
Host and Viral Factors in HIV-Mediated Bystander Apoptosis

Abstract
1. Introduction
2. Viral Factors
2.1. Env Glycoprotein
2.2. Mechanism of HIV Env-Mediated Apoptosis: Role of gp120 and gp41
2.3. HIV Env Glycoprotein Variability and Evolution
2.4. Signaling via HIV Env Glycoprotein
2.5. Targeting HIV gp41 Can Alter Bystander Apoptosis
2.6. Fas, TNF/TRAIL and Other Viral Proteins in Bystander Apoptosis
2.7. Effects of HIV Env on Cell Types Other Than CD4 T Cells
3. Host Factors
3.1. CCR5: Role of CCR5 in HIV Disease
3.2. CCR5 Polymorphisms
3.3. CCR5 Env Interaction in HIV-Mediated Apoptosis
3.4. CCR5 in Primate Models of SIV Infection
4. Immune Activation
4.1. Immune Activation in HIV Disease
4.2. Viremia and Immune Activation
4.3. Microbial Translocation, Th17 Depletion and Immune Activation
4.4. Toll-Like Receptors in Immune Activation
4.5. Immune Activation and Apoptosis
5. Other Pathways of Cell Death
5.1. Role of Autophagy in HIV-Mediated Cell Death
5.2. Role of Pyroptosis in HIV-Mediated Cell Death
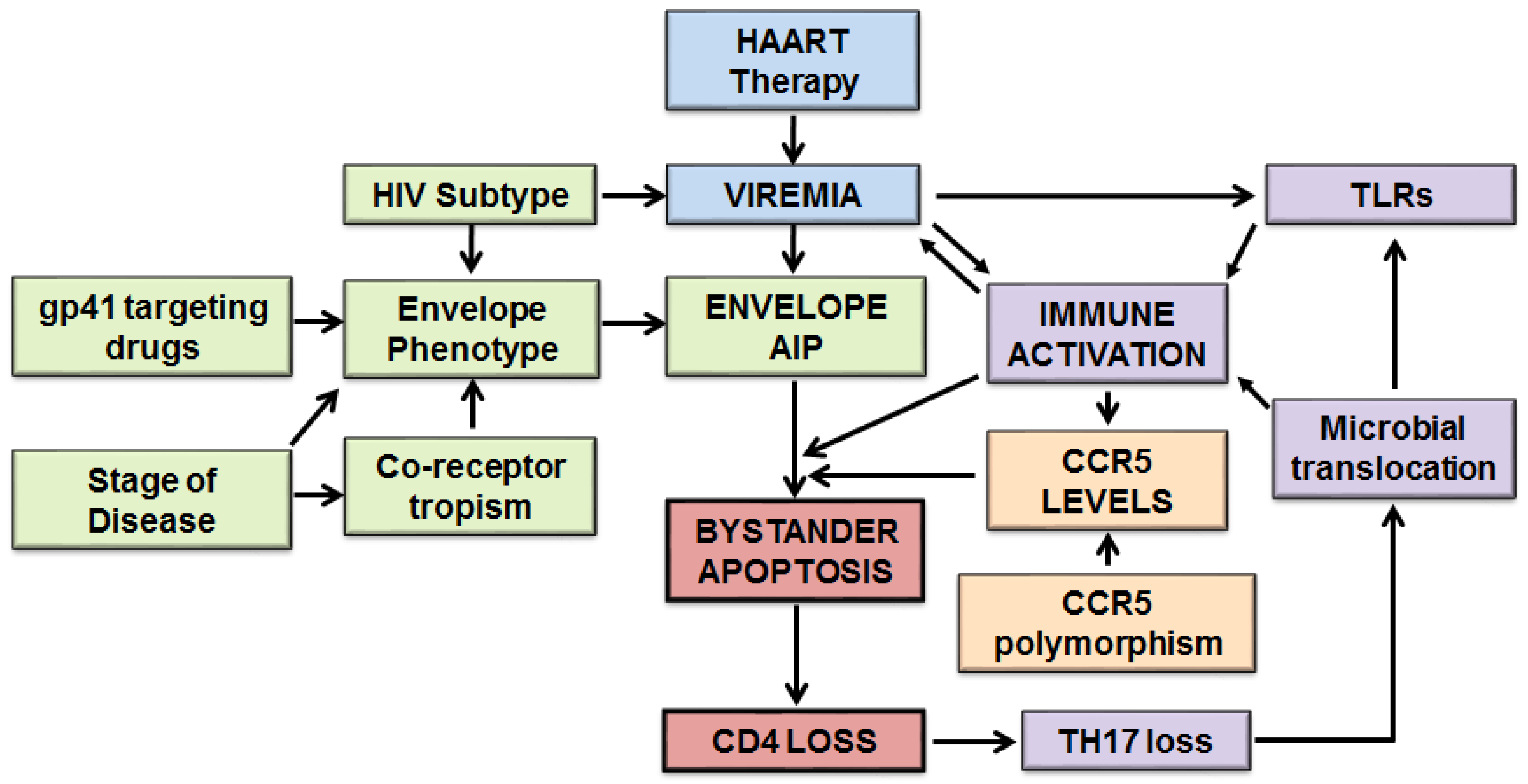
6. Model of HIV-Mediated Bystander Apoptosis
6.1. Detailed Model of Host and Viral Factors in HIV-Mediated Bystander Apoptosis
6.2. Host and Viral Factors Determining Differential CD4 Loss in HIV Infections
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pantaleo, G.; Fauci, A.S. Apoptosis in HIV infection. Nat. Med. 1995, 1, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Oyaizu, N.; Pahwa, S. Role of apoptosis in HIV disease pathogenesis. J. Clin. Immunol. 1995, 15, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, M.; Montagnier, L. Apoptosis in AIDS. Science 1993, 260, 1269–1270. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.; Sodora, D.; Koup, R.; Paiardini, M.; O’Neil, S.; McClure, H.; Staprans, S.; Feinberg, M. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 2003, 18, 441–452. [Google Scholar] [CrossRef]
- Meythaler, M.; Martinot, A.; Wang, Z.; Pryputniewicz, S.; Kasheta, M.; Ling, B.; Marx, P.A.; O’Neil, S.; Kaur, A. Differential CD4+ T-lymphocyte apoptosis and bystander T-cell activation in rhesus macaques and sooty mangabeys during acute simian immunodeficiency virus infection. J. Virol. 2009, 83, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Meythaler, M.; Pryputniewicz, S.; Kaur, A. Kinetics of T lymphocyte apoptosis and the cellular immune response in SIVmac239-infected rhesus macaques. J. Med. Primatol. 2008, 37, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Cumont, M.C.; Diop, O.; Vaslin, B.; Elbim, C.; Viollet, L.; Monceaux, V.; Lay, S.; Silvestri, G.; Le Grand, R.; Muller-Trutwin, M.; et al. Early divergence in lymphoid tissue apoptosis between pathogenic and nonpathogenic simian immunodeficiency virus infections of nonhuman primates. J. Virol. 2008, 82, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Reinberger, S.; Spring, M.; Nisslein, T.; Stahl-Hennig, C.; Hunsmann, G.; Dittmer, U. Kinetics of lymphocyte apoptosis in macaques infected with different simian immunodeficiency viruses or simian/human immunodeficiency hybrid viruses. Clin. Immunol. 1999, 90, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Ichimura, H.; Shimada, T.; Ibuki, K.; Ui, M.; Tamaru, K.; Kuwata, T.; Yonehara, S.; Imanishi, J.; Hayami, M. Role of apoptosis induction in both peripheral lymph nodes and thymus in progressive loss of CD4+ cells in SHIV-infected macaques. AIDS Res. Hum. Retrovir. 2000, 16, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Sedano, M.; Beauchamp, B.; Punke, E.B.; Mulla, Z.D.; Meza, A.; Alozie, O.K.; Mukherjee, D.; Garg, H. HIV-1 Env Glycoprotein phenotype along with immune activation determines CD4 T cell loss in HIV patients. J. Immunol. 2016, 196, 1768–1779. [Google Scholar] [CrossRef] [PubMed]
- Sternfeld, T.; Tischleder, A.; Schuster, M.; Bogner, J.R. Mitochondrial membrane potential and apoptosis of blood mononuclear cells in untreated HIV-1 infected patients. HIV Med. 2009, 10, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Oyaizu, N.; McCloskey, T.W.; Coronesi, M.; Chirmule, N.; Kalyanaraman, V.S.; Pahwa, S. Accelerated apoptosis in peripheral blood mononuclear cells (PBMCs) from human immunodeficiency virus type-1 infected patients and in CD4 cross-linked PBMCs from normal individuals. Blood 1993, 82, 3392–3400. [Google Scholar] [PubMed]
- Leng, Q.; Borkow, G.; Weisman, Z.; Stein, M.; Kalinkovich, A.; Bentwich, Z. Immune activation correlates better than HIV plasma viral load with CD4 T-cell decline during HIV infection. J. Acquir. Immune Defic. Syndr. 2001, 27, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.; Carneiro, J.; Meier-Schellersheim, M.; Grossman, Z.; Victorino, R. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J. Immunol. 2002, 169, 3400–3406. [Google Scholar] [CrossRef] [PubMed]
- Resino, S.; Seoane, E.; Gutiérrez, M.; León, J.; Muñoz-Fernández, M. CD4(+) T-cell immunodeficiency is more dependent on immune activation than viral load in HIV-infected children on highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2006, 42, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.J.; Tamma, S.L.; Kaplan, M.; Gersten, M.; Pahwa, S.G. Reduction in T cell apoptosis in patients with HIV disease following antiretroviral therapy. Clin. Immunol. 1999, 93, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Roger, P.M.; Breittmayer, J.P.; Arlotto, C.; Pugliese, P.; Pradier, C.; Bernard-Pomier, G.; Dellamonica, P.; Bernard, A. Highly active anti-retroviral therapy (HAART) is associated with a lower level of CD4+ T cell apoptosis in HIV-infected patients. Clin. Exp. Immunol. 1999, 118, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Roger, P.M.; Breittmayer, J.P.; Durant, J.; Sanderson, F.; Ceppi, C.; Brignone, C.; Cua, E.; Clevenbergh, P.; Fuzibet, J.G.; Pesce, A.; et al. Early CD4(+) T cell recovery in human immunodeficiency virus-infected patients receiving effective therapy is related to a down-regulation of apoptosis and not to proliferation. J. Infect. Dis. 2002, 185, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Mocroft, A.; Ledergerber, B.; Katlama, C.; Kirk, O.; Reiss, P.; d’Arminio Monforte, A.; Knysz, B.; Dietrich, M.; Phillips, A.N.; Lundgren, J.D. Decline in the AIDS and death rates in the EuroSIDA study: An observational study. Lancet 2003, 362, 22–29. [Google Scholar] [CrossRef]
