, W.C.; Zhang, W.; Pfister, M.; Griffen, S.C.; Li, T.; LaCreta, F.P.; Boulton, D.W. Lack of pharmacokinetic interaction between dapagliflozin, a novel sodium-glucose transporter 2 inhibitor, and metformin, pioglitazone, glimepiride or sitagliptin in healthy subjects. Diabetes Obes. Metab. 2011, 13, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Kasichayanula, S.; Chang, M.; Liu, X.; Shyu, W.C.; Griffen, S.C.; LaCreta, F.P.; Boulton, D.W. Lack of pharmacokinetic interactions between dapagliflozin and simvastatin, valsartan, warfarin, or digoxin. Adv. Ther. 2012, 29, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Devineni, D.; Vaccaro, N.; Murphy, J.; Curtin, C.; MamidiRao, N.; Weiner, S.; Wang, S.S.; Ariyawansa, J.; Stieltjes, H.; Wajs, E. Effects of rifampin, cyclosporine A, or probenecid on the pharmacokinetic profile of canagliflozin, a sodium glucose co-transporter 2 inhibitor, in healthy participants. Int. J. Clin. Pharmacol. Ther. 2015, 53, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Devineni, D.; Polidori, D. Clinical pharmacokinetic, pharmacodynamic, and drug–drug interaction profile of canagliflozin, a sodium-glucose co-transporter 2 inhibitor. Clin. Pharmacokinet. 2015, 54, 1027–1041. [Google Scholar] [CrossRef] [PubMed]
- Devineni, D.; Manitpisitkul, P.; Murphy, J.; Skee, D.; Wajs, E.; Mamidi, R.N.; Tian, H.; Vandebosch, A.; Wang, S.S.; Verhaeghe, T. Effect of canagliflozin on the pharmacokinetics of glyburide, metformin and simvastatin in healthy participants. Clin. Pharm. Drug Dev. 2015, 4, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical studies on drug-drug interactions involving metabolism and transport: Methodology, pitfalls, and interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Human | Dog | Monkey | Mouse | Rat |
|---|---|---|---|---|---|
| t1/2 (min) | 866.3 | 223.5 | 165.0 | 533.1 | 990.0 |
| Clint (mL/min/kg) | 5.7 | 42.7 | 32.3 | 61.4 | 9.8 |
| Clhep (mL/min/kg) | 4.5 | 17.9 | 18.5 | 36.5 | 8.3 |
| Hepatic extraction ratio | 0.22 | 0.56 | 0.43 | 0.41 | 0.15 |
| Compound | tR (min) | Elemental Composition | Observed [M+NH4]+ (m/z) | Mass Error (ppm) | Product Ions (m/z) | Biotransformation |
|---|---|---|---|---|---|---|
| DWP16001 | 9.4 | C24H31O6NCl | 464.1829 | −1.08 | 327.1140, 311.0675, 209.0361, 131.0854 | |
| M1 | 8.8 | C24H31O7NCl | 480.1781 | −0.62 | 427.1303, 409.1196, 325.0985, 309.0519, 207.0205, 131.0855 | Monohydroxylation |
| M2 | 8.0 | C24H31O7NCl | 480.1793 | 1.87 | 445.1405, 427.1301, 409.1193, 343.1093, 209.0361, 147.0803 | Monohydroxylation |
| M3 | 8.4 | C24H31O8NCl | 496.1727 | −1.21 | 479.1462, 443.1249, 425.1142, 383.1048, 341.0931, 131.0854 | Hydroxylation + oxidation |
| M4 | 7.4 | C24H31O8NCl | 496.1723 | −2.02 | 479.1468, 443.1257, 425.1151, 407.1046, 207.0208, 147.0804 | Dihydroxylation |
| M5 | 8.0 | C24H31O8NCl | 496.1728 | -1.01 | 479.1471, 443.1261, 425.0962, 359.1047, 163.0751 | Dihydroxylation |
| U1 | 8.1 | C30H39O12NCl | 640.2161 | 0.94 | 464.1826, 327.1149, 311.0678, 131.0856 | Glucuronidation |
| U2 | 9.1 | C30H39O12NCl | 640.2145 | −1.56 | 447.1571, 327.1149, 311.0680, 131.0857 | Glucuronidation |
| U3 | 8.6 | C30H39O12NCl | 640.2150 | −0.78 | 464.1833, 447.1566, 327.1143, 311.0678, 131.0854 | Glucuronidation |
| U4 | 8.1 | C30H39O13NCl | 656.2099 | −0.76 | 463.1516, 445.1410, 325.0987, 309.0522, 207.0205, 131.0855 | Hydroxylation + glucuronidation |
| U5 | 8.6 | C30H39O13NCl | 656.2099 | −0.76 | 445.1397, 325.0987, 309.0522, 207.0206, 131.0855 | Hydroxylation + glucuronidation |
| Enzymes | M1 Formation | M2 Formation | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Km (µM) | Vmax | Clint | n | Mode | Km (µM) | Vmax | Clint | n | Mode | |
| HLM | 150.1 | 980.3 | 6.531 | 0.9176 | Hill | 58.7 | 82.8 | 1.411 | 0.9773 | Hill |
| CYP3A4 | 471.4 | 28.9 | 0.061 | 0.7505 | Hill | 674.5 | 3.8 | 0.006 | 0.696 | Hill |
| CYP2C19 | 156.4 | 3.6 | 0.023 | - | Single enzyme | 35.4 | 0.688 | 0.019 | 1.3 | Hill |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-H.; Kim, D.K.; Choi, W.-G.; Ji, H.-Y.; Choi, J.-S.; Song, I.-S.; Lee, S.; Lee, H.S. In Vitro Metabolism of DWP16001, a Novel Sodium-Glucose Cotransporter 2 Inhibitor, in Human and Animal Hepatocytes. Pharmaceutics 2020, 12, 865. https://doi.org/10.3390/pharmaceutics12090865
Kim J-H, Kim DK, Choi W-G, Ji H-Y, Choi J-S, Song I-S, Lee S, Lee HS. In Vitro Metabolism of DWP16001, a Novel Sodium-Glucose Cotransporter 2 Inhibitor, in Human and Animal Hepatocytes. Pharmaceutics. 2020; 12(9):865. https://doi.org/10.3390/pharmaceutics12090865
Chicago/Turabian StyleKim, Ju-Hyun, Dong Kyun Kim, Won-Gu Choi, Hye-Young Ji, Ji-Soo Choi, Im-Sook Song, Sangkyu Lee, and Hye Suk Lee. 2020. "In Vitro Metabolism of DWP16001, a Novel Sodium-Glucose Cotransporter 2 Inhibitor, in Human and Animal Hepatocytes" Pharmaceutics 12, no. 9: 865. https://doi.org/10.3390/pharmaceutics12090865
APA StyleKim, J.-H., Kim, D. K., Choi, W.-G., Ji, H.-Y., Choi, J.-S., Song, I.-S., Lee, S., & Lee, H. S. (2020). In Vitro Metabolism of DWP16001, a Novel Sodium-Glucose Cotransporter 2 Inhibitor, in Human and Animal Hepatocytes. Pharmaceutics, 12(9), 865. https://doi.org/10.3390/pharmaceutics12090865