π-Donor/π-Acceptor Interactions for the Encapsulation of Neurotransmitters on Functionalized Polysilicon-Based Microparticles

 ,
,  , , and
, , and
Abstract
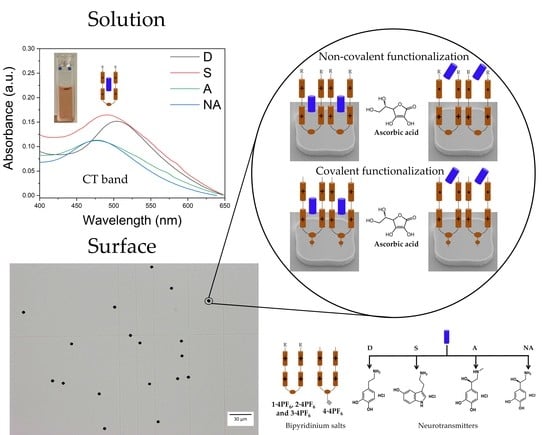
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
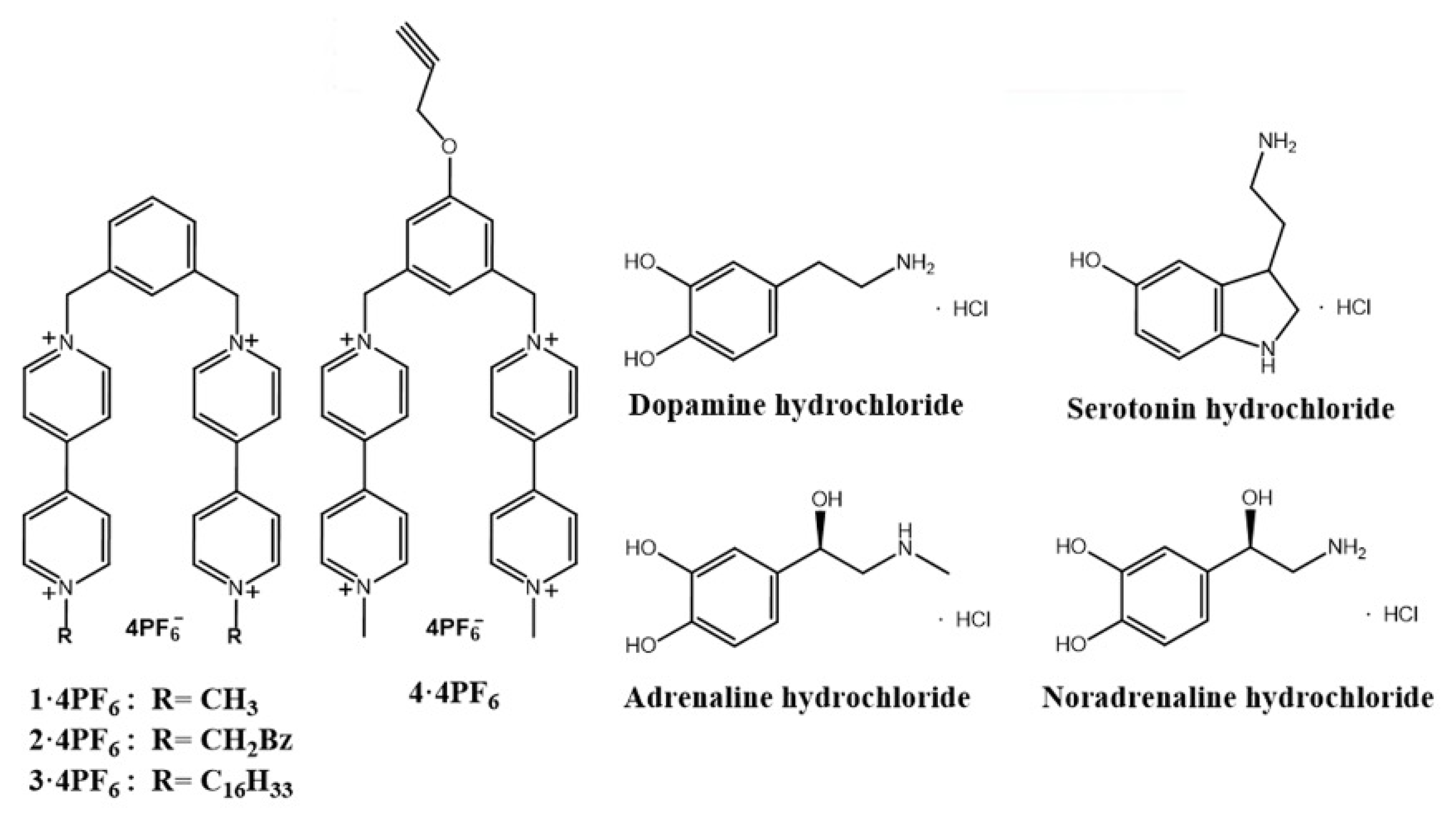
2.3. Synthesis and Characterization of Compounds Based on Bipyridinium Salts
2.3.1. Synthesis of 1,3-Bis(1′-methyl-4,4′-bipyridiniummethylene) Benzene-Tetrakis (Hexafluorophosphate) (1·4PF6)
2.3.2. Synthesis of 1,3-Bis(1′-hexadecyl-4,4′-bipyridiniummethylene) Benzene-Tetrakis (Hexafluorophosphate) (3·4PF6)
2.3.3. Synthesis of 1,3-Bis(1′-methyl-4,4′-bipyridiniummethylene)-5-propargyloxybenzene-tetrakis (Hexafluorophosphate) (4·4PF6)
2.4. Formation of π-Acceptor/π-Donor Complexes between 1·4PF6–4·4PF6 and Neurotransmitters: D, S, A and NA in Solution
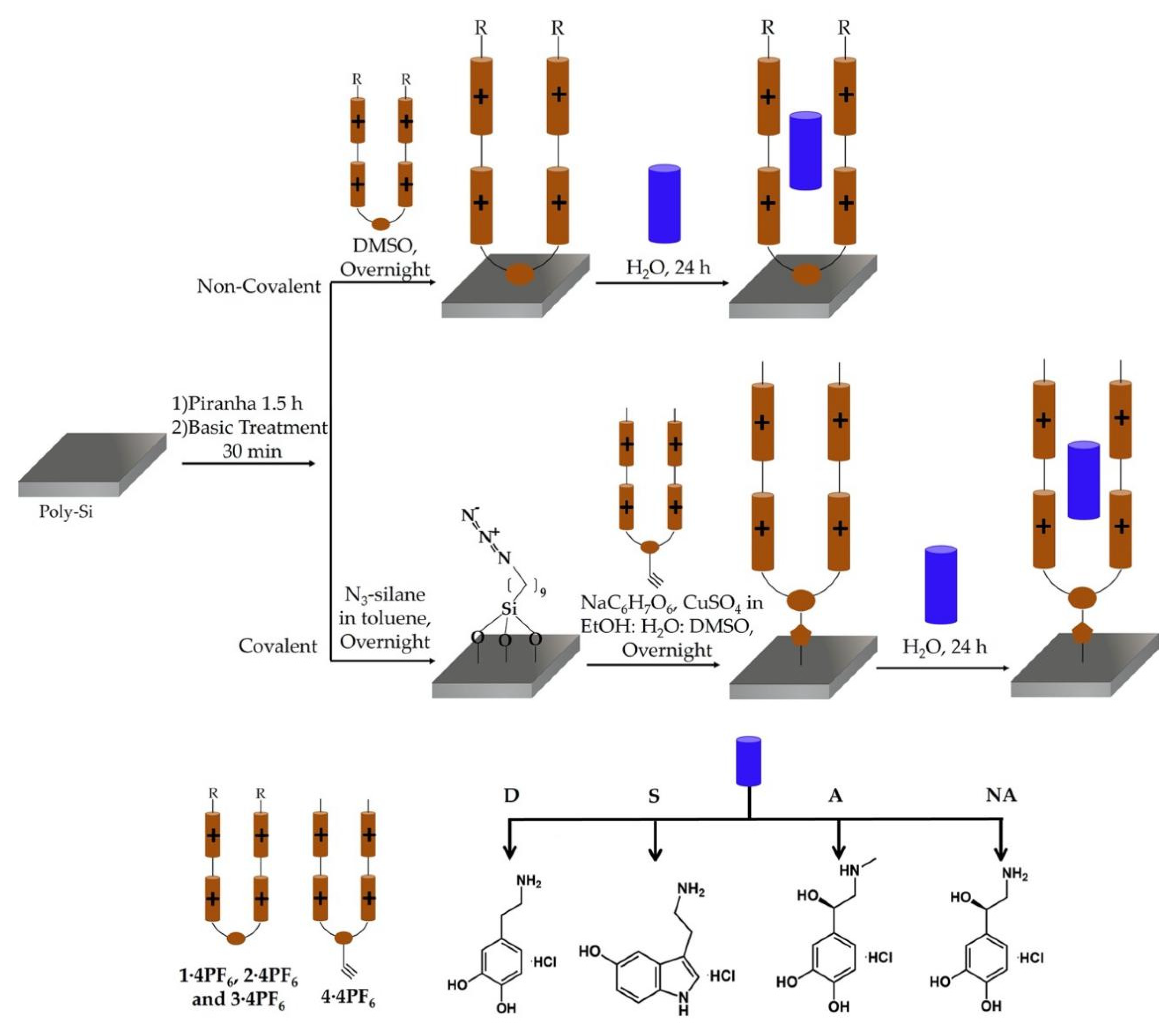
2.5. Functionalization of Polysilicon Surfaces (Poly-Surfs) with Bipyridinium Salts 1·4PF6–4·4PF6, and Incorporation of D, S, A and NA
2.5.1. Cleaning and Activation Surface Protocol
2.5.2. Non-Covalent Immobilization of 1·4PF6–3·4PF6 and Incorporation of D, S, A and NA
2.5.3. Covalent Immobilization of 4·4PF6 and Incorporation of D, S, A and NA
2.5.4. Control Surfaces with D, S, A and NA
2.6. Functionalization of Polysilicon Microparticles (Poly-Si μPs) with 1·4PF6 and 4·4PF6 and Incorporation of A
2.6.1. Cleaning and Activation Protocol of Poly-Si μPs
2.6.2. Non-Covalent Immobilization of 1·4PF6 on Poly-Si μPs
2.6.3. Covalent Immobilization of 4·4PF6 on Poly-Si μPs
2.6.4. Incorporation of A in 1·4PF6 and 4·4PF6 Functionalized Poly-Si μPs
2.7. Quantification of A Incorporated in Poly-Si μPs by HPLC Determination
2.8. Quantification of A Released from Poly-Si μPs by HPLC Determination
3. Results and Discussion
3.1. Synthesis of 1·4PF6–4·4PF6
3.2. Characterization of Neurotransmitters Complexation with 1·4PF6–4·4PF6 in Solution Using Ultraviolet-Visible Absorption and Fluorescence Spectroscopies
