
The Enigmatic Origin of Papillomavirus Protein Domains


Abstract
:1. Introduction
2. Materials and Methods
2.1. PfamA_28
2.2. HMMER “Hmmsearch”
2.3. Criteria for Considering PfamA_28 Database Hits and “Hmmsearch” Hits as True Positives
- Sequence annotation is valid (not showing evidence for viral contamination);
- The size and protein coding potential of the cellular contig/scaffold should exclude the possibility of viral contamination by small viruses (applied to complete genome/proteomes);
- “hmmscan” (protein sequence vs. profile-HMM database with HMMER) gives reciprocal best hit to query PfamA model; and
- 3D structure prediction by threading meta server LOMETS gives best modelling templates from PV structures at least with one algorithm [32].
2.4. Galaxy of Folds
2.5. SUPERFAMILY Database
2.6. Criteria for Considering Hits from SUPERFAMILY Database and from “Hmmsearch” as True Positives
- (1)
- Sequence annotation is correct (for UniProt data);
- (2)
- The size and protein coding potential of the cellular contig/scaffold exclude viral contamination by small viruses (applied to complete genomes);
- (3)
- “hmmscan” gives reciprocal best hit to query SF model; and
- (4)
- 3D structure prediction by threading meta server gives best modelling templates from respective SF at least with one algorithm.
3. Results
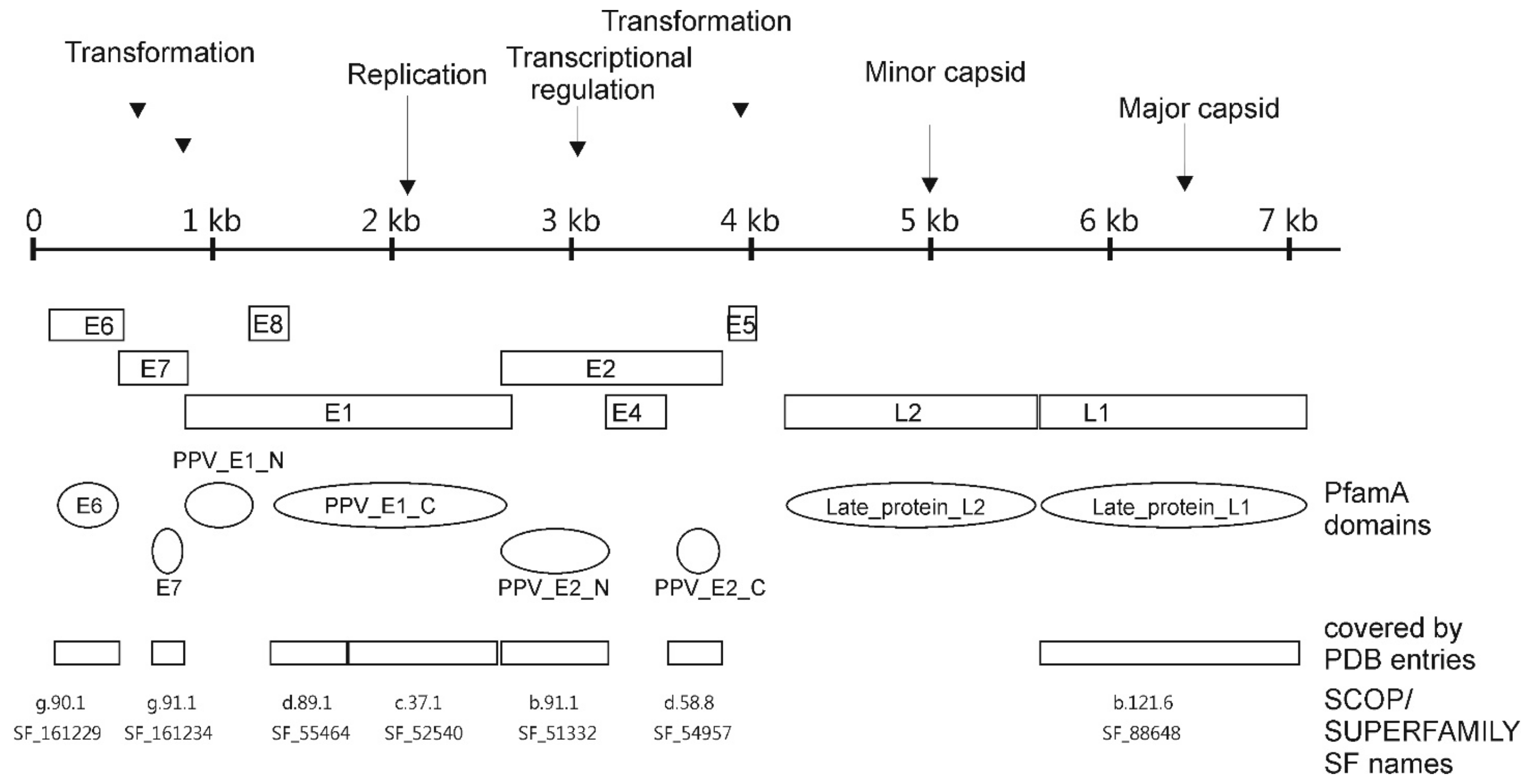
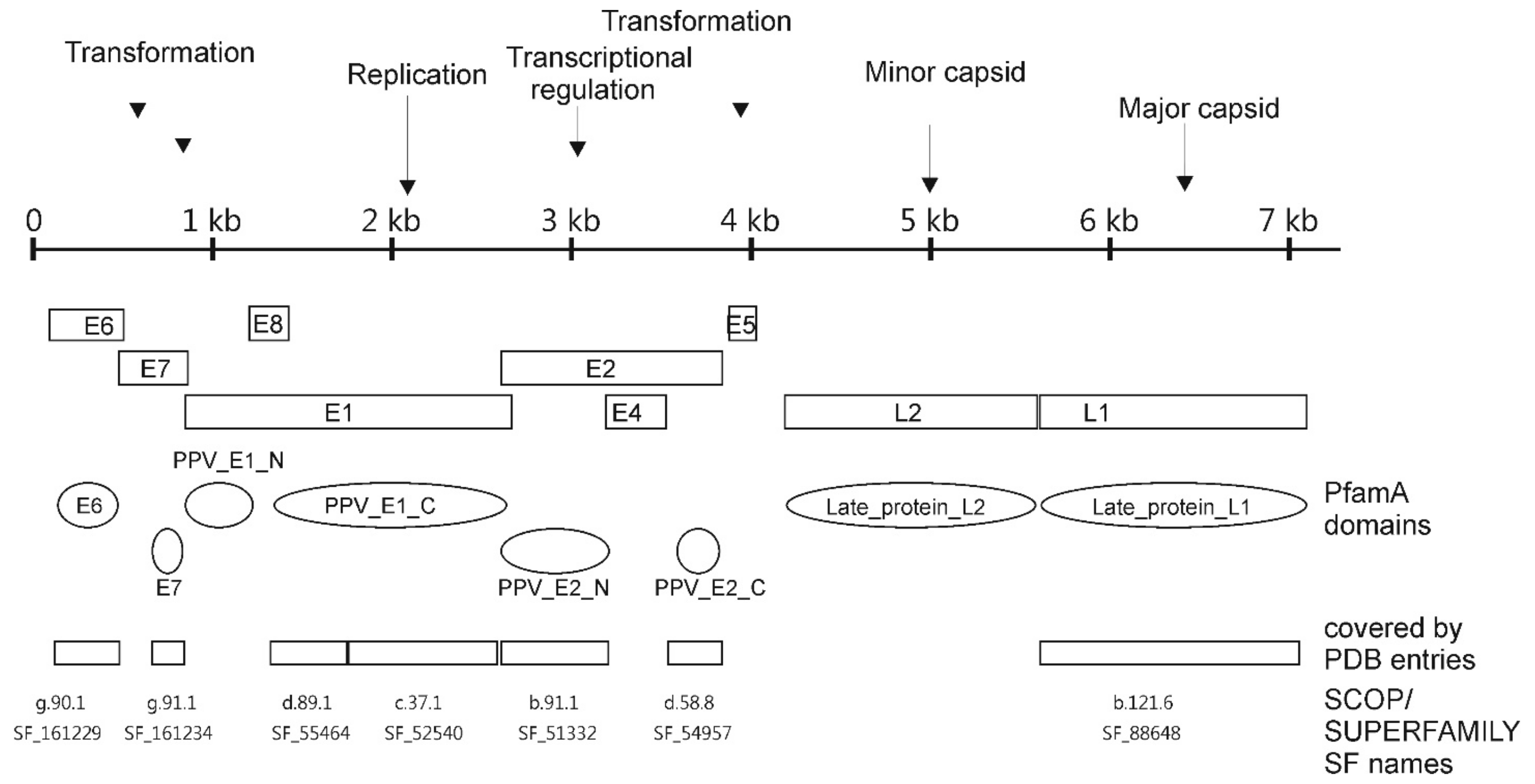
3.1. PfamA Protein Domains Found in PV
3.2. Relationships of PVs to the Sequenced Biosphere According to PfamA Domains
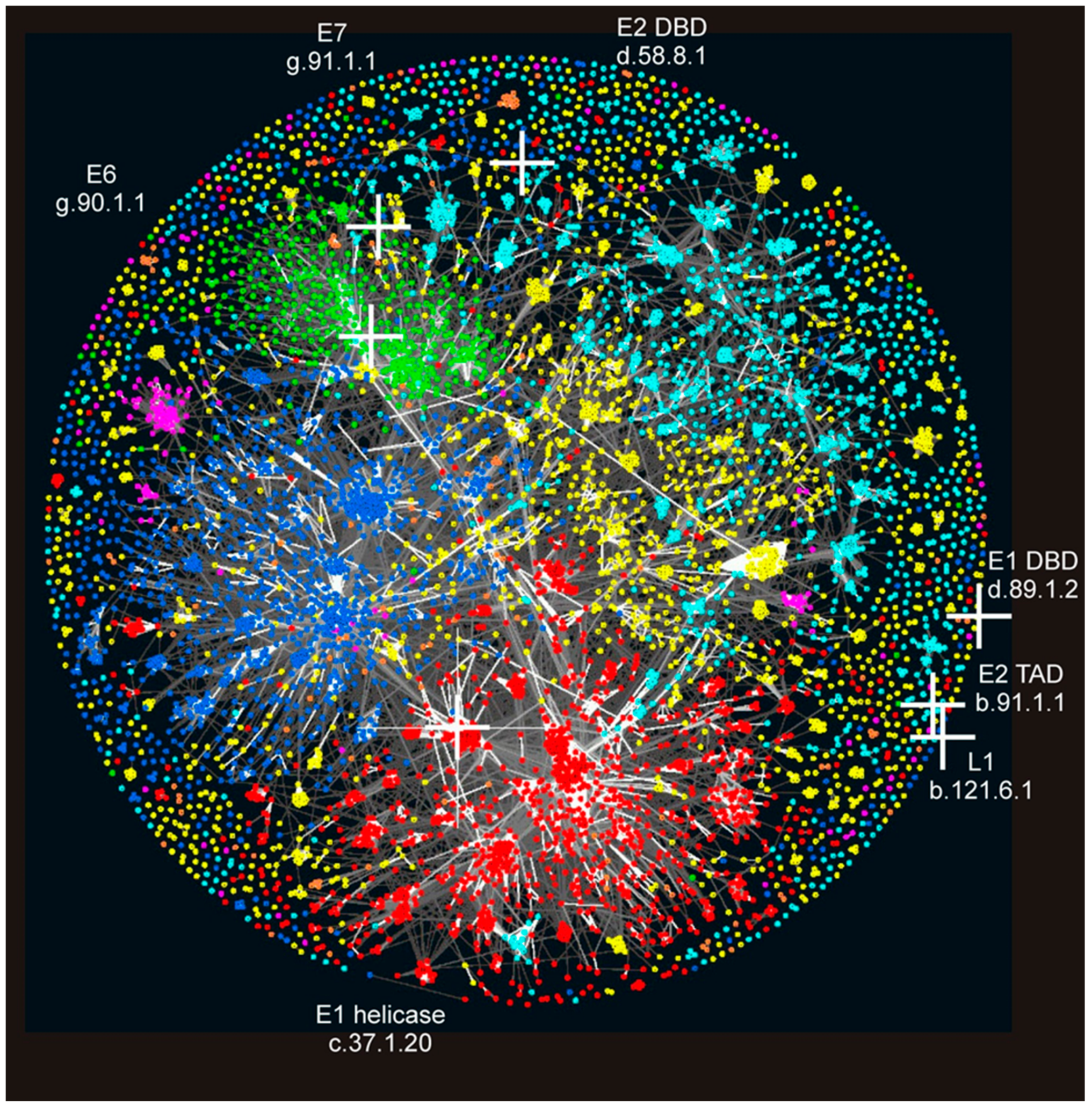
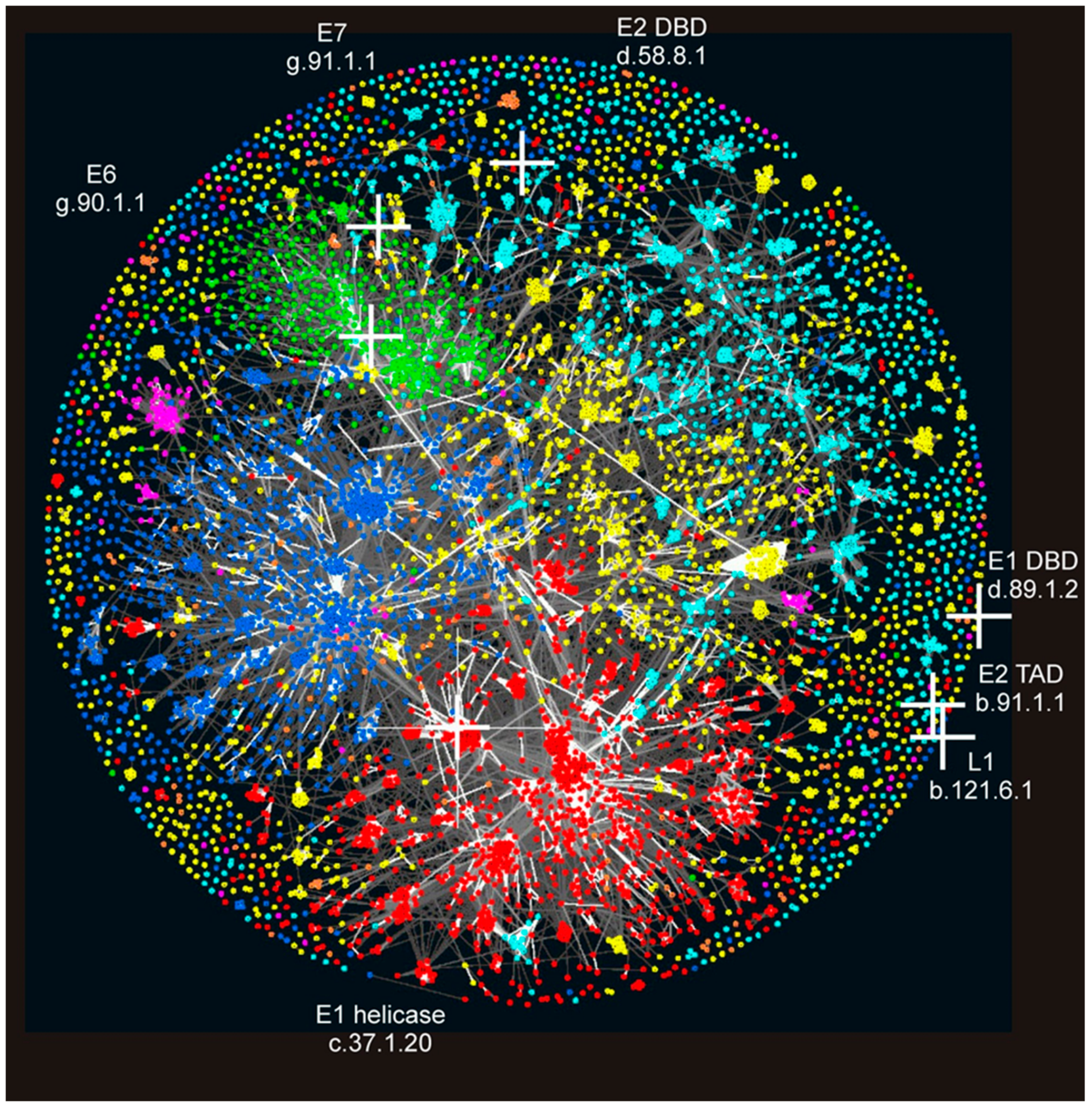
