Novel and Modified Neutrophil Elastase Inhibitor Loaded in Topical Formulations for Psoriasis Management

 ,
,  ,
,  ,
, 

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Synthesis of HNE Inhibitors
2.1.2. Formulation of HNE Inhibitor Loaded Emulsion and HNE Inhibitor Loaded Microemulsion
2.2. Methods
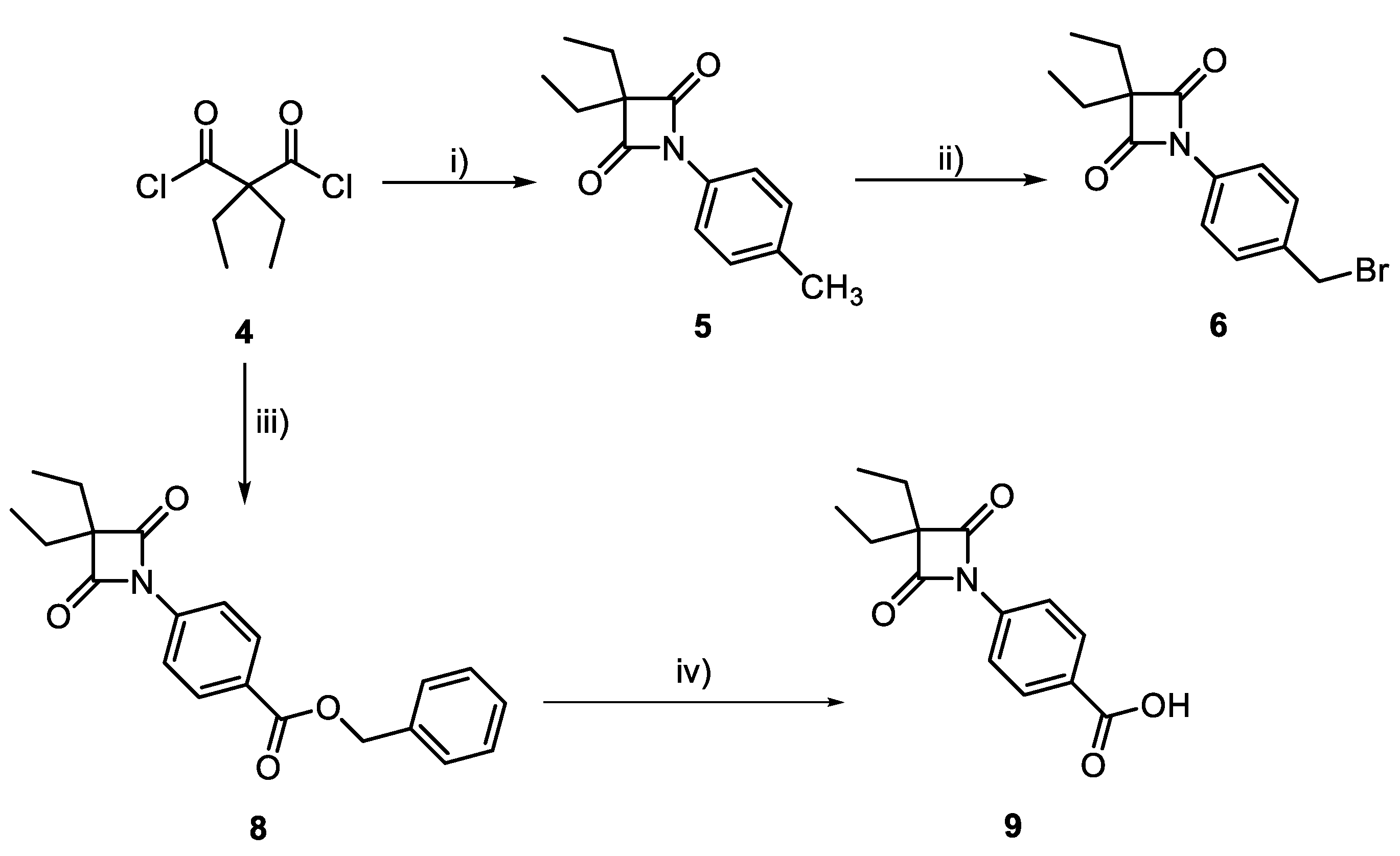
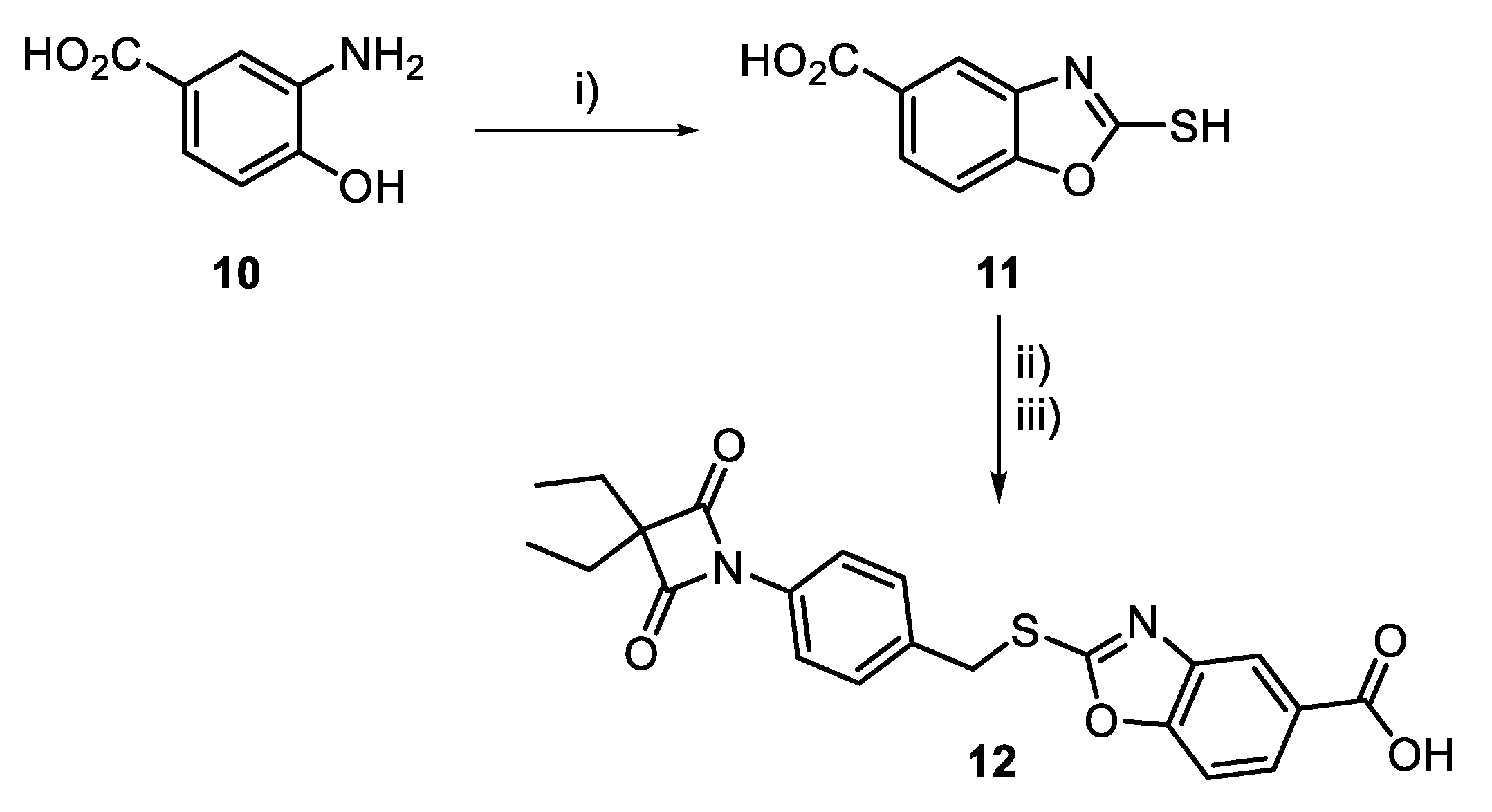
2.2.1. Synthesis and Characterization of HNE Inhibitors
2.2.2. Biological Assays of Synthesized HNE Inhibitors