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Blumenthal, R. Role of HIV Gp41 mediated fusion/hemifusion in bystander apoptosis. Cell. Mol. Life Sci. 2008, 65, 3134–3144. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Banda, N. Indirect mechanisms of HIV pathogenesis: How does HIV kill T cells? Curr. Opin. Immunol. 1994, 6, 605–615. [Google Scholar] [CrossRef]
- Finkel, T.; Tudor-Williams, G.; Banda, N.; Cotton, M.; Curiel, T.; Monks, C.; Baba, T.; Ruprecht, R.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Dunham, R.; Engram, J.; Estes, J.; Wang, Z.; Klatt, N.; Paiardini, M.; Pandrea, I.; Apetrei, C.; Sodora, D.; et al. Short-lived infected cells support virus replication in sooty mangabeys naturally infected with simian immunodeficiency virus: Implications for AIDS pathogenesis. J. Virol. 2008, 82, 3725–3735. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, M.; Colizzi, V.; Dalgleish, A.; Montagnier, L. New concepts in AIDS pathogenesis. AIDS Res. Hum. Retrovir. 1993, 9, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Joshi, A.; Ye, C.; Shankar, P.; Manjunath, N. Single amino acid change in gp41 region of HIV-1 alters bystander apoptosis and CD4 decline in humanized mice. Virol. J. 2011, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Holm, G.H.; Gabuzda, D. Distinct mechanisms of CD4+ and CD8+ T-cell activation and bystander apoptosis induced by human immunodeficiency virus type 1 virions. J. Virol. 2005, 79, 6299–6311. [Google Scholar] [CrossRef] [PubMed]
- Holm, G.H.; Zhang, C.; Gorry, P.R.; Peden, K.; Schols, D.; De Clercq, E.; Gabuzda, D. Apoptosis of bystander T cells induced by human immunodeficiency virus type 1 with increased envelope/receptor affinity and coreceptor binding site exposure. J. Virol. 2004, 78, 4541–4551. [Google Scholar] [CrossRef] [PubMed]
- Tsao, L.C.; Guo, H.; Jeffrey, J.; Hoxie, J.A.; Su, L. CCR5 interaction with HIV-1 Env contributes to Env-induced depletion of CD4 T cells In Vitro and In Vivo. Retrovirology 2016, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Ahr, B.; Robert-Hebmann, V.; Devaux, C.; Biard-Piechaczyk, M. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology 2004, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Perfettini, J.; Castedo, M.; Roumier, T.; Andreau, K.; Nardacci, R.; Piacentini, M.; Kroemer, G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005, 12, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Crawford, A.G.; Coccia, E.; Krust, B.; Hovanessian, A.G. Membrane-expressed HIV envelope glycoprotein heterodimer is a powerful inducer of cell death in uninfected CD4+ target cells. Res. Virol. 1995, 146, 5–17. [Google Scholar] [CrossRef]
- Laurent-Crawford, A.G.; Krust, B.; Riviere, Y.; Desgranges, C.; Muller, S.; Kieny, M.P.; Dauguet, C.; Hovanessian, A.G. Membrane expression of HIV envelope glycoproteins triggers apoptosis in CD4 cells. AIDS Res. Hum. Retrovir. 1993, 9, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Biard-Piechaczyk, M.; Robert-Hebmann, V.; Richard, V.; Roland, J.; Hipskind, R.; Devaux, C. Caspase-dependent apoptosis of cells expressing the chemokine receptor CXCR4 is induced by cell membrane-associated human immunodeficiency virus type 1 envelope glycoprotein (gp120). Virology 2000, 268, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Barretina, J.; Ferri, K.; Jacotot, E.; Gutiérrez, A.; Armand-Ugón, M.; Cabrera, C.; Kroemer, G.; Clotet, B.; Esté, J. Cell-surface-expressed HIV-1 envelope induces the death of CD4 T cells during GP41-mediated hemifusion-like events. Virology 2003, 305, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Blumenthal, R. HIV gp41-induced apoptosis is mediated by caspase-3-dependent mitochondrial depolarization, which is inhibited by HIV protease inhibitor nelfinavir. J. Leukoc. Biol. 2006, 79, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.; Sodroski, J. The HIV-1 envelope glycoproteins: Fusogens, antigens, and immunogens. Science 1998, 280, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.; Finnegan, C.; Viard, M.; Raviv, Y.; Dimitrov, A.; Rawat, S.; Puri, A.; Durell, S.; Blumenthal, R. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 2003, 1614, 36–50. [Google Scholar] [CrossRef]
- Wild, C.; Dubay, J.; Greenwell, T.; Baird, T.J.; Oas, T.; McDanal, C.; Hunter, E.; Matthews, T. Propensity for a leucine zipper-like domain of human immunodeficiency virus type 1 gp41 to form oligomers correlates with a role in virus-induced fusion rather than assembly of the glycoprotein complex. Proc. Natl. Acad. Sci. USA 1994, 91, 12676–12680. [Google Scholar] [CrossRef] [PubMed]
- Andreau, K.; Perfettini, J.; Castedo, M.; Métivier, D.; Scott, V.; Pierron, G.; Kroemer, G. Contagious apoptosis facilitated by the HIV-1 envelope: Fusion-induced cell-to-cell transmission of a lethal signal. J. Cell Sci. 2004, 117, 5643–5653. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Mohl, J.; Joshi, A. HIV-1 induced bystander apoptosis. Viruses 2012, 4, 3020–3043. [Google Scholar] [CrossRef] [PubMed]
- Biard-Piechaczyk, M.; Robert-Hebmann, V.; Roland, J.; Coudronniere, N.; Devaux, C. Role of CXCR4 in HIV-1-induced apoptosis of cells with a CD4+, CXCR4+ phenotype. Immunol. Lett. 1999, 70, 1–3. [Google Scholar] [CrossRef]