3.3. Location of PV Domains in the “Galaxy of Folds”
3.4. Structural Domains Found in PV Proteins According to SUPERFAMILY Analysis
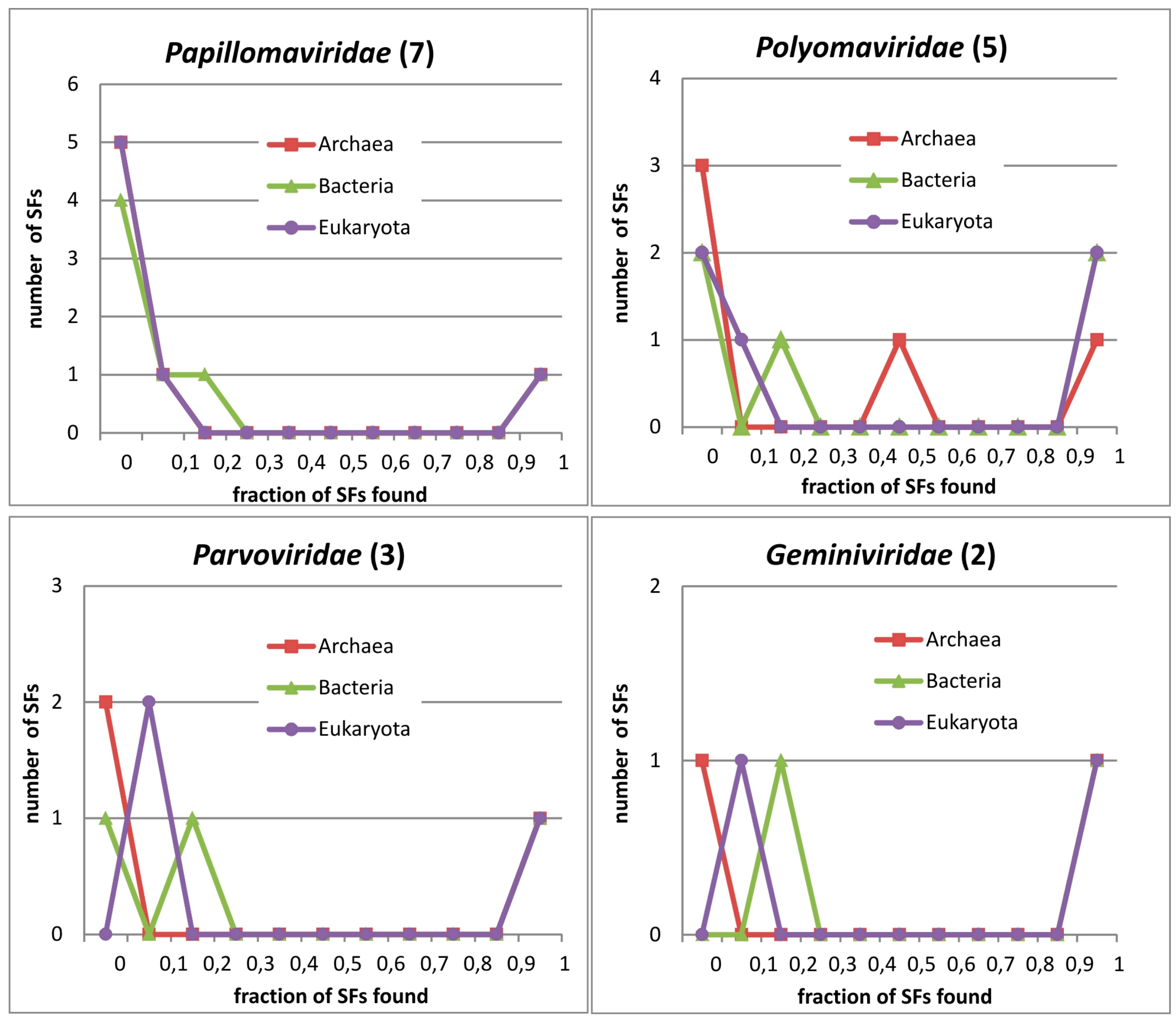
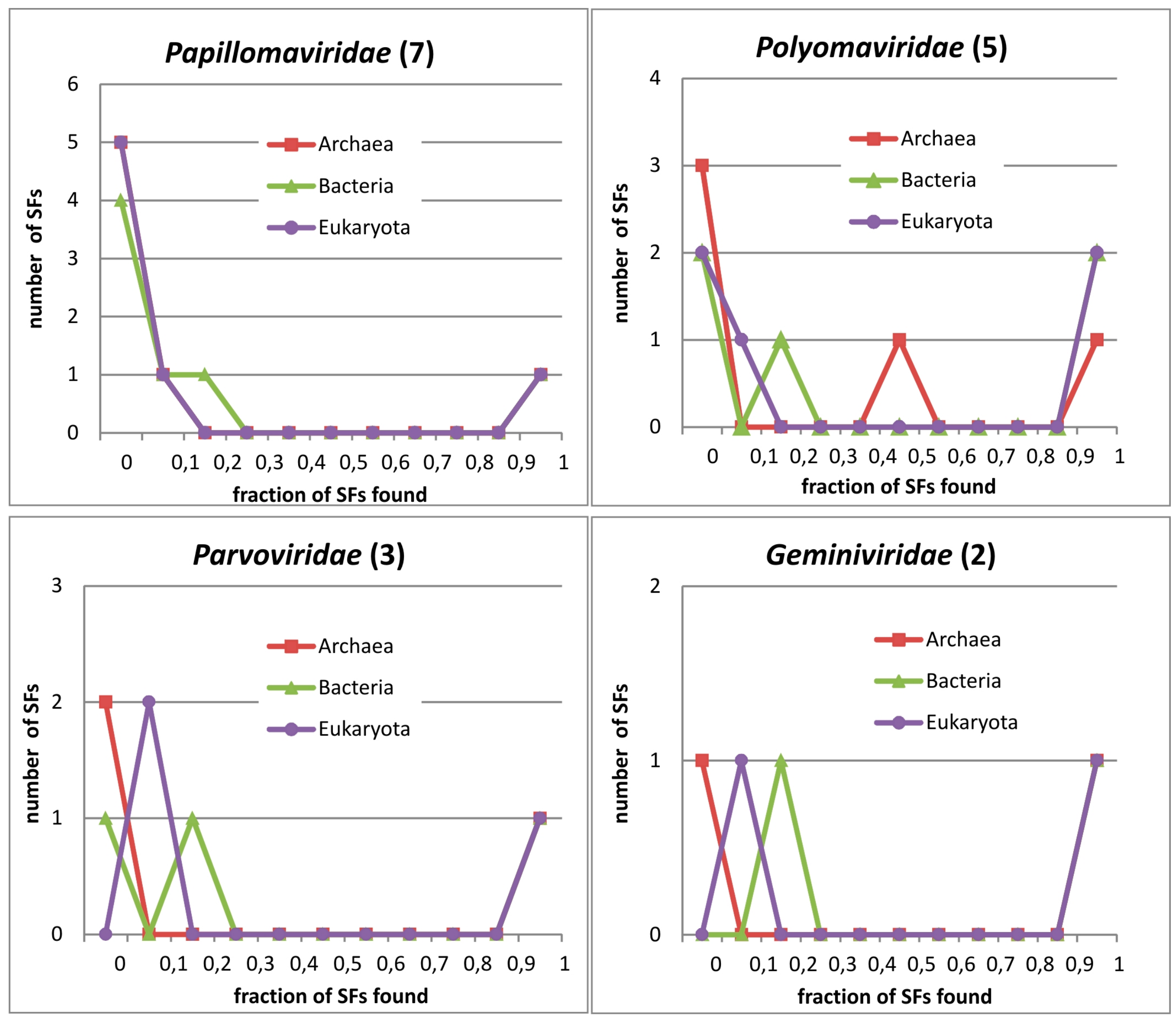
3.5. Phylogenetic Distribution of PV_SF Domains
3.6. Occurence of PV Protein Domains in Three Superkingdoms
3.7. Phylogenomic Distribution of the E1 SF_55464:SF_52540 Domain Pair
4. Discussion
4.1. SUPERFAMILY Limitations
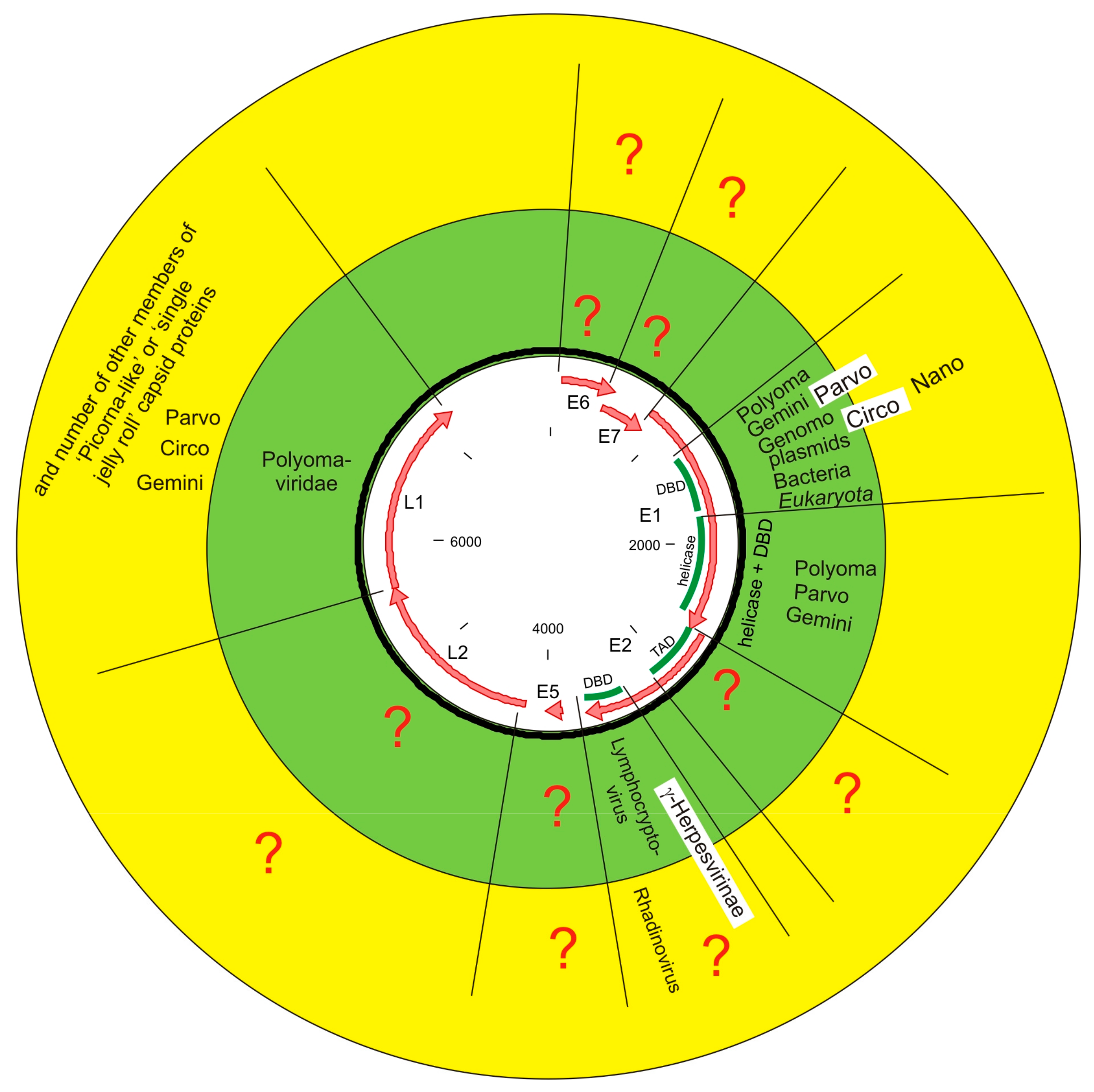
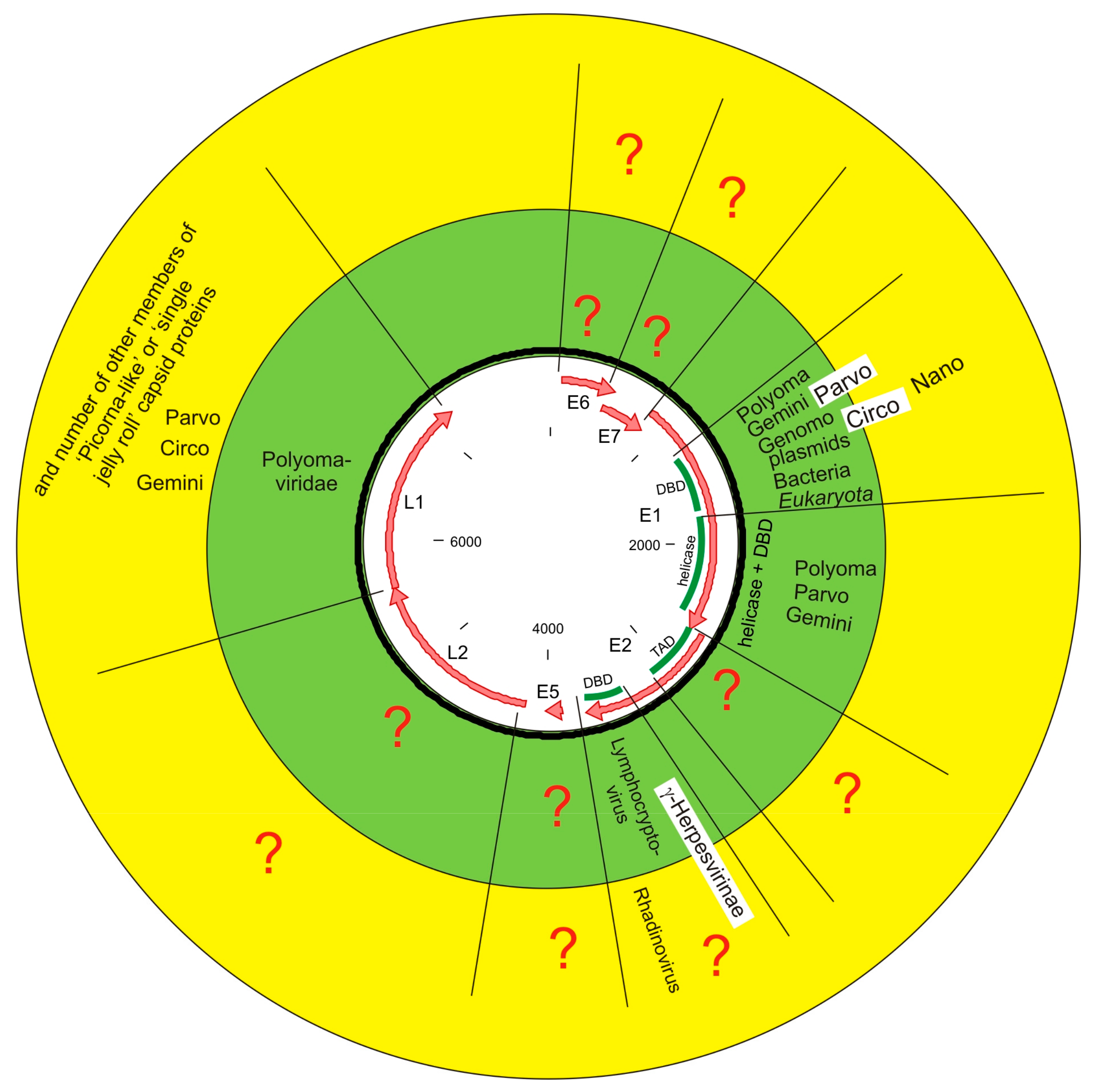
4.2. Capsid Protein Connects PVs with a Rest of the Virosphere
4.3. E2 DBD Most Likely Does Not Originate from Gammaherpesviruses
4.4. Replication Protein Connects PVs with a Rest of Biosphere
4.5. Closest Domain Pair of E1 Protein Is Found Far from Known PV Hosts
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shope, R.E.; Hurst, E.W. Infectious Papillomatosis of Rabbits: With a Note on the Histopathology. J. Exp. Med. 1933, 58, 607–624. [Google Scholar] [CrossRef] [PubMed]
- Papillomavirus Episteme. Available online: https://pave.niaid.nih.gov (accessed on 8 June 2017).