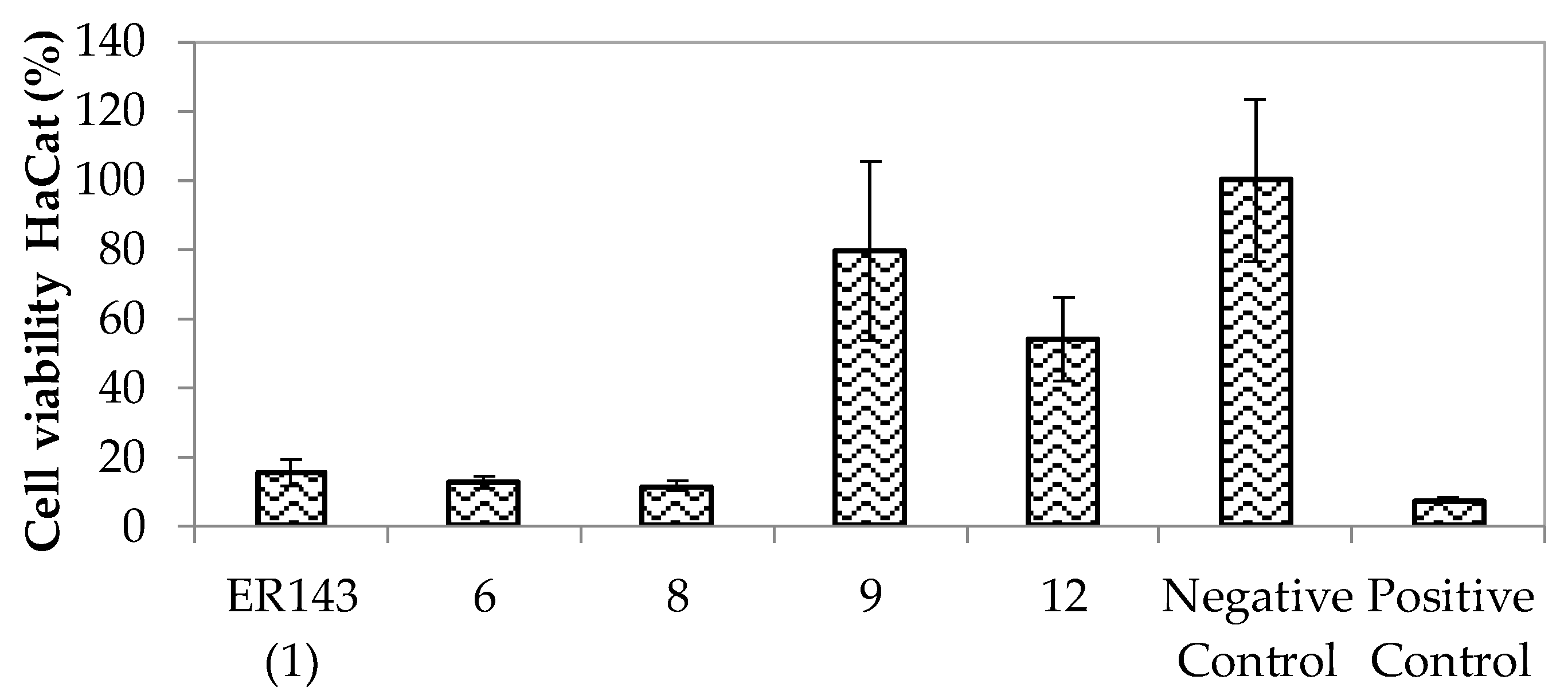
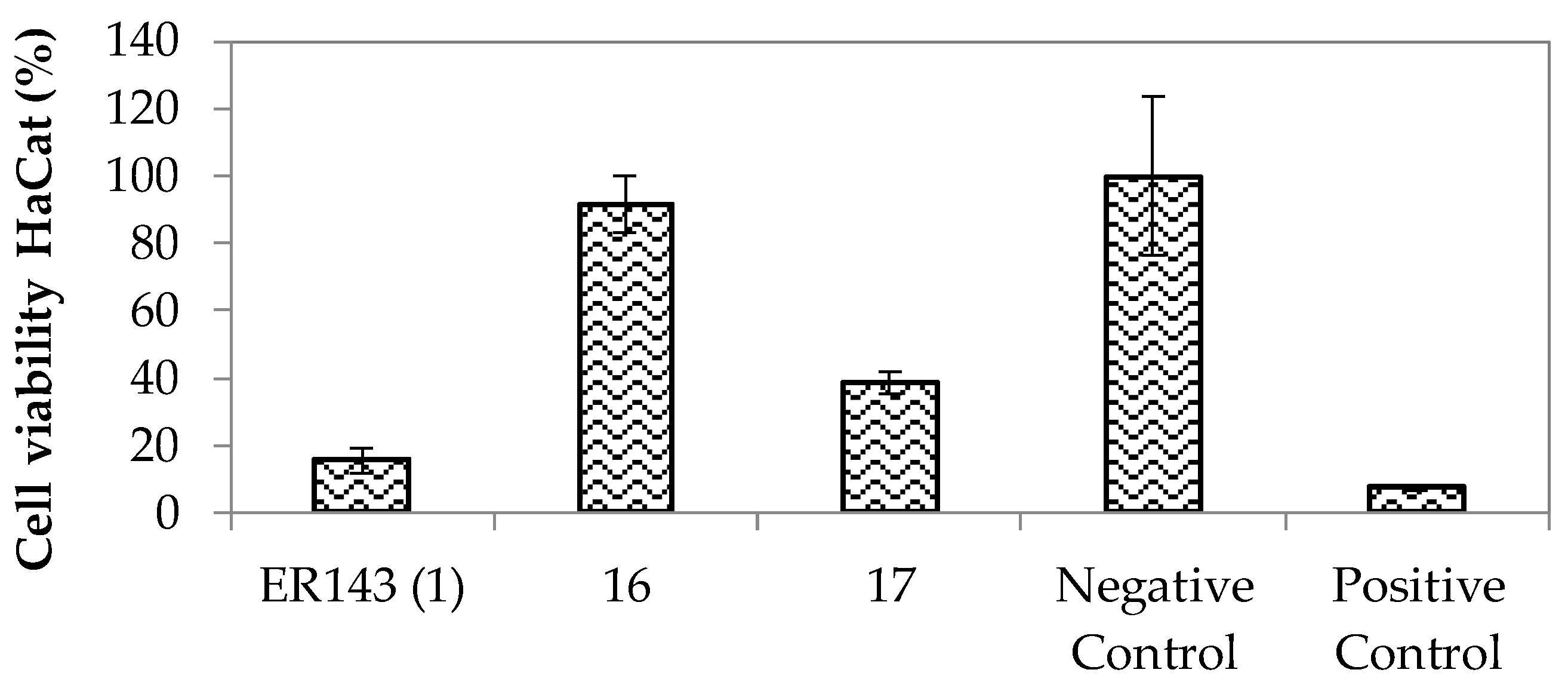
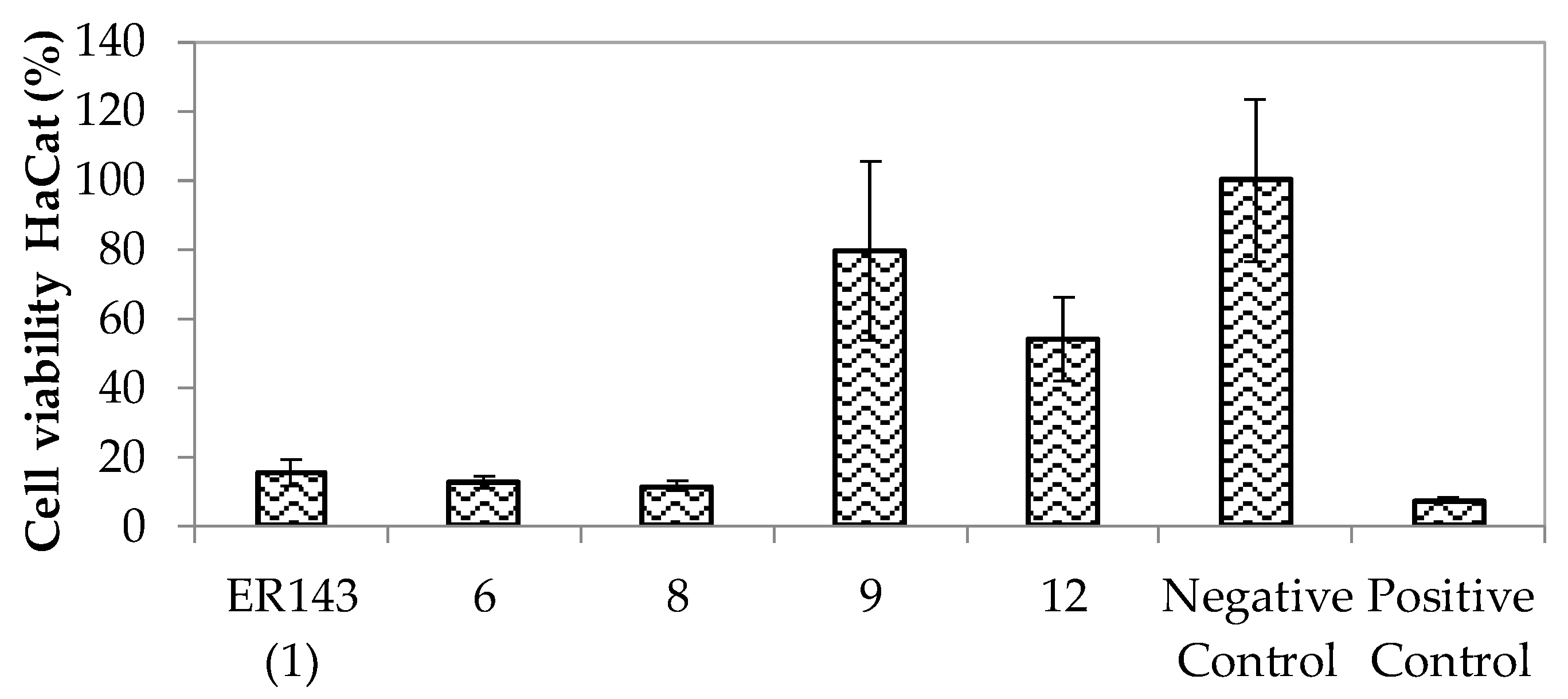
Enzymatic Inhibition Assays and In Vitro Cytotoxicity
2.2.3. HNE Inhibitor Topical Formulations and Characterization Studies
Solubility Studies
Preparation of HNE Inhibitor Loaded Emulsion and HNE Inhibitor Loaded Emulsion Microemulsion
Characterization Studies of HNE Inhibitor Loaded Emulsion and HNE Inhibitor Loaded Microemulsion
In Vitro Release Studies