- Blanco, J.; Barretina, J.; Clotet, B.; Este, J.A. R5 HIV gp120-mediated cellular contacts induce the death of single CCR5-expressing CD4 T cells by a gp41-dependent mechanism. J. Leukoc. Biol. 2004, 76, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Joshi, A.; Freed, E.; Blumenthal, R. Site-specific mutations in HIV-1 gp41 reveal a correlation between HIV-1-mediated bystander apoptosis and fusion/hemifusion. J. Biol. Chem. 2007, 282, 16899–16906. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L.; Kozlov, M. Membrane hemifusion: Crossing a chasm in two leaps. Cell 2005, 123, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Symeonides, M.; Lambele, M.; Roy, N.H.; Thali, M. Evidence showing that tetraspanins inhibit HIV-1-induced cell-cell fusion at a post-hemifusion stage. Viruses 2014, 6, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Bar, S.; Alizon, M. Role of the ectodomain of the gp41 transmembrane envelope protein of human immunodeficiency virus type 1 in late steps of the membrane fusion process. J. Virol. 2004, 78, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Cunyat, F.; Marfil, S.; Garcia, E.; Svicher, V.; Perez-Alvarez, N.; Curriu, M.; Perno, C.F.; Clotet, B.; Blanco, J.; Cabrera, C. The HR2 polymorphism N140I in the HIV-1 gp41 combined with the HR1 V38A mutation is associated with a less cytopathic phenotype. Retrovirology 2012, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Koot, M.; van’t Wout, A.; Kootstra, N.; de Goede, R.; Tersmette, M.; Schuitemaker, H. Relation between changes in cellular load, evolution of viral phenotype, and the clonal composition of virus populations in the course of human immunodeficiency virus type 1 infection. J. Infect. Dis. 1996, 173, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Spijkerman, I.; de Wolf, F.; Langendam, M.; Schuitemaker, H.; Coutinho, R. Emergence of syncytium-inducing human immunodeficiency virus type 1 variants coincides with a transient increase in viral RNA level and is an independent predictor for progression to AIDS. J. Infect. Dis. 1998, 178, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Schuitemaker, H.; Koot, M.; Kootstra, N.; Dercksen, M.; de Goede, R.; van Steenwijk, R.; Lange, J.; Schattenkerk, J.; Miedema, F.; Tersmette, M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: Progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 1992, 66, 1354–1360. [Google Scholar] [PubMed]
- Van Rij, R.P.; Blaak, H.; Visser, J.; Brouwer, M.; Rientsma, R.; Broersen, S.; de Roda Husman, A.M.; Schuitemaker, H. Differential coreceptor expression allows for independent evolution of non-syncytium-inducing and syncytium-inducing HIV-1. J. Clin. Investig. 2000, 106, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Kaneshima, H.; Su, L.; Bonyhadi, M.; Connor, R.; Ho, D.; McCune, J. Rapid-high, syncytium-inducing isolates of human immunodeficiency virus type 1 induce cytopathicity in the human thymus of the SCID-hu mouse. J. Virol. 1994, 68, 8188–8192. [Google Scholar] [PubMed]
- Etemad-Moghadam, B.; Rhone, D.; Steenbeke, T.; Sun, Y.; Manola, J.; Gelman, R.; Fanton, J.W.; Racz, P.; Tenner-Racz, K.; Axthelm, M.K.; et al. Understanding the basis of CD4(+) T-cell depletion in macaques infected by a simian-human immunodeficiency virus. Vaccine 2002, 20, 1934–1937. [Google Scholar] [CrossRef]
- Etemad-Moghadam, B.; Rhone, D.; Steenbeke, T.; Sun, Y.; Manola, J.; Gelman, R.; Fanton, J.W.; Racz, P.; Tenner-Racz, K.; Axthelm, M.K.; et al. Membrane-fusing capacity of the human immunodeficiency virus envelope proteins determines the efficiency of CD+ T-cell depletion in macaques infected by a simian-human immunodeficiency virus. J. Virol. 2001, 75, 5646–5655. [Google Scholar] [CrossRef] [PubMed]
- Etemad-Moghadam, B.; Sun, Y.; Nicholson, E.K.; Fernandes, M.; Liou, K.; Gomila, R.; Lee, J.; Sodroski, J. Envelope glycoprotein determinants of increased fusogenicity in a pathogenic simian-human immunodeficiency virus (SHIV-KB9) passaged in vivo. J. Virol. 2000, 74, 4433–4440. [Google Scholar] [CrossRef] [PubMed]
- Meissner, E.G.; Coffield, V.M.; Su, L. Thymic pathogenicity of an HIV-1 envelope is associated with increased CXCR4 binding efficiency and V5-gp41-dependent activity, but not V1/V2-associated CD4 binding efficiency and viral entry. Virology 2005, 336, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Sterjovski, J.; Churchill, M.; Ellett, A.; Gray, L.; Roche, M.; Dunfee, R.; Purcell, D.; Saksena, N.; Wang, B.; Sonza, S.; et al. Asn 362 in gp120 contributes to enhanced fusogenicity by CCR5-restricted HIV-1 envelope glycoprotein variants from patients with AIDS. Retrovirology 2007, 4, 89. [Google Scholar] [CrossRef] [PubMed]
- Meissner, E.G.; Zhang, L.; Jiang, S.; Su, L. Fusion-induced apoptosis contributes to thymocyte depletion by a pathogenic human immunodeficiency virus type 1 envelope in the human thymus. J. Virol. 2006, 80, 11019–11030. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Lee, R.T.; Mohl, J.; Sedano, M.; Khong, W.X.; Ng, O.T.; Maurer-Stroh, S.; Garg, H. Genetic signatures of HIV-1 envelope-mediated bystander apoptosis. J. Biol. Chem. 2014, 289, 2497–2514. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Gaschen, B.; Yusim, K.; Thakallapally, R.; Kesmir, C.; Detours, V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br. Med. Bull. 2001, 58, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Curlin, M.E.; Zioni, R.; Hawes, S.E.; Liu, Y.; Deng, W.; Gottlieb, G.S.; Zhu, T.; Mullins, J.I. HIV-1 envelope subregion length variation during disease progression. PLoS Pathog. 2010, 6, e1001228. [Google Scholar] [CrossRef] [PubMed]
- Camerini, D.; Su, H.P.; Gamez-Torre, G.; Johnson, M.L.; Zack, J.A.; Chen, I.S. Human immunodeficiency virus type 1 pathogenesis in SCID-hu mice correlates with syncytium-inducing phenotype and viral replication. J. Virol. 2000, 74, 3196–3204. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.; Mandalia, S.; Randell, P.; Wildfire, A.; Gazzard, B.; Moyle, G. The impact of HIV tropism on decreases in CD4 cell count, clinical progression, and subsequent response to a first antiretroviral therapy regimen. Clin. Infect. Dis. 2008, 46, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Nyakeriga, A.M.; Ravi, R.; Garg, H. HIV ENV glycoprotein-mediated bystander apoptosis depends on expression of the CCR5 co-receptor at the cell surface and ENV fusogenic activity. J. Biol. Chem. 2011, 286, 36404–36413. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Lee, R.T.; Maurer-Stroh, S.; Joshi, A. HIV-1 adaptation to low levels of CCR5 results in V3 and V2 loop changes that increase envelope pathogenicity, CCR5 affinity and decrease susceptibility to Maraviroc. Virology 2016, 493, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Scoggins, R.M.; Taylor, J.R., Jr.; Patrie, J.; van’t Wout, A.B.; Schuitemaker, H.; Camerini, D. Pathogenesis of primary R5 human immunodeficiency virus type 1 clones in SCID-hu mice. J. Virol. 