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Danos, O.; Katinka, M.; Yaniv, M. Human papillomavirus 1a complete DNA sequence: A novel type of genome organization among papovaviridae. EMBO J. 1982, 1, 231–236. [Google Scholar] [PubMed]
- Chen, E.Y.; Howley, P.M.; Levinson, A.D.; Seeburg, P.H. The primary structure and genetic organization of the bovine papillomavirus type 1 genome. Nature 1982, 299, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Danos, O.; Engel, L.W.; Chen, E.Y.; Yaniv, M.; Howley, P.M. Comparative analysis of the human type 1a and bovine type 1 papillomavirus genomes. J. Virol. 1983, 46, 557–566. [Google Scholar] [PubMed]
- Clertant, P.; Seif, I. A common function for polyoma virus large-T and papillomavirus E1 proteins? Nature 1984, 311, 276–279. [Google Scholar] [CrossRef]
- Karlin, S.; Ghandour, G.; Foulser, D.E.; Korn, L.J. Comparative analysis of human and bovine papillomaviruses. Mol. Biol. Evol. 1984, 1, 357–370. [Google Scholar] [PubMed]
- Fuchs, P.G.; Iftner, T.; Weninger, J.; Pfister, H. Epidermodysplasia verruciformis-associated human papillomavirus 8: Genomic sequence and comparative analysis. J. Virol. 1986, 58, 626–634. [Google Scholar] [PubMed]
- Campione-Piccardo, J.; Montpetit, M.L.; Grégoire, L.; Arella, M. A highly conserved nucleotide string shared by all genomes of human papillomaviruses. Virus Genes 1991, 5, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.D.; Doorbar, J.; Goldstein, R.A. Analysis of host-parasite incongruence in papillomavirus evolution using importance sampling. Mol. Biol. Evol. 2010, 27, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, M.; Stamatakis, A.; Nindl, I.; Stockfleth, E.; Alonso, A.; Bravo, I.G. Multiple evolutionary mechanisms drive papillomavirus diversification. Mol. Biol. Evol. 2007, 24, 1242–1258. [Google Scholar] [CrossRef] [PubMed]
- Pimenoff, V.N.; de Oliveira, C.M.; Bravo, I.G. Transmission between archaic and modern human ancestors during the evolution of the oncogenic human papillomavirus 16. Mol. Biol. Evol. 2017, 34, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K. Evolution of the Papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; McBride, A.A. Molecular archeological evidence in support of the repeated loss of a papillomavirus gene. Sci. Rep. 2016, 6, 33028. [Google Scholar] [CrossRef] [PubMed]
- UniProt Rereference Proteomes. Available online: http://www.uniprot.org/help/reference_proteome (accessed on 8 June 2017).
- UniProt Proteomes. Available online: http://www.uniprot.org/help/proteome (accessed on 8 June 2017).
- Herbst, L.H.; Lenz, J.; van Doorslaer, K.; Chen, Z.; Stacy, B.A.; Wellehan, J.F.X.; Manire, C.A.; Burk, R.D. Genomic characterization of two novel reptilian papillomaviruses, Chelonia mydas papillomavirus 1 and Caretta caretta papillomavirus 1. Virology 2009, 383, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Rector, A.; van Ranst, M. Animal papillomaviruses. Virology 2013, 445, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Rector, A.; Lemey, P.; Tachezy, R.; Mostmans, S.; Ghim, S.-J.; van Doorslaer, K.; Roelke, M.; Bush, M.; Montali, R.J.; Joslin, J.; et al. Ancient papillomavirus-host co-speciation in Felidae. Genome Biol. 2007, 8, R57. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Subramanian, S. Mutation rates in mammalian genomes. Proc. Natl. Acad. Sci. USA. 2002, 99, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Katzourakis, A. Time-Dependent Rate Phenomenon in Viruses. J. Virol. 2016, 90, 7184–7195. [Google Scholar] [CrossRef] [PubMed]
- Bravo, I.G.; Felez-Sanchez, M. Papillomaviruses: Viral evolution, cancer and evolutionary medicine. Evol. Med. Public Heal. 2015, 2015, 32–51. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [PubMed]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure is three to ten times more conserved than sequence—A study of structural response in protein cores. Proteins Struct. Funct. Bioinform. 2009, 77, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Challis, C.J.; Schmidler, S.C. A Stochastic Evolutionary Model for Protein Structure Alignment and Phylogeny. Mol. Biol. Evol. 2012, 29, 3575–3587. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.L.; Challis, C.J.; Novák, Á.; Hein, J.; Schmidler, S.C. Simultaneous Bayesian estimation of alignment and phylogeny under a joint model of protein sequence and structure. Mol. Biol. Evol. 2014, 31, 2251–2266. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Stahlhacke, J.; Vavoulis, D.V.; Smithers, B.; Rackham, O.J.L.; Sardar, A.J.; Zaucha, J.; Thurlby, N.; Fang, H.; Gough, J. The SUPERFAMILY 1.75 database in 2014: A doubling of data. Nucleic Acids Res. 2015, 43, 227–233. [Google Scholar] [CrossRef] [PubMed]
- HMMER: Biosequence Analysis Using Profile Hidden Markov Models. Available online: http://www.hmmer.org/ (accessed on 8 June 2017).