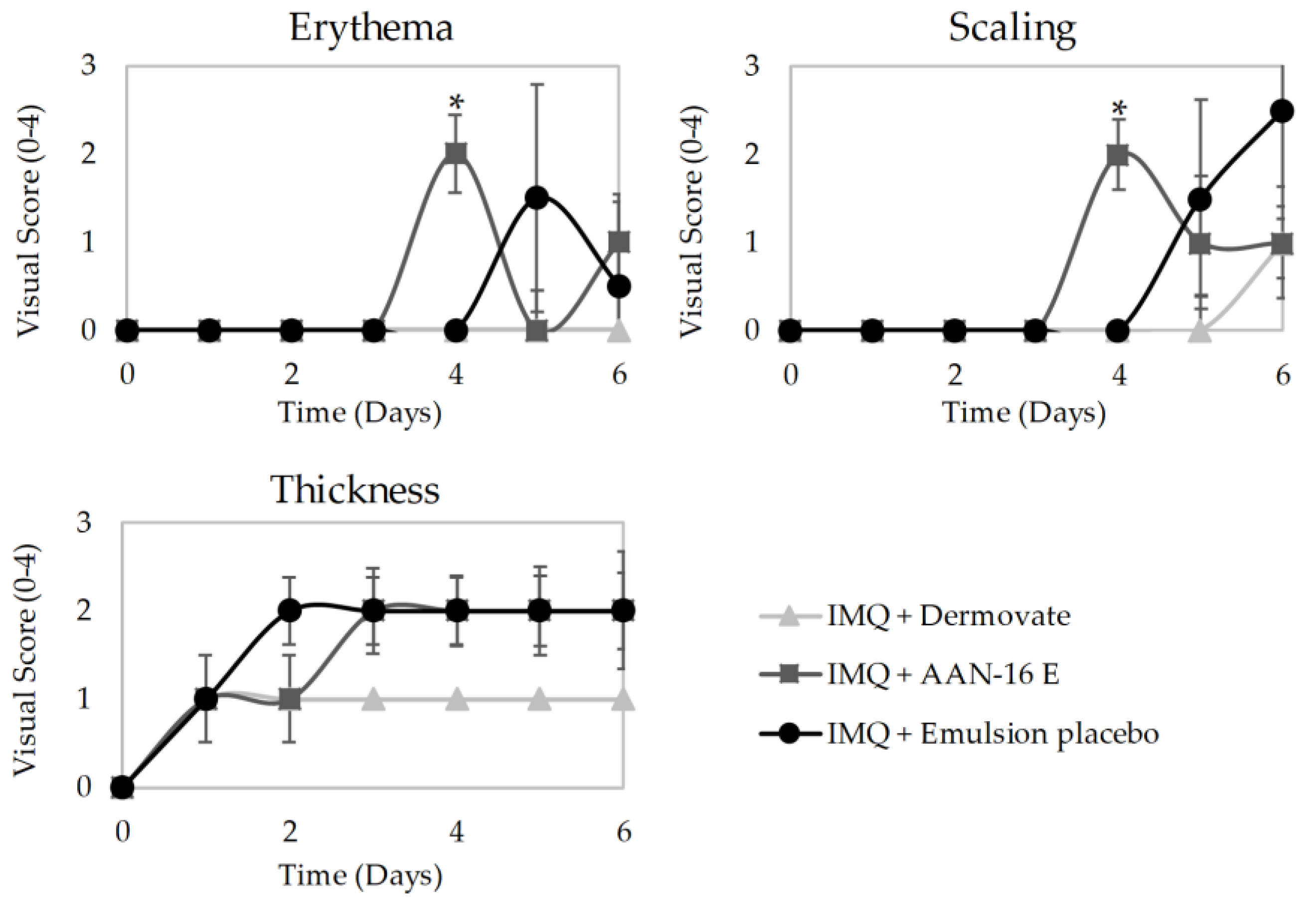
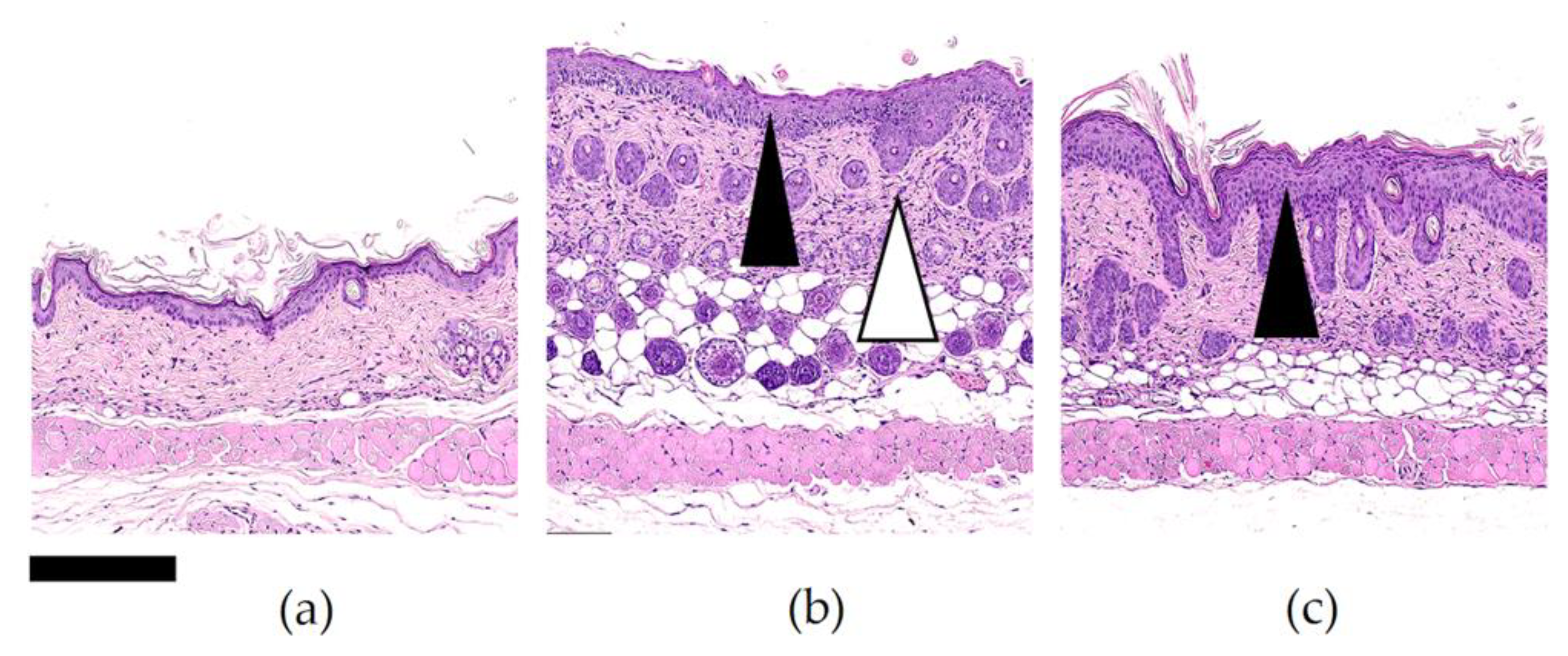
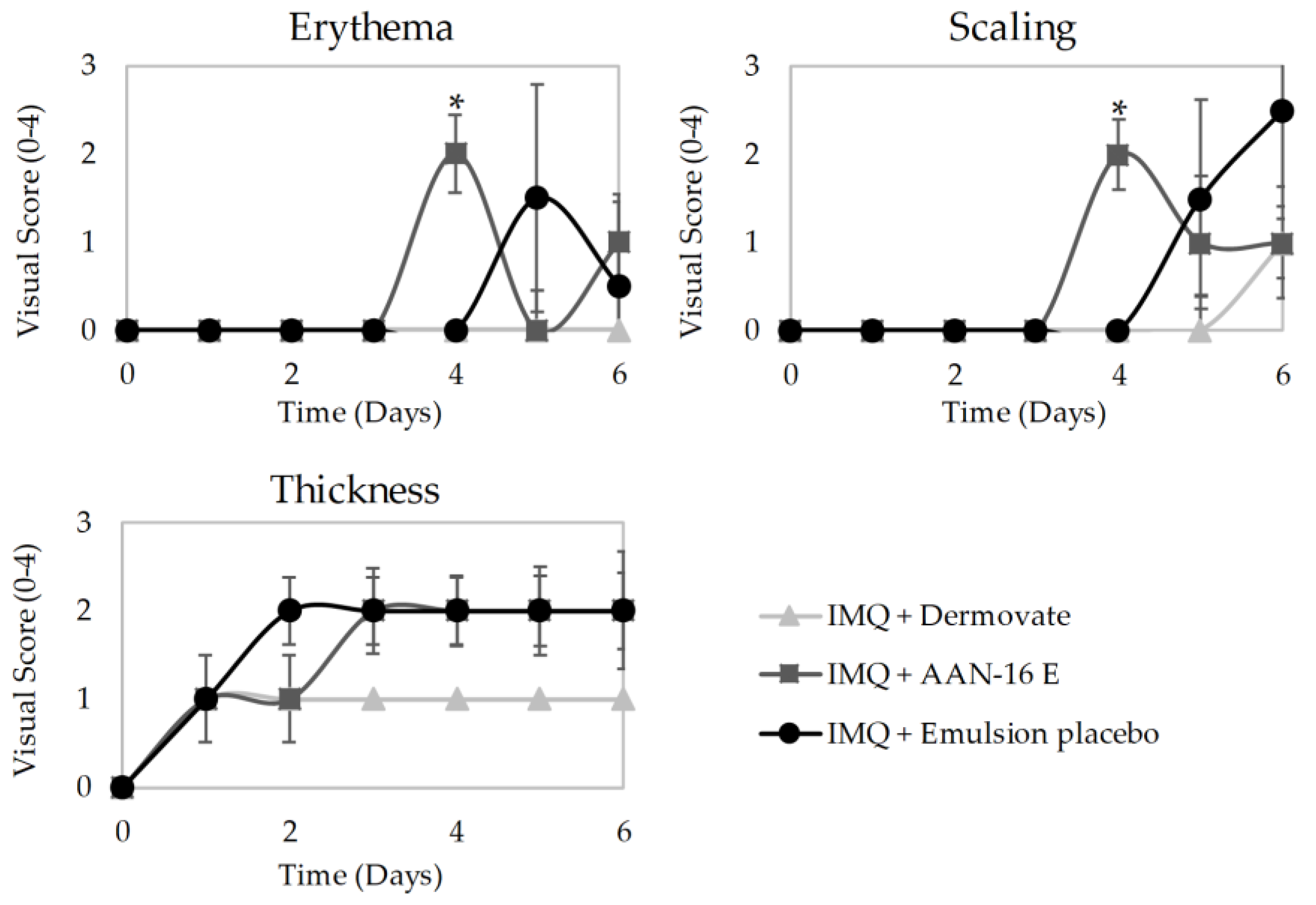
In Vivo Antipsoriatic Activity
- Group 1 (n = 5) was treated with 60 mg of Dermovate® (GSK, Algés, Portugal) (containing 0.05% clobetasol propionate) as a positive control of inflammation inhibition;
- Group 2 (n = 4) was treated with 60 mg of emulsion placebo;
- Group 3 (n = 4) was treated with 60 mg of microemulsion placebo;
- Group 4 (n = 5) was treated with 60 mg of AAN-16 E;
- Group 5 (n = 5) was treated with 60 mg of AAN-16 ME;
- Group 6 (n = 2) was used for control of non-induced and non-treated skin.
2.2.4. Statistical Analysis
3. Results and Discussion
3.1. Synthesis of 4-oxo-β-Lactams
3.2. Biological Assays of Synthesized HNE Inhibitors
Enzymatic Inhibition Assays and In Vitro Cytotoxicity
3.3. HNE Inhibitor Loaded Emulsion and HNE Inhibitor Loaded Microemulsion
3.3.1. Solubility Studies
3.3.2. Characterization Studies of AAN-16 Loaded Emulsion and AAN-16 Loaded Microemulsion
3.3.3. In Vitro Release Studies
3.3.4. In Vivo Antipsoriatic Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, R.; Symmons, D.P.M.; Griffiths, C.E.M.; Ashcroft, D.M. Global epidemiology of psoriasis: A systematic review of incidence and prevalence. J. Investig. Dermatol. 2013, 133, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noncommunicable Diseases and Their Risk Factors. Psoriasis. Available online: https://www.who.int/ncds/management/psoriasis/en/ (accessed on 22 April 2019).