2000, 74, 3205–3216. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.K.; Choudhary, N.R.; Kimbrell, K.C.; Colasanti, J.; Ziogas, A.; Kwa, D.; Schuitemaker, H.; Camerini, D. R5 human immunodeficiency virus type 1 infection of fetal thymic organ culture induces cytokine and CCR5 expression. J. Virol. 2005, 79, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, K.; Scoggins, R.; Bor, Y.; Matthews, A.; Mark, D.; Taylor, J.J.; Chernauskas, D.; Hammarskjöld, M.; Rekosh, D.; Camerini, D. The envelope gene is a cytopathic determinant of CCR5 tropic HIV-1. Virology 2007, 358, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Roggero, R.; Robert-Hebmann, V.; Harrington, S.; Roland, J.; Vergne, L.; Jaleco, S.; Devaux, C.; Biard-Piechaczyk, M. Binding of human immunodeficiency virus type 1 gp120 to CXCR4 induces mitochondrial transmembrane depolarization and cytochrome c-mediated apoptosis independently of Fas signaling. J. Virol. 2001, 75, 7637–7650. [Google Scholar] [CrossRef] [PubMed]
- Ferri, K.F.; Jacotot, E.; Blanco, J.; Este, J.A.; Kroemer, G. Mitochondrial control of cell death induced by HIV-1-encoded proteins. Ann. N. Y. Acad. Sci. 2000, 926, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Ohnimus, H.; Heinkelein, M.; Jassoy, C. Apoptotic cell death upon contact of CD4+ T lymphocytes with HIV glycoprotein-expressing cells is mediated by caspases but bypasses CD95 (Fas/Apo-1) and TNF receptor 1. J. Immunol. 1997, 159, 5246–5252. [Google Scholar] [PubMed]
- Gandhi, R.; Chen, B.; Straus, S.; Dale, J.; Lenardo, M.; Baltimore, D. HIV-1 directly kills CD4+ T cells by a Fas-independent mechanism. J. Exp. Med. 1998, 187, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Nadeau, P.E.; Lo, Y.T.; Mergia, A. Caveolin-1 modulates HIV-1 envelope-induced bystander apoptosis through gp41. J. Virol. 2010, 84, 6515–6526. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.; Oliveira, M.; Lerner, S.; Tao, Y.; Brenner, B.G. Modulation of stress protein (hsp27 and hsp70) expression in CD4+ lymphocytic cells following acute infection with human immunodeficiency virus type-1. Virology 1997, 233, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Molina, L.; Grimaldi, M.; Robert-Hebmann, V.; Espert, L.; Varbanov, M.; Devaux, C.; Granier, C.; Biard-Piechaczyk, M. Proteomic analysis of the cellular responses induced in uninfected immune cells by cell-expressed X4 HIV-1 envelope. Proteomics 2007, 7, 3116–3130. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Viard, M.; Jacobs, A.; Blumenthal, R. Targeting HIV-1 gp41-induced fusion and pathogenesis for anti-viral therapy. Curr. Top. Med. Chem. 2011, 11, 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Blanco, J.; Bonjoch, A.; Llano, A.; Clotet, B.; Esté, J. Immunological and virological study of enfuvirtide-treated HIV-positive patients. AIDS 2004, 18, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.; Lee, F.; Miamidian, J.; Jabara, C.; Juntilla, M.; Doms, R. Enfuvirtide resistance mutations: Impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J. Virol. 2005, 79, 4991–4999. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, S.; D’Arrigo, R.; Svicher, V.; Perri, G.; Caputo, S.; Visco-Comandini, U.; Santoro, M.; Bertoli, A.; Mazzotta, F.; Bonora, S.; et al. Specific mutations in HIV-1 gp41 are associated with immunological success in HIV-1-infected patients receiving enfuvirtide treatment. J. Antimicrob. Chemother. 2006, 58, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Melby, T.; Despirito, M.; Demasi, R.; Heilek, G.; Thommes, J.; Greenberg, M.; Graham, N. Association between specific enfuvirtide resistance mutations and CD4 cell response during enfuvirtide-based therapy. AIDS 2007, 21, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Joshi, A.; Blumenthal, R. Altered bystander apoptosis induction and pathogenesis of enfuvirtide-resistant HIV type 1 Env mutants. AIDS Res. Hum. Retrovir. 2009, 25, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Steiner, M.; Lynch, D.H.; Staiano-Coico, L.; Laurence, J. HIV-1 upregulates Fas ligand expression in CD4+ T cells In Vitro and In Vivo: Association with Fas-mediated apoptosis and modulation by aurintricarboxylic acid. Immunology 1996, 87, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Badley, A.D.; Dockrell, D.; Simpson, M.; Schut, R.; Lynch, D.H.; Leibson, P.; Paya, C.V. Macrophage-dependent apoptosis of CD4+ T lymphocytes from HIV-infected individuals is mediated by FasL and tumor necrosis factor. J. Exp. Med. 1997, 185, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tikhonov, I.; Ruckwardt, T.J.; Djavani, M.; Zapata, J.C.; Pauza, C.D.; Salvato, M.S. Monocytes treated with human immunodeficiency virus Tat kill uninfected CD4(+) cells by a tumor necrosis factor-related apoptosis-induced ligand-mediated mechanism. J. Virol. 2003, 77, 6700–6708. [Google Scholar] [CrossRef] [PubMed]
- Herbein, G.; Khan, K.A. Is HIV infection a TNF receptor signalling-driven disease? Trends Immunol. 2008, 29, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Bernier, J.; Kurahara, D.K.; Kurrle, R.; Haigwood, N.; Sekaly, R.P.; Finkel, T.H. Crosslinking CD4 by human immunodeficiency virus gp120 primes T cells for activation-induced apoptosis. J. Exp. Med. 1992, 176, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; Gibellini, D.; Secchiero, P.; Dutartre, H.; Olive, D.; Capitani, S.; Collette, Y. Human immunodeficiency virus type 1 Nef protein sensitizes CD4(+) T lymphoid cells to apoptosis via functional upregulation of the CD95/CD95 ligand pathway. Blood 1999, 93, 1000–1010. [Google Scholar] [PubMed]
- Westendorp, M.O.; Frank, R.; Ochsenbauer, C.; Stricker, K.; Dhein, J.; Walczak, H.; Debatin, K.M.; Krammer, P.H. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 1995, 375, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Jeremias, I.; Herr, I.; Boehler, T.; Debatin, K.M. TRAIL/Apo-2-ligand-induced apoptosis in human T cells. Eur. J. Immunol. 1998, 28, 143–152. [Google Scholar] [CrossRef]