3.3. Functionalization and Characterization of Polysilicon Surfaces and Microparticles with 1·4PF6–4·4PF6 and Neurotransmitter Incorporation
3.4. Release Studies from A Encapsulated in Microparticles
3.5. Cytotoxicity and Genotoxicity Assay of 1·4PF6
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bah, M.G.; Bilal, H.M.; Wang, J. Fabrication and application of complex microcapsules: A review. Soft Matter 2020, 16, 570–590. [Google Scholar] [CrossRef] [PubMed]
- Panwar, N.; Soehartono, A.M.; Chan, K.K.; Zeng, S.; Xu, G.; Qu, J.; Coquet, P.; Yong, K.T.; Chen, X. Nanocarbons for Biology and Medicine: Sensing, Imaging, and Drug Delivery. Chem. Rev. 2019, 119, 9559–9656. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Vallet-Regí, M. Mesoporous Silica Nanoparticles for Drug Delivery. Adv. Funct. Mater. 2020, 30, 3–5. [Google Scholar] [CrossRef]
- Shalek, A.K.; Robinson, J.T.; Karp, E.S.; Lee, J.S.; Ahn, D.R.; Yoon, M.H.; Sutton, A.; Jorgolli, M.; Gertner, R.S.; Gujral, T.S.; et al. Vertical silicon nanowires as a universal platform for delivering biomolecules into living cells. Proc. Natl. Acad. Sci. USA 2010, 107, 1870–1875. [Google Scholar] [CrossRef]
- Siqueira, J.R.; Caseli, L.; Crespilho, F.N.; Zucolotto, V.; Oliveira, O.N. Immobilization of biomolecules on nanostructured films for biosensing. Biosens. Bioelectron. 2010, 25, 1254–1263. [Google Scholar] [CrossRef]
- Cheng, F.; Shang, J.; Ratner, D.M. A versatile method for functionalizing surfaces with bioactive glycans. Bioconjug. Chem. 2011, 22, 50–57. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, N.; Comini, E. The role of self-assembled monolayers in electronic devices. J. Mater. Chem. C 2020, 8, 3938–3955. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Z.; Das, J.; Labib, M.; Ahmed, S.; Sargent, E.H.; Kelley, S.O. Potential-Responsive Surfaces for Manipulation of Cell Adhesion, Release, and Differentiation. Angew. Chem. Int. Ed. 2019, 58, 14519–14523. [Google Scholar] [CrossRef]
- Qiu, M.; Singh, A.; Wang, D.; Qu, J.; Swihart, M.; Zhang, H.; Prasad, P.N. Biocompatible and biodegradable inorganic nanostructures for nanomedicine: Silicon and black phosphorus. Nano Today 2019, 25, 135–155. [Google Scholar] [CrossRef]