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; van Doorslaer, K.; Peretti, A.; Geoghegan, E.M.; Tisza, M.J.; An, P.; Katz, J.P.; Pipas, J.M.; McBride, A.A.; Camus, A.C.; et al. The Ancient Evolutionary History of Polyomaviruses. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alva, V.; Remmert, M.; Biegert, A.; Lupas, A.N.; Söding, J. A galaxy of folds. Protein Sci. 2010, 19, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Galaxy of Folds. Available online: https://toolkit.tuebingen.mpg.de/hhcluster/ (accessed on 27 May 2016).
- Gough, J.; Karplus, K.; Hughey, R.; Chothia, C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 2001, 313, 903–919. [Google Scholar] [CrossRef] [PubMed]
- Astell, C.R.; Mol, C.D.; Anderson, W.F. Structural and functional homology of parvovirus and papovavirus polypeptides. J. Gen. Virol. 1987, 68, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Abroi, A. A protein domain-based view of the virosphere-host relationship. Biochimie 2015, 119, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, I.; Lewis, T.E.; Cuff, A.; Das, S.; Ashford, P.; Dawson, N.L.; Furnham, N.; Laskowski, R.A.; Lee, D.; Lees, J.G.; et al. CATH: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015, 43, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Gough, J. Convergent evolution of domain architectures (is rare). Bioinformatics 2005, 21, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.; de la Cruz, F.; Dyda, F.; Hickman, A.B.; Moncalian, G.; Ton-Hoang, B. Breaking and joining single-stranded DNA: The HUH endonuclease superfamily. Nat. Rev. Microbiol. 2013, 11, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Ghabrial, S.A.; Jiang, D.; Varsani, A. Genomoviridae: A new family of widespread single-stranded DNA viruses. Arch. Virol. 2016, 161, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Abroi, A.; Gough, J. Are viruses a source of new protein folds for organisms?—Virosphere structure space and evolution. BioEssays 2011, 33, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Howorth, D.; Chothia, C.; Kulesha, E.; Murzin, A.G. SCOP2 prototype: A new approach to protein structure mining. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.K.; Brenner, S.E.; Chandonia, J.-M. SCOPe: Structural Classification of Proteins—Extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014, 42, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rocha, S.; Gronenborn, B.; Gronenborn, A.; Campos-Olivas, R. Solution structure of the endonuclease domain from the master replication initiator protein of the nanovirus faba bean necrotic yellows virus and comparison with the corresponding geminivirus and circovirus structures. Biochemistry 2007, 46, 6201–6212. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, J.; Krupovic, M.; Koonin, E.V. The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Breitburd, F.; Croissant, O.; Orth, G. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J. Virol. 1977, 21, 1205–1209. [Google Scholar] [PubMed]
- Friedmann, T.; David, D. Structural roles of polyoma virus proteins. J. Virol. 1972, 10, 776–782. [Google Scholar] [PubMed]
- Krupovic, M.; Koonin, E.V. Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef] [PubMed]
- Abrescia, N.G.A.; Bamford, D.H.; Grimes, J.M.; Stuart, D.I. Structure Unifies the Viral Universe. Annu. Rev. Biochem. 2012, 81, 795–822. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.; Cerqueira, S.A.; Beauchemin, C.; Pires de Miranda, M.; Li, S.; Ponnusamy, R.; Rodrigues, L.; Schneider, T.R.; Carrondo, M.A.; Kaye, K.M.; et al. Crystal Structure of the γ-2 Herpesvirus LANA DNA Binding Domain Identifies Charged Surface Residues Which Impact Viral Latency. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Hellert, J.; Weidner-Glunde, M.; Krausze, J.; Richter, U.; Adler, H.; Fedorov, R.; Pietrek, M.; Rückert, J.; Ritter, C.; Schulz, T.F.; et al. A Structural Basis for BRD2/4-Mediated Host Chromatin Interaction and Oligomer Assembly of Kaposi Sarcoma-Associated Herpesvirus and Murine γherpesvirus LANA Proteins. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Domsic, J.F.; Chen, H.S.; Lu, F.; Marmorstein, R.; Lieberman, P.M. Molecular Basis for Oligomeric-DNA Binding and Episome Maintenance by KSHV LANA. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, R.; Petoukhov, M.V.; Correia, B.; Custodio, T.F.; Juillard, F.; Tan, M.; Pires De Miranda, M.; Carrondo, M.A.; Simas, J.P.; Kaye, K.M.; et al. KSHV but not MHV-68 LANA induces a strong bend upon binding to terminal repeat viral DNA. Nucleic Acids Res. 2015, 43, 10039–10054. [Google Scholar] [CrossRef] [PubMed]
- Grose, C. Pangaea and the Out-of-Africa Model of Varicella-Zoster Virus Evolution and Phylogeography. J. Virol. 2012, 86, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef] [PubMed]
- López-Bueno, A.; Mavian, C.; Labella, A.M.; Castro, D.; Borrego, J.J.; Alcami, A.; Alejo, A. Concurrence of Iridovirus, Polyomavirus, and a Unique Member of a New Group of Fish Papillomaviruses in Lymphocystis Disease-Affected Gilthead Sea Bream. J. Virol. 2016, 90, 8768–8779. [Google Scholar] [CrossRef] [PubMed]