- Zhang, M.; Li, X.; Pang, X.; Ding, L.; Wood, O.; Clouse, K.; Hewlett, I.; Dayton, A.I. Identification of a potential HIV-induced source of bystander-mediated apoptosis in T cells: Upregulation of trail in primary human macrophages by HIV-1 tat. J. Biomed. Sci. 2001, 8, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Than, S.; Oyaizu, N.; Pahwa, R.N.; Kalyanaraman, V.S.; Pahwa, S. Effect of human immunodeficiency virus type-1 envelope glycoprotein gp160 on cytokine production from cord-blood T cells. Blood 1994, 84, 184–188. [Google Scholar] [PubMed]
- Chirmule, N.; Pahwa, S. Envelope glycoproteins of human immunodeficiency virus type 1: Profound influences on immune functions. Microbiol. Rev. 1996, 60, 386–406. [Google Scholar] [PubMed]
- Su, L.; Kaneshima, H.; Bonyhadi, M.; Salimi, S.; Kraft, D.; Rabin, L.; McCune, J.M. HIV-1-induced thymocyte depletion is associated with indirect cytopathogenicity and infection of progenitor cells in vivo. Immunity 1995, 2, 25–36. [Google Scholar] [CrossRef]
- Robey, E.; Axel, R. CD4: Collaborator in immune recognition and HIV infection. Cell 1990, 60, 697–700. [Google Scholar] [CrossRef]
- Chirmule, N.; Oyaizu, N.; Kalyanaraman, V.S.; Pahwa, S. Inhibition of normal B-cell function by human immunodeficiency virus envelope glycoprotein, gp120. Blood 1992, 79, 1245–1254. [Google Scholar] [PubMed]
- Chirmule, N.; McCloskey, T.W.; Hu, R.; Kalyanaraman, V.S.; Pahwa, S. HIV gp120 inhibits T cell activation by interfering with expression of costimulatory molecules CD40 ligand and CD80 (B71). J. Immunol. 1995, 155, 917–924. [Google Scholar] [PubMed]
- Meyaard, L.; Schuitemaker, H.; Miedema, F. T-cell dysfunction in HIV infection: Anergy due to defective antigen-presenting cell function? Immunol. Today 1993, 14, 161–164. [Google Scholar] [CrossRef]
- Silverstein, P.S.; Shah, A.; Weemhoff, J.; Kumar, S.; Singh, D.P.; Kumar, A. HIV-1 gp120 and drugs of abuse: Interactions in the central nervous system. Curr. HIV Res. 2012, 10, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Van’t Wout, A.B.; Kootstra, N.A.; Mulder-Kampinga, G.A.; Albrecht-van Lent, N.; Scherpbier, H.J.; Veenstra, J.; Boer, K.; Coutinho, R.A.; Miedema, F.; Schuitemaker, H. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J. Clin. Investig. 1994, 94, 2060–2067. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Bailes, E.; Pham, K.T.; Salazar, M.G.; Guffey, M.B.; Keele, B.F.; Derdeyn, C.A.; Farmer, P.; Hunter, E.; Allen, S.; et al. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 2008, 82, 3952–3970. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.M.; Hunter, E. HIV transmission. Cold Spring Harb. Perspect. Med. 2012, 2, a006965. [Google Scholar] [CrossRef] [PubMed]
- Boesecke, C.; Pett, S.L. Clinical studies with chemokine receptor-5 (CCR5)-inhibitors. Curr. Opin. HIV AIDS 2012, 7, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Woollard, S.M.; Kanmogne, G.D. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Dev. Ther. 2015, 9, 5447–5468. [Google Scholar]
- Mummidi, S.; Ahuja, S.S.; Gonzalez, E.; Anderson, S.A.; Santiago, E.N.; Stephan, K.T.; Craig, F.E.; O’Connell, P.; Tryon, V.; Clark, R.A.; et al. Genealogy of the CCR5 locus and chemokine system gene variants associated with altered rates of HIV-1 disease progression. Nat. Med. 1998, 4, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Punke, E.B.; Sedano, M.; Beauchamp, B.; Patel, R.; Hossenlopp, C.; Alozie, O.K.; Gupta, J.; Mukherjee, D.; Garg, H. CCR5 promoter activity correlates with HIV disease progression by regulating CCR5 cell surface expression and CD4 T cell apoptosis. Sci. Rep. 2017, 7, 232. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 1996, 273, 1856–1862. [Google Scholar] [PubMed]
- Huang, Y.; Paxton, W.A.; Wolinsky, S.M.; Neumann, A.U.; Zhang, L.; He, T.; Kang, S.; Ceradini, D.; Jin, Z.; Yazdanbakhsh, K.; et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat. Med. 1996, 2, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Paxton, W.; Choe, S.; Ceradini, D.; Martin, S.; Horuk, R.; MacDonald, M.; Stuhlmann, H.; Koup, R.; Landau, N. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Paxton, W.A.; Kassam, N.; Ruffing, N.; Rottman, J.B.; Sullivan, N.; Choe, H.; Sodroski, J.; Newman, W.; Koup, R.A.; et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med. 1997, 185, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Dean, M.; Smith, M.W.; Winkler, C.; Gerrard, B.; Michael, N.L.; Lee, B.; Doms, R.W.; Margolick, J.; Buchbinder, S.; et al. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science 1998, 282, 1907–1911. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Bamshad, M.; Sato, N.; Mummidi, S.; Dhanda, R.; Catano, G.; Cabrera, S.; McBride, M.; Cao, X.H.; Merrill, G.; et al. Race-specific HIV-1 disease-modifying effects associated with CCR5 haplotypes. Proc. Natl. Acad. Sci. USA 1999, 96, 12004–12009. [Google Scholar] [CrossRef] [PubMed]
- Gornalusse, G.G.; Mummidi, S.; Gaitan, A.A.; Jimenez, F.; Ramsuran, V.; Picton, A.; Rogers, K.; Manoharan, M.S.; Avadhanam, N.; Murthy, K.K.; et al. Epigenetic mechanisms, T-cell activation, and CCR5 genetics interact to regulate T-cell expression of CCR5, the major HIV-1 coreceptor. Proc. Natl. Acad. Sci. USA 2015, 112, E4762–E4771. [Google Scholar] [CrossRef] [PubMed]
- Platt, E.; Wehrly, K.; Kuhmann, S.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [PubMed]
- Reeves, J.; Gallo, S.; Ahmad, N.; Miamidian, J.; Harvey, P.; Sharron, M.; Pohlmann, S.; Sfakianos, J.; Derdeyn, C.; Blumenthal, R.; et al. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 16249–16254. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Apetrei, C.; Gordon, S.; Barbercheck, J.; Dufour, J.; Bohm, R.; Sumpter, B.; Roques, P.; Marx, P.A.; Hirsch, V.M.; et al. Paucity of CD4+ CCR5+ T cells is a typical feature of natural SIV hosts. Blood 2007, 109, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Onanga, R.; Souquiere, S.; Mouinga-Ondeme, A.; Bourry, O.; Makuwa, M.; Rouquet, P.; Silvestri, G.; Simon, F.; Roques, P.; et al. Paucity of CD4+ CCR5+ T cells may prevent transmission of simian immunodeficiency virus in natural nonhuman primate hosts by breast-feeding. J. Virol. 2008, 82, 5501–5509. [Google Scholar] [CrossRef] [PubMed]