- Koch, B.; Rubino, I.; Quan, F.S.; Yoo, B.; Choi, H.J. Microfabrication for drug delivery. Materials 2016, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, X.; Kundu, S.; Nag, A.; Afsarimanesh, N.; Sapra, S.; Mukhopadhyay, S.C.; Han, T. Silicon-Based Sensors for Biomedical Applications: A Review. Sensors 2019, 19, 2908. [Google Scholar] [CrossRef] [PubMed]
- Bruce, G.; Samperi, M.; Amabilino, D.B.; Duch, M.; Plaza, J.A.; Pérez-García, L. Singlet oxygen generation from porphyrin-functionalized hexahedral polysilicon microparticles. J. Porphyr. Phthalocyanines 2019, 23, 223–233. [Google Scholar] [CrossRef]
- Penon, O.; Siapkas, D.; Novo, S.; Durán, S.; Oncins, G.; Errachid, A.; Barrios, L.; Nogués, C.; Duch, M.; Plaza, J.A.; et al. Optimized immobilization of lectins using self-assembled monolayers on polysilicon encoded materials for cell tagging. Colloids Surf. B Biointerfaces 2014, 116, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Penon, O.; Novo, S.; Durán, S.; Ibañez, E.; Nogués, C.; Samitier, J.; Duch, M.; Plaza, J.A.; Pérez-García, L. Efficient biofunctionalization of polysilicon barcodes for adhesion to the zona pellucida of mouse embryos. Bioconjug. Chem. 2012, 23, 2392–2402. [Google Scholar] [CrossRef]
- Fernández-Rosas, E.; Gómez, R.; Ibañez, E.; Barrios, L.; Duch, M.; Esteve, J.; Plaza, J.A.; Nogués, C. Internalization and cytotoxicity analysis of silicon-based microparticles in macrophages and embryos. Biomed. Microdevices 2010, 12, 371–379. [Google Scholar] [CrossRef]
- Patiño, T.; Soriano, J.; Amirthalingam, E.; Durán, S.; González-Campo, A.; Duch, M.; Ibáñez, E.; Barrios, L.; Plaza, J.A.; Pérez-García, L.; et al. Polysilicon-chromium-gold intracellular chips for multi-functional biomedical applications. Nanoscale 2016, 8, 8773–8783. [Google Scholar] [CrossRef]
- Durán, S.; Duch, M.; Gómez-Martínez, R.; Fernández-Regúlez, M.; Agusil, J.P.; Reina, M.; Müller, C.; Paulo, Á.S.; Esteve, J.; Castel, S.; et al. Internalization and viability studies of suspended nanowire silicon chips in hela cells. Nanomaterials 2020, 10, 893. [Google Scholar]