- Deng, Y.; Chang, C.; Lu, Q. The Inflammatory Response in Psoriasis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 50, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Krueger, J.G. The Immunopathogenesis of Psoriasis. Dermatol. Clin. 2015, 33, 13–23. [Google Scholar] [CrossRef]
- Liang, Y.; Sarkar, M.K.; Tsoi, L.C.; Gudjonsson, J.E. Psoriasis: A mixed autoimmune and autoinflammatory disease. Curr. Opin. Immunol. 2017, 49, 1–8. [Google Scholar] [CrossRef]
- Current and Potential New Therapies for the Treatment of Psoriasis. Available online: https://www.pharmaceutical-journal.com/news-and-analysis/news/current-and-potential-new-therapies-for-the-treatment-of-psoriasis/11013061.article?firstPass=false (accessed on 19 July 2019).
- Hansen, G.; Gielen-haertwig, H.; Reinemer, P.; Schomburg, D.; Harrenga, A.; Niefind, K. Unexpected Active-Site Flexibility in the Structure of Human Neutrophil Elastase in Complex with a New Dihydropyrimidone Inhibitor. J. Mol. Biol. 2011, 409, 681–691. [Google Scholar] [CrossRef]
- Shreder, K.R.; Cajica, J.; Du, L.; Fraser, A.; Hu, Y.; Kohno, Y.; Lin, E.C.; Liu, S.J.; Okerberg, E.; Pham, L.; et al. Synthesis and optimization of 2-pyridin-3-yl-benzo[d][1,3]oxazin-4-one based inhibitors of human neutrophil elastase. Bioorganic Med. Chem. Lett. 2009, 19, 4743–4746. [Google Scholar] [CrossRef]
- Lucas, S.D.; Costa, E.; Guedes, R.C.; Moreira, R. Targeting COPD: Advances on Low-Molecular-Weight Inhibitors of Human Neutrophil Elastase. Med. Res. Rev. 2013, 33, E73–E101. [Google Scholar] [CrossRef]
- Heutinck, K.M.; ten Berge, I.J.M.; Hack, C.E.; Hamann, J.; Rowshani, A.T. Serine proteases of the human immune system in health and disease. Mol. Immunol. 2010, 47, 1943–1955. [Google Scholar] [CrossRef]
- Crocetti, L.; Giovannoni, M.P.; Schepetkin, I.A.; Quinn, M.T.; Khlebnikov, A.I.; Cilibrizzi, A.; Dal Piaz, V.; Graziano, A.; Vergelli, C. Design, synthesis and evaluation of N-benzoylindazole derivatives and analogues as inhibitors of human neutrophil elastase. Bioorganic Med. Chem. 2011, 19, 4460–4472. [Google Scholar] [CrossRef]
- Giovannoni, M.P.; Schepetkin, I.A.; Quinn, M.T.; Cantini, N.; Crocetti, L.; Guerrini, G.; Iacovone, A.; Paoli, P.; Rossi, P.; Bartolucci, G.; et al. Synthesis, biological evaluation, and molecular modelling studies of potent human neutrophil elastase (HNE) inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 1108–1124. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santana, A.B.; Lucas, S.D.; Gonçalves, L.M.; Correia, H.F.; Cardote, T.A.F.; Guedes, R.C.; Iley, J.; Moreira, R. N-Acyl and N-sulfonyloxazolidine-2,4-diones are pseudo-irreversible inhibitors of serine proteases. Bioorg. Med. Chem. Lett. 2012, 22, 3993–3997. [Google Scholar] [CrossRef]
- Yang, X.; Yan, H.; Zhai, Z.; Hao, F.; Ye, Q.; Zhong, B. Neutrophil elastase promotes proliferation of HaCaT cell line and transwell psoriasis organ culture model. Int. J. Dermatol. 2010, 49, 1068–1074. [Google Scholar] [CrossRef]
- Zhong, J.; Groutas, W. Recent Developments in the Design of Mechanism-based and Alternate Substrate Inhibitors of Serine Proteases. Curr. Top. Med. Chem. 2005, 4, 1203–1216. [Google Scholar] [CrossRef]
- Leung, D.; Abbenante, G.; Fairlie, D.P. Protease inhibitors: Current status and future prospects. J. Med.Chem. 2000, 43, 305–341. [Google Scholar] [CrossRef]
- Guyot, N.; Butler, M.W.; McNally, P.; Weldon, S.; Greene, C.M.; Levine, R.L.; O’Neill, S.J.; Taggart, C.C.; McElvaney, N.G. Elafin, an elastase-specific inhibitor, is cleaved by its cognate enzyme neutrophil elastase in sputum from individuals with cystic fibrosis. J. Biol. Chem. 2008, 283, 32377–32385. [Google Scholar] [CrossRef] [Green Version]
- Powers, J.C.; Asgian, J.L.; Ekici, O.D.; James, K.E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002, 102, 4639–4750. [Google Scholar] [CrossRef]
- Marto, J.; Ruivo, E.; Lucas, S.D.; Gonçalves, L.M.; Simões, S.; Gouveia, L.F.; Felix, R.; Moreira, R.; Ribeiro, H.M.; Almeida, A.J. Starch nanocapsules containing a novel neutrophil elastase inhibitor with improved pharmaceutical performance. Eur. J. Pharm. Biopharm. 2018, 127, 1–11. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Costanzo, M.J. Inhibitors of proteases and amide hydrolases that employ an α-ketoheterocycle as a key enabling functionality. Bioorg. Med. Chem. 2008, 16, 1562–1595. [Google Scholar] [CrossRef]
- Groutas, W.C.; Dou, D.; Alliston, K.R. Neutrophil Elastase Inhibitors. Expert Opin. Ther. Pat. 2012, 21, 339–354. [Google Scholar] [CrossRef] [Green Version]
- Mulchande, J.; Oliveira, R.; Carrasco, M.; Gouveia, L.; Guedes, R.C.; Iley, J.; Moreira, R. 4-Oxo-β-lactams (Azetidine-2, 4-diones) Are Potent and Selective Inhibitors of Human Leukocyte Elastase. J. Med. Chem. 2010, 53, 241–253. [Google Scholar] [CrossRef]
- Mulchande, J.; Simões, S.I.; Gaspar, M.M.; Eleutério, C.V.; Oliveira, R.; Cruz, M.E.M.; Moreira, R.; Iley, J. Synthesis, stability, biochemical and pharmacokinetic properties of a new potent and selective 4-oxo-β-lactam inhibitor of human leukocyte elastase. J. Enzyme Inhib. Med. Chem. 2011, 26, 169–175. [Google Scholar] [CrossRef]