- Paiardini, M.; Cervasi, B.; Reyes-Aviles, E.; Micci, L.; Ortiz, A.M.; Chahroudi, A.; Vinton, C.; Gordon, S.N.; Bosinger, S.E.; Francella, N.; et al. Low levels of SIV infection in sooty mangabey central memory CD(4)(+) T cells are associated with limited CCR5 expression. Nat. Med. 2011, 17, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Muenchhoff, M.; Adland, E.; Karimanzira, O.; Crowther, C.; Pace, M.; Csala, A.; Leitman, E.; Moonsamy, A.; McGregor, C.; Hurst, J.; et al. Nonprogressing HIV-infected children share fundamental immunological features of nonpathogenic SIV infection. Sci. Transl. Med. 2016, 8, 358ra125. [Google Scholar] [CrossRef] [PubMed]
- Grossman, Z.; Meier-Schellersheim, M.; Paul, W.; Picker, L. Pathogenesis of HIV infection: What the virus spares is as important as what it destroys. Nat. Med. 2006, 12, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Grossman, Z.; Meier-Schellersheim, M.; Sousa, A.; Victorino, R.; Paul, W. CD4+ T-cell depletion in HIV infection: Are we closer to understanding the cause? Nat. Med. 2002, 8, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Bentwich, Z.; Kalinkovich, A.; Weisman, Z.; Grossman, Z. Immune activation in the context of HIV infection. Clin. Exp. Immunol. 1998, 111, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Al-Harthi, L.; MaWhinney, S.; Connick, E.; Schooley, R.; Forster, J.; Benson, C.; Thompson, M.; Judson, F.; Palella, F.; Landay, A. Immunophenotypic alterations in acute and early HIV infection. Clin. Immunol. 2007, 125, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Biancotto, A.; Grivel, J.; Iglehart, S.; Vanpouille, C.; Lisco, A.; Sieg, S.; Debernardo, R.; Garate, K.; Rodriguez, B.; Margolis, L.; et al. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood 2007, 109, 4272–4279. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, M. T cell apoptosis as a consequence of chronic activation of the immune system in HIV infection. Adv. Exp. Med. Biol. 1995, 374, 121–127. [Google Scholar] [PubMed]
- Sodora, D.; Silvestri, G. Immune activation and AIDS pathogenesis. AIDS 2008, 22, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Estes, J.; Gordon, S.; Zeng, M.; Chahroudi, A.; Dunham, R.; Staprans, S.; Reilly, C.; Silvestri, G.; Haase, A. Early resolution of acute immune activation and induction of PD-1 in SIV-infected sooty mangabeys distinguishes nonpathogenic from pathogenic infection in rhesus macaques. J. Immunol. 2008, 180, 6798–6807. [Google Scholar] [CrossRef] [PubMed]
- Brainard, D.M.; Seung, E.; Frahm, N.; Cariappa, A.; Bailey, C.C.; Hart, W.K.; Shin, H.S.; Brooks, S.F.; Knight, H.L.; Eichbaum, Q.; et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J. Virol. 2009, 83, 7305–7321. [Google Scholar] [CrossRef] [PubMed]
- Paiardini, M.; Muller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101. [Google Scholar] [CrossRef] [PubMed]
- Ploquin, M.J.; Silvestri, G.; Muller-Trutwin, M. Immune activation in HIV infection: What can the natural hosts of simian immunodeficiency virus teach us? Curr. Opin. HIV AIDS 2016, 11, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Cockerham, L.R.; Siliciano, J.D.; Sinclair, E.; O’Doherty, U.; Palmer, S.; Yukl, S.A.; Strain, M.C.; Chomont, N.; Hecht, F.M.; Siliciano, R.F.; et al. CD4+ and CD8+ T cell activation are associated with HIV DNA in resting CD4+ T cells. PLoS ONE 2014, 9, e110731. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Taiwo, B.; Gandhi, R.T.; Hunt, P.W.; Collier, A.C.; Flexner, C.; Bosch, R.J. Factors associated with CD8+ T-cell activation in HIV-1-infected patients on long-term antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2014, 67, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Stuart, J.W.; Otto, S.A.; Borleffs, J.C.; Boucher, C.A.; de Boer, R.J.; Miedema, F.; Hamann, D. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: A longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 2000, 95, 249–255. [Google Scholar] [PubMed]
- Hunt, P.W.; Martin, J.N.; Sinclair, E.; Bredt, B.; Hagos, E.; Lampiris, H.; Deeks, S.G. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J. Infect. Dis. 2003, 187, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Mohri, H.; Perelson, A.S.; Tung, K.; Ribeiro, R.M.; Ramratnam, B.; Markowitz, M.; Kost, R.; Hurley, A.; Weinberger, L.; Cesar, D.; et al. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 2001, 194, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, A.; Deleage, C.; Sereti, I.; Rerknimitr, R.; Phanuphak, N.; Phuang-Ngern, Y.; Estes, J.D.; Sandler, N.G.; Sukhumvittaya, S.; Marovich, M.; et al. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog. 2014, 10, e1004543. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Massanella, M.; Llibre, J.M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Gatell, J.M.; Domingo, P.; Paredes, R.; Sharkey, M.; et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat. Med. 2010, 16, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Biancotto, A.; Iglehart, S.; Vanpouille, C.; Condack, C.; Lisco, A.; Ruecker, E.; Hirsch, I.; Margolis, L.; Grivel, J. HIV-1 induced activation of CD4+ T cells creates new targets for HIV-1 infection in human lymphoid tissue ex vivo. Blood 2008, 111, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Cheng, M.; Nunoya, J.; Cheng, L.; Guo, H.; Yu, H.; Liu, Y.J.; Su, L.; Zhang, L. Plasmacytoid dendritic cells suppress HIV-1 replication but contribute to HIV-1 induced immunopathogenesis in humanized mice. PLoS Pathog. 2014, 10, e1004291. [Google Scholar] [CrossRef] [PubMed]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yu, H.; Li, G.; Li, F.; Ma, J.; Li, J.; Chi, L.; Zhang, L.; Su, L. Type I interferons suppress viral replication but contribute to T cell depletion and dysfunction during chronic HIV-1 infection. JCI Insight 2017, 2, 94366. [Google Scholar] [CrossRef] [PubMed]
- Douek, D. HIV disease progression: Immune activation, microbes, and a leaky gut. Top. HIV Med. 2007, 15, 114–117. [Google Scholar] [PubMed]