- Li, Z.; Song, N.; Yang, Y.W. Stimuli-Responsive Drug-Delivery Systems Based on Supramolecular Nanovalves. Matter 2019, 1, 345–368. [Google Scholar] [CrossRef]
- Goodnow, T.T.; Reddington, M.V.; Stoddart, J.F.; Kaifer, A.E. Cyclobis(paraquat-p-phenylene): A Novel Synthetic Receptor for Amino Acids with Electron-Rich Aromatic Moieties. J. Am. Chem. Soc. 1991, 113, 4335–4337. [Google Scholar] [CrossRef]
- Bernardo, A.R.; Stoddart, J.F.; Kaifer, A.E. Cyclobis(paraquat-p-phenylene) as a Synthetic Receptor for Electron-Rich Aromatic Compounds: Electrochemical and Spectroscopic Studies of Neurotransmitter Binding. J. Am. Chem. Soc. 1992, 114, 10624–10631. [Google Scholar] [CrossRef]
- Raymo, F.M.; Cejas, M.A. Supramolecular association of dopamine with immobilized fluorescent probes. Org. Lett. 2002, 4, 3183–3185. [Google Scholar] [CrossRef] [PubMed]
- Cejas, M.A.; Raymo, F.M. Fluorescent diazapyrenium films and their response to dopamine. Langmuir 2005, 21, 5795–5802. [Google Scholar] [CrossRef] [PubMed]
- Anelli, P.L.; Ashton, P.R.; Philp, D.; Pietraszkiewicz, M.; Reddington, M.V.; Spencer, N.; Stoddart, J.F.; Vicent, C.; Ballardini, R.; Balzani, V.; et al. Molecular Meccano. 1. [2]Rotaxanes and a [2]Catenane Made to Order. J. Am. Chem. Soc. 1992, 114, 193–218. [Google Scholar] [CrossRef]
- Amabilino, D.B.; Ashton, P.R.; Brown, C.L.; Newton, S.P.; Pietraszkiewicz, M.; Philp, D.; Raymo, F.M.; Reder, A.S.; Rutland, M.T.; Spencer, N.; et al. Molecular Meccano. 2 Self-Assembly of [n] Catenanes. J. Am. Chem. Soc. 1995, 117, 1271–1293. [Google Scholar] [CrossRef]
- Ashton, P.R.; Pérez-García, L.; Stoddart, J.F.; Ballardini, R.; Balzani, V.; Credi, A.; Gandolfi, M.T.; Prodi, L.; Venturi, M.; Menzer, S.; et al. Molecular Meccano. 4. The Self-Assembly of [2] Catenanes Incorporating Photoactive and Electroactive π-Extended Systems. J. Am. Chem. Soc. 1995, 117, 11171–11197. [Google Scholar]