- Mulchande, J.; Guedes, R.C.; Tsang, W.; Page, M.I.; Moreira, R.; Iley, J. Azetidine-2,4-diones (4-Oxo-β-lactams ) as Scaffolds for Designing Elastase Inhibitors. J. Med. Chem. 2008, 51, 1783–1790. [Google Scholar] [CrossRef]
- Carneiro, R.; Salgado, A.; Raposo, S.; Marto, J.; Simões, S.; Urbano, M.; Ribeiro, H.M. Topical emulsions containing ceramides: Effects on the skin barrier function and anti-inflammatory properties. Eur. J. Lipid Sci. Technol. 2011, 113, 961–966. [Google Scholar] [CrossRef]
- Raposo, S.C.; Simões, S.D.; Almeida, A.J.; Ribeiro, H.M. Advanced systems for glucocorticoids’ dermal delivery. Expert Opin. Drug Deliv. 2013, 10, 857–877. [Google Scholar] [CrossRef]
- Simões, S.; Ribeiro, H.M.; Almeida, A.J. Microemulsões e nanoemulsões. In Novas Formas Farmacêuticas para Administração de Fármacos, 1st ed.; Souto, E.B., Lopes, C.M., Eds.; Edições Universidade Fernando Pessoa: Porto, Portugal, 2011; pp. 271–296. [Google Scholar]
- Sakamoto, K.; Lochhead, R.Y.; Maibach, H.I.; Yamashita, Y. Cosmetic Science and Technology: Theoretical Principles and Applications, 1st ed.; Elseivier: Amsterdam, The Netherlands, 2017; pp. 1–835. [Google Scholar] [CrossRef]
- Marto, J.; Ascenso, A.; Simões, S.; Almeida, A.; Ribeiro, H. Pickering emulsions: Challenges and opportunities in topical delivery. Expert Opin. Drug Deliv. 2016, 13, 1093–1107. [Google Scholar] [CrossRef]
- Anton, N.; Vandamme, T.F. Nano-emulsions and micro-emulsions: Clarifications of the critical differences. Pharm. Res. 2011, 28, 978–985. [Google Scholar] [CrossRef]
- Kogan, A.; Garti, N. Microemulsions as transdermal drug delivery vehicles. Adv. Colloid Interface Sci. 2006, 123, 369–385. [Google Scholar] [CrossRef]
- Lopes, L.B. Overcoming the cutaneous barrier with microemulsions. Pharmaceutics 2014, 6, 52–77. [Google Scholar] [CrossRef] [Green Version]
- Tadros, T.F. Microemulsions. In Applied Surfactants: Principles and Applications, 1st ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 309–334. [Google Scholar] [CrossRef]
- Virtual Computational Chemistry Laboratory. Available online: http://www.virtuallaboratory.org/lab/alogps/ (accessed on 24 March 2020).
- Lucas, S.D.; Gonçalves, L.M.; Cardote, T.A.F.; Correia, H.F.; Moreira, R.; Guedes, R.C. Structure Based Virtual Screening for Discovery of Novel Human Neutrophil Elastase Inhibitors. Med. Chem. Commun. 2012, 3, 1299–1304. [Google Scholar] [CrossRef]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Mateus, D.; Marto, J.; Trindade, P.; Gonçalves, H.; Salgado, A.; Machado, P.; Melo-Gouveia, A.; Ribeiro, H.M.; Almeida, A.J. Improved morphine-loaded hydrogels for wound-related pain relief. Pharmaceutics 2019, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Pivetta, T.P.; Simões, S.; Araújo, M.M.; Carvalho, T.; Arruda, C.; Marcato, P.D. Development of nanoparticles from natural lipids for topical delivery of thymol: Investigation of its anti-inflammatory properties. Colloids Surf. B Biointerfaces 2018, 164, 281–290. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention: Facts about Bromine. Available online: https://emergency.cdc.gov/agent/bromine/basics/facts.asp (accessed on 26 July 2019).
- Baroni, A.; Buommino, E.; De Gregorio, V.; Ruocco, E.; Ruocco, V.; Wolf, R. Structure and function of the epidermis related to barrier properties. Clin. Dermatol. 2012, 30, 257–262. [Google Scholar] [CrossRef]
- Lambers, H.; Piessens, S.; Bloem, A.; Pronk, H.; Finkel, P. Natural skin surface pH is on average below 5, which is beneficial for its resident flora. Int. J. Cosmet. Sci. 2006, 28, 359–370. [Google Scholar] [CrossRef]
- Barnes, H.A.; Hutton, J.F.; Walters, K. An Introduction to Rheology, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 1989; pp. 1–200. [Google Scholar]
- Schramm, G. A Practical Approach to Rheology and Rheometry, 2nd ed.; Thermo Electron: Waltham, MA, USA, 2004; pp. 1–259. [Google Scholar]
- Gilbert, L.; Picard, C.; Savary, G.; Grisel, M. Rheological and textural characterization of cosmetic emulsions containing natural and synthetic polymers: Relationships between both data. Colloids Surf. A Physicochem. Eng. Asp. 2013, 421, 150–163. [Google Scholar] [CrossRef]
- Eccleston, G.M. Emulsions and microemulsions. In Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, 1st ed.; Mishra, M., Ed.; CRC Press: Boca Raton, FL, USA, 2015; Volume 11, pp. 3259–3276. [Google Scholar] [CrossRef]
- Ribeiro, H.M.; Morais, J.A.; Eccleston, G.M. Structure and rheology of semisolid o/w creams containing cetyl alcohol/non-ionic surfactant mixed emulsifier and different polymers. Int. J. Cosmet. Sci. 2004, 26, 47–59. [Google Scholar] [CrossRef]
- Callender, S.P.; Mathews, J.A.; Kobernyk, K.; Wettig, S.D. Microemulsion utility in pharmaceuticals: Implications for multi-drug delivery. Int. J. Pharm. 2017, 526, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Lippacher, A.; Müller, R.H.; Mäder, K. Liquid and semisolid SLNTM dispersions for topical application: Rheological characterization. Eur. J. Pharm. Biopharm. 2004, 58, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Otto, A.; Du Plessis, J.; Wiechers, J.W. Formulation effects of topical emulsions on transdermal and dermal delivery. Int. J. Cosmet. Sci. 2009, 31, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, U.G.; Zatz, J.L. Effect of nonionic surfactants on penetration of dissolved benzocaine through hairless mouse skin. J. Soc. Cosmet. Chem. 1981, 32, 87–94. [Google Scholar]