- Tincati, C.; Douek, D.C.; Marchetti, G. Gut barrier structure, mucosal immunity and intestinal microbiota in the pathogenesis and treatment of HIV infection. AIDS Res. Ther. 2016, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, G.; Tincati, C.; Silvestri, G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin. Microbiol. Rev. 2013, 26, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.; Price, D.; Schacker, T.; Asher, T.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Lederman, M.M.; Hunt, P.; Sieg, S.F.; Haley, K.; Rodriguez, B.; Landay, A.; Martin, J.; Sinclair, E.; Asher, A.I.; et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J. Infect. Dis. 2009, 199, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, G.; Bellistri, G.M.; Borghi, E.; Tincati, C.; Ferramosca, S.; La Francesca, M.; Morace, G.; Gori, A.; Monforte, A.D. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. AIDS 2008, 22, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- Merlini, E.; Bai, F.; Bellistri, G.M.; Tincati, C.; d’Arminio Monforte, A.; Marchetti, G. Evidence for polymicrobic flora translocating in peripheral blood of HIV-infected patients with poor immune response to antiretroviral therapy. PLoS ONE 2011, 6, e18580. [Google Scholar] [CrossRef] [PubMed]
- Bukh, A.R.; Melchjorsen, J.; Offersen, R.; Jensen, J.M.; Toft, L.; Stovring, H.; Ostergaard, L.; Tolstrup, M.; Sogaard, O.S. Endotoxemia is associated with altered innate and adaptive immune responses in untreated HIV-1 infected individuals. PLoS ONE 2011, 6, e21275. [Google Scholar] [CrossRef] [PubMed]
- Nowroozalizadeh, S.; Mansson, F.; da Silva, Z.; Repits, J.; Dabo, B.; Pereira, C.; Biague, A.; Albert, J.; Nielsen, J.; Aaby, P.; et al. Microbial translocation correlates with the severity of both HIV-1 and HIV-2 infections. J. Infect. Dis. 2010, 201, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Guadalupe, M.; Reay, E.; Sankaran, S.; Prindiville, T.; Flamm, J.; McNeil, A.; Dandekar, S. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 2003, 77, 11708–11717. [Google Scholar] [CrossRef] [PubMed]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Horowitz, A.; Hurley, A.; Hogan, C.; Boden, D.; Racz, P.; Markowitz, M. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 2004, 200, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Mehandru, S.; Poles, M.A.; Tenner-Racz, K.; Manuelli, V.; Jean-Pierre, P.; Lopez, P.; Shet, A.; Low, A.; Mohri, H.; Boden, D.; et al. Mechanisms of gastrointestinal CD4+ T-cell depletion during acute and early human immunodeficiency virus type 1 infection. J. Virol. 2007, 81, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Duan, L.; Estes, J.D.; Ma, Z.M.; Rourke, T.; Wang, Y.; Reilly, C.; Carlis, J.; Miller, C.J.; Haase, A.T. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 2005, 434, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, S.; George, M.D.; Reay, E.; Guadalupe, M.; Flamm, J.; Prindiville, T.; Dandekar, S. Rapid onset of intestinal epithelial barrier dysfunction in primary human immunodeficiency virus infection is driven by an imbalance between immune response and mucosal repair and regeneration. J. Virol. 2008, 82, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Paiardini, M.; Knox, K.S.; Asher, A.I.; Cervasi, B.; Asher, T.E.; Scheinberg, P.; Price, D.A.; Hage, C.A.; Kholi, L.M.; et al. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 2008, 112, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Omenetti, S. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Favre, D.; Lederer, S.; Kanwar, B.; Ma, Z.M.; Proll, S.; Kasakow, Z.; Mold, J.; Swainson, L.; Barbour, J.D.; Baskin, C.R.; et al. Critical loss of the balance between Th17 and T regulatory cell populations in pathogenic SIV infection. PLoS Pathog. 2009, 5, e1000295. [Google Scholar] [CrossRef] [PubMed]
- Raffatellu, M.; Santos, R.L.; Verhoeven, D.E.; George, M.D.; Wilson, R.P.; Winter, S.E.; Godinez, I.; Sankaran, S.; Paixao, T.A.; Gordon, M.A.; et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat. Med. 2008, 14, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Mudd, J.C.; Brenchley, J.M. Gut mucosal barrier dysfunction, microbial dysbiosis, and their role in HIV-1 disease progression. J. Infect. Dis. 2016, 214, S58–S66. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Aranami, T.; Yamamura, T. Cutting edge: Human Th17 cells are identified as bearing CCR2+CCR5- phenotype. J. Immunol. 2007, 178, 7525–7529. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.M.; Klase, Z.A.; DiNapoli, S.R.; Vujkovic-Cvijin, I.; Carmack, K.; Perkins, M.R.; Calantone, N.; Vinton, C.L.; Riddick, N.E.; Gallagher, J.; et al. IL-21 and probiotic therapy improve Th17 frequencies, microbial translocation, and microbiome in ARV-treated, SIV-infected macaques. Mucosal Immunol. 2016, 9, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Mandl, J.N.; Barry, A.P.; Vanderford, T.H.; Kozyr, N.; Chavan, R.; Klucking, S.; Barrat, F.J.; Coffman, R.L.; Staprans, S.I.; Feinberg, M.B. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat. Med. 2008, 14, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Manches, O.; Wilen, C.; Gopal, R.; Huq, R.; Wu, V.; Sunseri, N.; Bhardwaj, N. CD4 receptor is a key determinant of divergent HIV-1 sensing by plasmacytoid dendritic cells. PLoS Pathog. 2016, 12, e1005553. [Google Scholar] [CrossRef] [PubMed]
- Funderburg, N.; Luciano, A.A.; Jiang, W.; Rodriguez, B.; Sieg, S.F.; Lederman, M.M. Toll-like receptor ligands induce human T cell activation and death, a model for HIV pathogenesis. PLoS ONE 2008, 3, e1915. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.M.; Down, C.M.; Boulware, D.R.; Stauffer, W.M.; Cavert, W.P.; Schacker, T.W.; Brenchley, J.M.; Douek, D.C. Reduction of immune activation with chloroquine therapy during chronic HIV infection. J. Virol. 2010, 84, 12082–12086. [Google Scholar] [CrossRef] [PubMed]
- Piconi, S.; Parisotto, S.; Rizzardini, G.; Passerini, S.; Terzi, R.; Argenteri, B.; Meraviglia, P.; Capetti, A.; Biasin, M.; Trabattoni, D.; et al. Hydroxychloroquine drastically reduces immune activation in HIV-infected, antiretroviral therapy-treated immunologic nonresponders. Blood 2011, 118, 3263–3272. [Google Scholar] [CrossRef] [PubMed]