- Jordão, N.; Cruz, H.; Branco, A.; Pina, F.; Branco, L.C. Bis(bipyridinium) Salts as Multicolored Electrochromic Devices. Chempluschem 2017, 82, 1211–1217. [Google Scholar] [CrossRef]
- Clarke, D.E.; Olesińska, M.; Mönch, T.; Schoenaers, B.; Stesmans, A.; Scherman, O.A. Aryl-viologen pentapeptide self-assembled conductive nanofibers. Chem. Commun. 2019, 55, 7354–7357. [Google Scholar] [CrossRef]
- Škorjanc, T.; Shetty, D.; Olson, M.A.; Trabolsi, A. Design strategies and redox-dependent applications of insoluble viologen-based covalent organic polymers. ACS Appl. Mater. Interfaces 2019, 11, 6705–6716. [Google Scholar] [CrossRef]
- Guerrini, L.; Garcia-Ramos, J.V.; Domingo, C.; Sanchez-Cortes, S. Nanosensors based on viologen functionalized silver nanoparticles: Few molecules surface-enhanced Raman spectroscopy detection of polycyclic aromatic hydrocarbons in interparticle hot spots. Anal. Chem. 2009, 81, 1418–1425. [Google Scholar] [CrossRef]
- Sultanova, E.D.; Krasnova, E.G.; Kharlamov, S.V.; Nasybullina, G.R.; Yanilkin, V.V.; Nizameev, I.R.; Kadirov, M.K.; Mukhitova, R.K.; Zakharova, L.Y.; Ziganshina, A.Y.; et al. Thermoresponsive polymer nanoparticles based on viologen cavitands. Chempluschem 2015, 80, 217–222. [Google Scholar] [CrossRef]
- Ryu, J.H.; Lee, J.H.; Han, S.J.; Suh, K. Do Influence of viologen lengths on the response time of the reflective electrochromic display prepared by monodisperse viologen-modified polymeric microspheres. Colloids Surf. A Physicochem. Eng. Asp. 2008, 315, 31–37. [Google Scholar] [CrossRef]
- Liu, A.; Han, S.; Che, H.; Hua, L. Fluorescent hybrid with electron acceptor methylene viologen units inside the pore walls-of mesoporous MCM-48 silica. Langmuir 2010, 26, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Yamanoi, Y.; Nishihara, H. Assembly of nanosize metallic particles and molecular wires on electrode surfaces. Chem. Commun. 2007, 3983–3989. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ne, K.G.; Kang, E.T. Viologen-Functionalized Conductive Surfaces: Physicochemical and Electrochemical Characteristics, and Stability. Langmuir 2002, 18, 9041–9047. [Google Scholar] [CrossRef]