- Florence, A.T.; Gillan, J.M.N. Biological implications of the use of surfactants in medicines and the biphasic effects of surfactants in biological systems. Pestic. Sci. 1975, 6, 429–439. [Google Scholar] [CrossRef]
- Sarpotdar, P.P.; Zatz, J.L. Evaluation of penetration enhancement of lidocaine by nonionic surfactants through hairless mouse skin in vitro. J. Pharm. Sci. 1986, 75, 176–181. [Google Scholar] [CrossRef]
- Mohammed, D.; Hirata, K.; Hadgraft, J.; Lane, M.E. Influence of skin penetration enhancers on skin barrier function and skin protease activity. Eur. J. Pharm. Sci. 2014, 51, 118–122. [Google Scholar] [CrossRef]
- Lin, Y.K.; Yang, S.H.; Chen, C.C.; Kao, H.C.; Fang, J.Y. Using imiquimod-induced psoriasis-like skin as a model to measure the skin penetration of anti-psoriatic drugs. PLoS ONE 2015, 10, e0137890. [Google Scholar] [CrossRef] [Green Version]
- Van der Fits, L.; Mourits, S.; Voerman, J.S.A.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-Induced Psoriasis-Like Skin Inflammation in Mice Is Mediated via the IL-23/IL-17 Axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef]
- Alvarez, P.; Jensen, L.E. Imiquimod Treatment Causes Systemic Disease in Mice Resembling Generalized Pustular Psoriasis in an IL-1 and IL-36 Dependent Manner. Mediators Inflamm. 2016, 2016, 6756138. [Google Scholar] [CrossRef]
- Jang, H.J.; Shin, C.Y.; Kim, K.B. Safety Evaluation of Polyethylene Glycol (PEG) Compounds for Cosmetic Use. Toxicol. Res. 2015, 31, 105–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lémery, E.; Briançon, S.; Chevalier, Y.; Bordes, C.; Oddos, T.; Gohier, A.; Bolzinger, M.A. Skin toxicity of surfactants: Structure/toxicity relationships. Colloids Surf. A Physicochem. Eng. Asp. 2015, 469, 166–179. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Y.; Hu, J. Curcumin Inhibits Imiquimod-Induced Psoriasis-Like Inflammation by Inhibiting IL-1beta and IL-6 Production in Mice. PLoS ONE 2013, 8, e67078. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Wu, C.S.; Chao, Y.H.; Lin, C.C.; Tsai, H.Y.; Li, Y.R.; Chen, Y.Z.; Tsai, W.H.; Chen, Y.K. Lactobacillus pentosus GMNL-77 inhibits skin lesions in imiquimod-induced psoriasis-like mice. J. Food Drug Anal. 2017, 25, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. Dual Inhibition of TNFR1 and IFNAR1 in Imiquimod-Induced Psoriasiform Skin Inflammation in Mice. J. Immunol. 2015, 194, 5094–5102. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disperse Phase | Ingredients (INCI) | Formulations (%, w/w) | |
|---|---|---|---|
| AAN-16 E | AAN-16 ME | ||
| Aqueous phase | Water | 72.79 | 5.97 |
| Hydrogenated lecithin (and) C12-16 alcohols (and) palmitic acid | 6.00 | - | |
| Glycerin | 5.00 | - | |
| PEG-20 glyceryl triisostearate | - | 40.27 | |
| Propylene glycol | - | 5.97 | |
| Methylparaben | 0.18 | 0.11 | |
| Propylparaben | 0.02 | 0.01 | |
| Oily Phase | Caprylic/capric triglycerides | 2.50 | 15.90 |
| C15-19 alkane | - | 15.90 | |
| C21-C28 alkane | - | 15.90 | |
| C10–18 triglycerides | 4.00 | - | |
| Isopropyl myristate | 5.00 | - | |
| Oleic acid | 1.00 | - | |
| Alpha bisabolol | 1.00 | - | |
| Cetyl alcohol | 2.50 | - | |
| Compound 16 | 0.01 | 0.01 | |
| Compounds | Structure | IC50 ± SD (nM) |
|---|---|---|
| 6 |  | 0.64 ± 0.03 |
| ER 143 (1) | 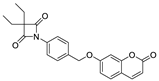 | 0.42 ± 0.09 |
| 8 |  | 0.96 ± 0.13 |
| 9 | 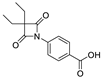 | 14.71 ± 5.55 |
| 12 | 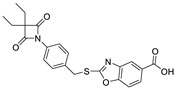 | 0.38 ± 0.05 |
| Compounds | Structure | IC50 ± SD (nM) | IC50 ± SD (µM) | |||
|---|---|---|---|---|---|---|
| HNE | Trypsin | Chymotrypsin | Urokinase | Kallikrein | ||
| ER143 (1) | 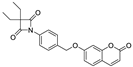 | 0.42 ± 0.09 | >1 | >8 | >50 | >8 |
| 16 | 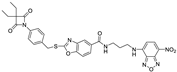 | 0.85 ± 0.10 | >1 | >1 | >2 | >5 |
| 17 |  | 0.64 ± 0.03 | >1 | >0.5 | >2 | >3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes, A.; Marto, J.; Gonçalves, L.M.; Simões, S.; Félix, R.; Ascenso, A.; Lopes, F.; Ribeiro, H.M. Novel and Modified Neutrophil Elastase Inhibitor Loaded in Topical Formulations for Psoriasis Management. Pharmaceutics 2020, 12, 358. https://doi.org/10.3390/pharmaceutics12040358
Nunes A, Marto J, Gonçalves LM, Simões S, Félix R, Ascenso A, Lopes F, Ribeiro HM. Novel and Modified Neutrophil Elastase Inhibitor Loaded in Topical Formulations for Psoriasis Management. Pharmaceutics. 2020; 12(4):358. https://doi.org/10.3390/pharmaceutics12040358
Chicago/Turabian StyleNunes, Andreia, Joana Marto, Lídia Maria Gonçalves, Sandra Simões, Rita Félix, Andreia Ascenso, Francisca Lopes, and Helena Margarida Ribeiro. 2020. "Novel and Modified Neutrophil Elastase Inhibitor Loaded in Topical Formulations for Psoriasis Management" Pharmaceutics 12, no. 4: 358. https://doi.org/10.3390/pharmaceutics12040358
APA StyleNunes, A., Marto, J., Gonçalves, L. M., Simões, S., Félix, R., Ascenso, A., Lopes, F., & Ribeiro, H. M. (2020). Novel and Modified Neutrophil Elastase Inhibitor Loaded in Topical Formulations for Psoriasis Management. Pharmaceutics, 12(4), 358. https://doi.org/10.3390/pharmaceutics12040358