- Saccardo, F.; Cettul, E.; Palmano, S.; Noris, E.; Firrao, G. On the alleged origin of geminiviruses from extrachromosomal DNAs of phytoplasmas. BMC Evol. Biol. 2011, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Ravantti, J.J.; Bamford, D.H. Geminiviruses: A tale of a plasmid becoming a virus. BMC Evol. Biol. 2009, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.J.; Weindler, F.W.; Gray, D.; Schwaab, V.; Heilbronn, R. Human herpesvirus-6 (HHV-6) is a helper virus for adenoassociated virus type-2 (AAV-2) and the AAV-2 Rep gene homolog in HHV-6 can mediate AAV-2 DNA-replication and regulate gene-expression. Virology 1994, 204, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.J.; Efstathiou, S.; Honess, R.W. Acquisition of the Human Adenoassociated Virus Type-2 Rep Gene By Human Herpesvirus Type-6. Nature 1991, 351, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Barott, K. Viral information. Biol. Philos. 2013, 28, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PfamA_28 * | SUPERFAMILY | |||||
|---|---|---|---|---|---|---|
| Sequence Coverage 1 | Residue Coverage 2 | No. of Genomes | Sequence Coverage 1 | Residue Coverage 2 | No. of Genomes | |
| Archaea | 73.8 | 58.0 | 182 | 64.4 | 61.1 | 122 |
| Bacteria | 82.0 | 63.3 | 3513 | 67.6 | 62.6 | 1153 |
| Eukaryota | 67.9 | 38.6 | 422 | 56.9 | 38.8 | 440 |
| Viruses | 84.4 | 65.7 | 1198 | 34.3 | 28.1 | 4041 |
| dsDNA viruses | 62.5 | 52.9 | 270 | 24.8 | 25.4 | 1758 |
| Papillomaviridae | 90.8 | 83.8 | 76 | 69.5 | 57.5 | 125 |
| Polyomaviridae | 92.5 | 70.3 | 10 | 60.2 | 65.3 | 50 |
| Parvoviridae | 74.7 | 56.3 | 23 | 69.5 | 55.0 | 81 |
| Geminiviridae | 97.0 | 79.9 | 34 | 18.5 | 15.1 | 332 |
| Herpesviridae | 74.2 | 53.6 | 28 | 27.6 | 20.7 | 57 |
| Papillomaviridae 1,5 | PDB PfamA_28 2 | PfamA Domain Length 3 | PDB PfamA_31 2 | Best Coverage of PfamA by PDB (% aa) | Eukaryota (Proteomes) 1 | Bacteria (Proteomes) 1 | Archaea (Proteomes) 1 | Viruses 1,4 | Eukaryota (Full up) 1 | Bacteria (Full up) 1 | Archaea (Full up) 1 | Viruses (Full up) 1,4,6 | HMMER E 1 | HMMER B 1 | HMMER A 1 | HMMER V 1,6 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PF00500 | Late_protein_L1 | 76 | 10 | 498 | 18 | 0.96 | - | - | - | - | - | - | - | - | - | - | - | - |
| PF00508 | PPV_E2_N | 76 | 8 | 200 | 8 | 0.98 | - | - | - | - | - | - | - | - | - | - | - | - |
| PF00511 | PPV_E2_C | 76 | 16 | 80 | 16 | 0.96 | - | - | - | - | - | - | - | - | - | - | - | - |
| PF00513 | Late_protein_L2 | 76 | 0 | 525 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | |
| PF00518 | E6 | 71 | 7 | 110 | 8 | 0.99 | - | - | - | - | - | - | - | - | - | - | - | - |
| PF00519 | PPV_E1_C | 74 | 7 | 432 | 8 | 0.96 | - | 1 | - | - | - | 20 | - | 1 | - | 1 | - | 1 |
| PF00524 | PPV_E1_N | 72 | 0 | 121 | 0 | 2 | - | - | - | 4 | - | - | - | - | - | - | - | |
| PF00527 | E7 | 71 | 3 | 93 | 4 | 0.50 | - | - | - | - | - | - | - | - | - | - | - | - |
| PF02711 | Pap_E4 | 25 | 0 | 95 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | |
| PF03025 | Papilloma_E5 | 9 | 0 | 72 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | |
| PF05776 | Papilloma_E5A | 5 | 0 | 91 | 0 | - | - | - | - | - | - | - | - | - | - | - | - | |
| PF08135 | EPV_E5 | 3 | 0 | 43 | 0 | - | - | - | - | - | - | - | - | - | - | - | - |
| SCOP/SF ID | Classification | SF/FOLD | Families/SF | Description | PV | Viruses 1 | Plasmids 2 | Archaea | Bacteria | Eukaryota | HMMER A | HMMER B | HMMER E | HMMER V 1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 55464 | d.89.1 | 1 | 5 | Origin of replication-binding domain, RBD-like (E1 DBD) | 123 | 424/15 * | 420 | - | 134 | 8 | - | 4038 | 32 | 1563/169 * |
| 52540 | c.37.1 | 1 | 24 | P-loop containing nucleoside triphosphate hydrolases (E1 helicase) | 123 | 2346 | 19971 | 122 | 1153 | 440 | ND | ND | ND | ND |
| 51332 | b.91.1 | 1 | 1 | E2 regulatory, transactivation domain (E2 TAD) | 123 | - | - | - | - | - | - | - | - | - |
| 54957 | d.58.8 | 59 | 1 | Viral DNA-binding domain (E2 DBD) | 123 | 4 | - | - | - | - | - | - | - | 6 |
| 88648 | b.121.6 | 7 | 1 | Group I dsDNA viruses (L1) | 123 | 50/- * | - | - | - | - | - | - | - | 170/- * |
| 161229 | g.90.1 | 1 | 1 | E6 C-terminal domain-like | 115 | - | - | - | - | - | - | 1? | - | - |
| 161234 | g.91.1 | 1 | 1 | E7 C-terminal domain-like | 108 | - | - | - | - | - | - | - | - | - |
| 55464:52540 | DBD + helicase | 123 | 7 | 356 | - | 119 | 5 | - | ND | 10 | ||||
| No. of 52540 Domains | PV 1 123 * | Polyomaviridae 2 50 * | Parvoviridae 81 * | Geminiviridae 3 332 * | Other Viruses | Plasmids | Bacteria | Eukaryota |
|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 0 | 0 | 350 | 10 | 64 | 35 | 4 |
| 1 | 122 | 49 | 33 | 14 | 6 | 20 | 20 | 5 |
| 2 | 0 | 0 | 0 | 0 | 1 | 334 | 183 | 0 |
| 3 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puustusmaa, M.; Kirsip, H.; Gaston, K.; Abroi, A. The Enigmatic Origin of Papillomavirus Protein Domains. Viruses 2017, 9, 240. https://doi.org/10.3390/v9090240
Puustusmaa M, Kirsip H, Gaston K, Abroi A. The Enigmatic Origin of Papillomavirus Protein Domains. Viruses. 2017; 9(9):240. https://doi.org/10.3390/v9090240
Chicago/Turabian StylePuustusmaa, Mikk, Heleri Kirsip, Kevin Gaston, and Aare Abroi. 2017. "The Enigmatic Origin of Papillomavirus Protein Domains" Viruses 9, no. 9: 240. https://doi.org/10.3390/v9090240
APA StylePuustusmaa, M., Kirsip, H., Gaston, K., & Abroi, A. (2017). The Enigmatic Origin of Papillomavirus Protein Domains. Viruses, 9(9), 240. https://doi.org/10.3390/v9090240