- Sou, K.; Le, D.L.; Sato, H. Nanocapsules for Programmed Neurotransmitter Release: Toward Artificial Extracellular Synaptic Vesicles. Small 2019, 15, 1–12. [Google Scholar] [CrossRef]
- Si, B.; Song, E. Recent advances in the detection of neurotransmitters. Chemosensors 2018, 6, 1. [Google Scholar] [CrossRef]
- Niyonambaza, S.D.; Kumar, P.; Xing, P.; Mathault, J.; De Koninck, P.; Boisselier, E.; Boukadoum, M.; Miled, A. A Review of neurotransmitters sensing methods for neuro-engineering research. Appl. Sci. 2019, 9, 4719. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Chircov, C.; Grumezescu, A.M.; Teleanu, R.I. Neuronanomedicine: An up-to-date overview. Pharmaceutics 2019, 11, 101. [Google Scholar] [CrossRef]
- Le, D.L.; Ferdinandus; Tnee, C.K.; Vo Doan, T.T.; Arai, S.; Suzuki, M.; Sou, K.; Sato, H. Neurotransmitter-Loaded Nanocapsule Triggers On-Demand Muscle Relaxation in Living Organism. ACS Appl. Mater. Interfaces 2018, 10, 37812–37819. [Google Scholar] [CrossRef]
- Ragusa, A.; Priore, P.; Giudetti, A.M.; Ciccarella, G.; Gaballo, A. Neuroprotective investigation of Chitosan nanoparticles for dopamine delivery. Appl. Sci. 2018, 8, 474. [Google Scholar] [CrossRef]
- Lai, C.Y.; Trewyn, B.G.; Jeftinija, D.M.; Jeftinija, K.; Xu, S.; Jeftinija, S.; Lin, V.S.Y. A mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, G.; Notti, A.; Pappalardo, S.; Parisi, M.F.; Pisagatti, I.; Patanè, S. Encapsulation of monoamine neurotransmitters and trace amines by amphiphilic anionic calix[5]arene micelles. New J. Chem. 2014, 38, 5983–5990. [Google Scholar] [CrossRef]
- Geuder, W.; Hünig, S.; Suchy, A. Single and double bridged viologenes and intramolecular pimerization of their cation radicals. Tetrahedron 1986, 42, 1665–1677. [Google Scholar] [CrossRef]
- Lamberto, M.; Rastede, E.E.; Decker, J.; Raymo, F.M. Microwave-assisted synthesis of symmetric and asymmetric viologens. Tetrahedron Lett. 2010, 51, 5618–5620. [Google Scholar] [CrossRef]