- Ameisen, J.C.; Capron, A. Cell dysfunction and depletion in AIDS: The programmed cell death hypothesis. Immunol. Today 1991, 12, 102–105. [Google Scholar] [CrossRef]
- Bao, R.; Zhuang, K.; Liu, J.; Wu, J.; Li, J.; Wang, X.; Ho, W.Z. Lipopolysaccharide induces immune activation and SIV replication in rhesus macaques of Chinese origin. PLoS ONE 2014, 9, e98636. [Google Scholar] [CrossRef] [PubMed]
- Hofer, U.; Schlaepfer, E.; Baenziger, S.; Nischang, M.; Regenass, S.; Schwendener, R.; Kempf, W.; Nadal, D.; Speck, R.F. Inadequate clearance of translocated bacterial products in HIV-infected humanized mice. PLoS Pathog. 2010, 6, e1000867. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Gaufin, T.; Brenchley, J.M.; Gautam, R.; Monjure, C.; Gautam, A.; Coleman, C.; Lackner, A.A.; Ribeiro, R.M.; Douek, D.C.; et al. Cutting edge: Experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J. Immunol. 2008, 181, 6687–6691. [Google Scholar] [CrossRef] [PubMed]
- Koning, F.; Otto, S.; Hazenberg, M.; Dekker, L.; Prins, M.; Miedema, F.; Schuitemaker, H. Low-level CD4+ T cell activation is associated with low susceptibility to HIV-1 infection. J. Immunol. 2005, 175, 6117–6122. [Google Scholar] [CrossRef] [PubMed]
- Juffermans, N.P.; Paxton, W.A.; Dekkers, P.E.; Verbon, A.; de Jonge, E.; Speelman, P.; van Deventer, S.J.; van der Poll, T. Up-regulation of HIV coreceptors CXCR4 and CCR5 on CD4(+) T cells during human endotoxemia and after stimulation with (myco)bacterial antigens: The role of cytokines. Blood 2000, 96, 2649–2654. [Google Scholar] [PubMed]
- Espert, L.; Codogno, P.; Biard-Piechaczyk, M. What is the role of autophagy in HIV-1 infection? Autophagy 2008, 4, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy and CD4+ T lymphocyte destruction by HIV-1. Autophagy 2007, 3, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, T. Autophagy: A regulated bulk degradation process inside cells. Biochem. Biophys. Res. Commun. 2004, 313, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Biard-Piechaczyk, M. Autophagy in HIV-induced T cell death. Curr. Top. Microbiol. Immunol. 2009, 335, 307–321. [Google Scholar] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, K.; Taylor, M.P.; Jackson, W.T. Cellular autophagy: Surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2004, 2, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Garcia, E.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell. Signal. 2014, 26, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Maiuri, M.C.; Kroemer, G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 2010, 29, 1717–1719. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Greene, W.C. Dissecting how CD4 T cells are lost during HIV infection. Cell Host Microbe 2016, 19, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Munoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Galloway, N.L.; Doitsh, G.; Monroe, K.M.; Yang, Z.; Munoz-Arias, I.; Levy, D.N.; Greene, W.C. Cell-to-Cell Transmission of HIV-1 Is Required to Trigger Pyroptotic Death of Lymphoid-Tissue-Derived CD4 T Cells. Cell Rep. 2015, 12, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Poropatich, K.; Sullivan, D.J., Jr. Human immunodeficiency virus type 1 long-term non-progressors: The viral, genetic and immunological basis for disease non-progression. J. Gen. Virol. 2011, 92, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.K.; Vrisekoop, N.; Jansen, C.A.; Otto, S.A.; Schuitemaker, H.; Miedema, F.; Camerini, D. Low immune activation despite high levels of pathogenic human immunodeficiency virus type 1 results in long-term asymptomatic disease. J. Virol. 2007, 81, 8838–8842. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Bosinger, S.E.; Peck, M.; Richert-Spuhler, L.E.; Heigele, A.; Gile, J.P.; Patel, N.; Taaffe, J.; Julg, B.; Camerini, D.; et al. Limited HIV infection of central memory and stem cell memory CD4+ T cells is associated with lack of progression in viremic individuals. PLoS Pathog. 2014, 10, e1004345. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, V.; Zhang, L.; Meissner, E.G.; Jeffrey, J.L.; Su, L. The heptad repeat 2 domain is a major determinant for enhanced human immunodeficiency virus type 1 (HIV-1) fusion and pathogenicity of a highly pathogenic HIV-1 Env. J. Virol. 2009, 83, 11715–11725. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Rotger, M.; Erkizia, I.; Rauch, A.; Reche, P.; Pino, M.; Esteve, A.; Palou, E.; Brander, C.; Paredes, R.; et al. Highly pathogenic adapted HIV-1 strains limit host immunity and dictate rapid disease progression. AIDS 2014, 28, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Si, Z.; Gorry, P.; Babcock, G.; Owens, C.M.; Cayabyab, M.; Phan, N.; Sodroski, J. Envelope glycoprotein determinants of increased entry in a pathogenic simian-human immunodeficiency virus (SHIV-HXBc2P 3.2) passaged in monkeys. AIDS Res. Hum. Retrovir. 2004, 20, 163–173. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Factors Limiting Bystander Apoptosis | Factors Enhancing Bystander Apoptosis |
|---|---|
| ➢ Poor virus replication | ➢ High virus replication |
| ➢ Low AIP phenotype | ➢ High AIP phenotype |
| ➢ Low CCR5 levels | ➢ High CCR5 levels |
| ➢ Low immune activation | ➢ High immune activation |
| ➢ Low Env CCR5 binding affinity | ➢ High Env CCR5 binding affinity |
| ➢ Less fusogenic Env | ➢ Highly fusogenic Env |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garg, H.; Joshi, A. Host and Viral Factors in HIV-Mediated Bystander Apoptosis. Viruses 2017, 9, 237. https://doi.org/10.3390/v9080237
Garg H, Joshi A. Host and Viral Factors in HIV-Mediated Bystander Apoptosis. Viruses. 2017; 9(8):237. https://doi.org/10.3390/v9080237
Chicago/Turabian StyleGarg, Himanshu, and Anjali Joshi. 2017. "Host and Viral Factors in HIV-Mediated Bystander Apoptosis" Viruses 9, no. 8: 237. https://doi.org/10.3390/v9080237
APA StyleGarg, H., & Joshi, A. (2017). Host and Viral Factors in HIV-Mediated Bystander Apoptosis. Viruses, 9(8), 237. https://doi.org/10.3390/v9080237