- He, C.; Li, L.W.; He, W.D.; Jiang, W.X.; Wu, C. “Click” long seesaw-type A~~B~A chains together into huge defect-free hyperbranched polymer chains with uniform subchains. Macromolecules 2011, 44, 6233–6236. [Google Scholar] [CrossRef]
- Gómez-Martínez, R.; Vázquez, P.; Duch, M.; Muriano, A.; Pinacho, D.; Sanvicens, N.; Sánchez-Baeza, F.; Boya, P.; De La Rosa, E.J.; Esteve, J.; et al. Intracellular silicon chips in living cells. Small 2010, 6, 499–502. [Google Scholar] [CrossRef]
- Prakash, S.; Long, T.M.; Selby, J.C.; Moore, J.S.; Shannon, M.A. “Click” modification of silica surfaces and glass microfluidic channels. Anal. Chem. 2007, 79, 1661–1667. [Google Scholar] [CrossRef]
- Tian, F.; Cheng, N.; Nouvel, N.; Geng, J.; Scherman, O.A. Site-selective immobilization of colloids on au substrates via a noncovalent supramolecular “handcuff”. Langmuir 2010, 26, 5323–5328. [Google Scholar] [CrossRef]
- Sun, Z.; Xi, L.; Zheng, K.; Zhang, Z.; Baldridge, K.K.; Olson, M.A. Classical and non-classical melatonin receptor agonist-directed micellization of bipyridinium- based supramolecular amphiphiles in water. Soft Matter 2020, 16, 4788–4799. [Google Scholar] [CrossRef]
- Janghel, E.K.; Gupta, V.K.; Rai, M.K.; Rai, J.K. Micro determination of ascorbic acid using methyl viologen. Talanta 2007, 72, 1013–1016. [Google Scholar] [CrossRef]
- Watanabe, T.; Honda, K. Measurement of the extinction coefficient of the methyl viologen cation radical and the efficiency of its formation by semiconductor photocatalysis. J. Phys. Chem. 1982, 86, 2617–2619. [Google Scholar] [CrossRef]
- Bradshaw, M.P.; Prenzler, P.D.; Scollary, G.R. Ascorbic acid-induced browning of (+)-catechin in a model wine system. J. Agric. Food Chem. 2001, 49, 934–939. [Google Scholar] [CrossRef] [PubMed]
- ASTM-E2186. Standard Guide for Determining DNA Single-Strand Damage in Eukaryotic Cells Using the Comet Assay; ASTM International: West Conshohocken, PA, USA, 2003; Volume 11, pp. 1–12. Available online: https://www.astm.org/DATABASE.CART/HISTORICAL/E2186-02A.htm (accessed on 25 July 2016).
- Kui, W.; Guo, D.S.; Zhang, H.Q.; Dong, L.; Zheng, X.L.; Yu, L. Highly effective binding of viologens by p-sulfonatocalixarenes for the treatment of viologen poisoning. J. Med. Chem. 2009, 52, 6402–6412. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average Contact Angles (θ) ± Standard Deviation (SD) (°) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Polysilicon Surfaces | 44 ± 3 | ||||||||
| After Piranha | 16 ± 2 | ||||||||
| After Basic Treatment | 10 ± 1 | ||||||||
| 1·4PF6 | 2·4PF6 | 3·4PF6 | 4·4PF6 | Controls | |||||
| Surface | θ ± SD | Surface | θ ± SD | Surface | θ ± SD | Surface | θ ± SD | Surface | θ ± SD |
| 1·4PF6 | 57 ± 1 | 2·4PF6 | 66 ± 1 | 3·4PF6 | 89± 1 | 4·4PF6 | 55 ± 4 | - | |
| 1·4PF6:D | 48 ± 1 | 2·4PF6:D | 57 ± 1 | 3·4PF6:D | 73 ± 2 | 4·4PF6:D | 44 ± 3 | D | 31 ± 3 |
| 1·4PF6:S | 44 ± 1 | 2·4PF6:S | 55 ± 1 | 3·4PF6:S | 69 ± 2 | 4·4PF6:S | 45 ± 2 | S | 32 ± 1 |
| 1·4PF6:A | 40 ± 1 | 2·4PF6:A | 50 ± 2 | 3·4PF6:A | 70 ± 1 | 4·4PF6:A | 48 ± 2 | A | 30 ± 1 |
| 1·4PF6:NA | 43 ± 1 | 2·4PF6:NA | 52 ± 2 | 3·4PF6:NA | 66 ± 1 | 4·4PF6:NA | 43 ± 1 | NA | 30 ± 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giraldo, S.; Alea-Reyes, M.E.; Limón, D.; González, A.; Duch, M.; Plaza, J.A.; Ramos-López, D.; de Lapuente, J.; González-Campo, A.; Pérez-García, L. π-Donor/π-Acceptor Interactions for the Encapsulation of Neurotransmitters on Functionalized Polysilicon-Based Microparticles. Pharmaceutics 2020, 12, 724. https://doi.org/10.3390/pharmaceutics12080724
Giraldo S, Alea-Reyes ME, Limón D, González A, Duch M, Plaza JA, Ramos-López D, de Lapuente J, González-Campo A, Pérez-García L. π-Donor/π-Acceptor Interactions for the Encapsulation of Neurotransmitters on Functionalized Polysilicon-Based Microparticles. Pharmaceutics. 2020; 12(8):724. https://doi.org/10.3390/pharmaceutics12080724
Chicago/Turabian StyleGiraldo, Sandra, María E. Alea-Reyes, David Limón, Asensio González, Marta Duch, José A. Plaza, David Ramos-López, Joaquín de Lapuente, Arántzazu González-Campo, and Lluïsa Pérez-García. 2020. "π-Donor/π-Acceptor Interactions for the Encapsulation of Neurotransmitters on Functionalized Polysilicon-Based Microparticles" Pharmaceutics 12, no. 8: 724. https://doi.org/10.3390/pharmaceutics12080724
APA StyleGiraldo, S., Alea-Reyes, M. E., Limón, D., González, A., Duch, M., Plaza, J. A., Ramos-López, D., de Lapuente, J., González-Campo, A., & Pérez-García, L. (2020). π-Donor/π-Acceptor Interactions for the Encapsulation of Neurotransmitters on Functionalized Polysilicon-Based Microparticles. Pharmaceutics, 12(8), 724. https://doi.org/10.3390/pharmaceutics12080724