
Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression


Abstract
1. Introduction
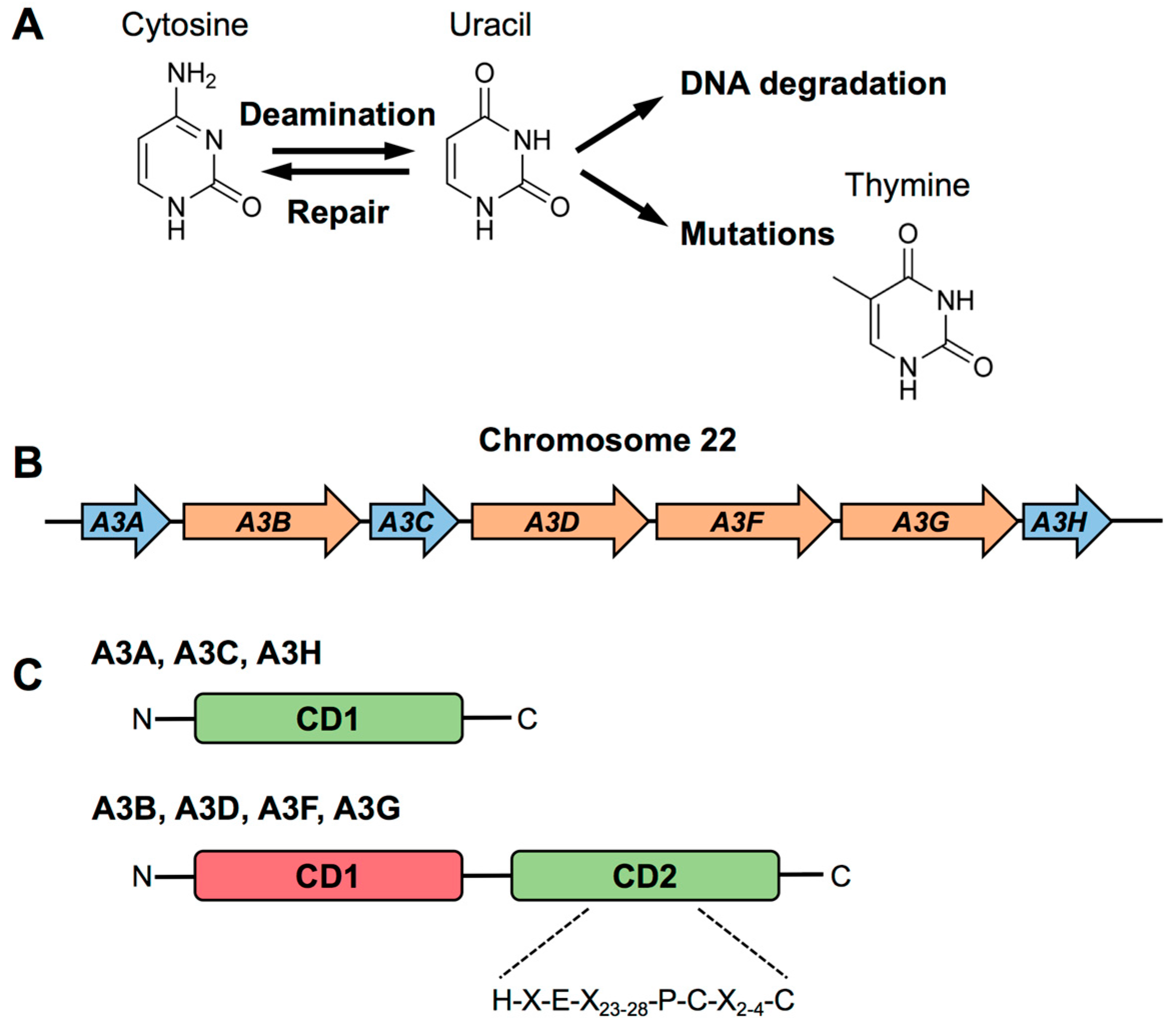
2. Biology of APOBEC3
2.1. Structural Features of APOBEC3s
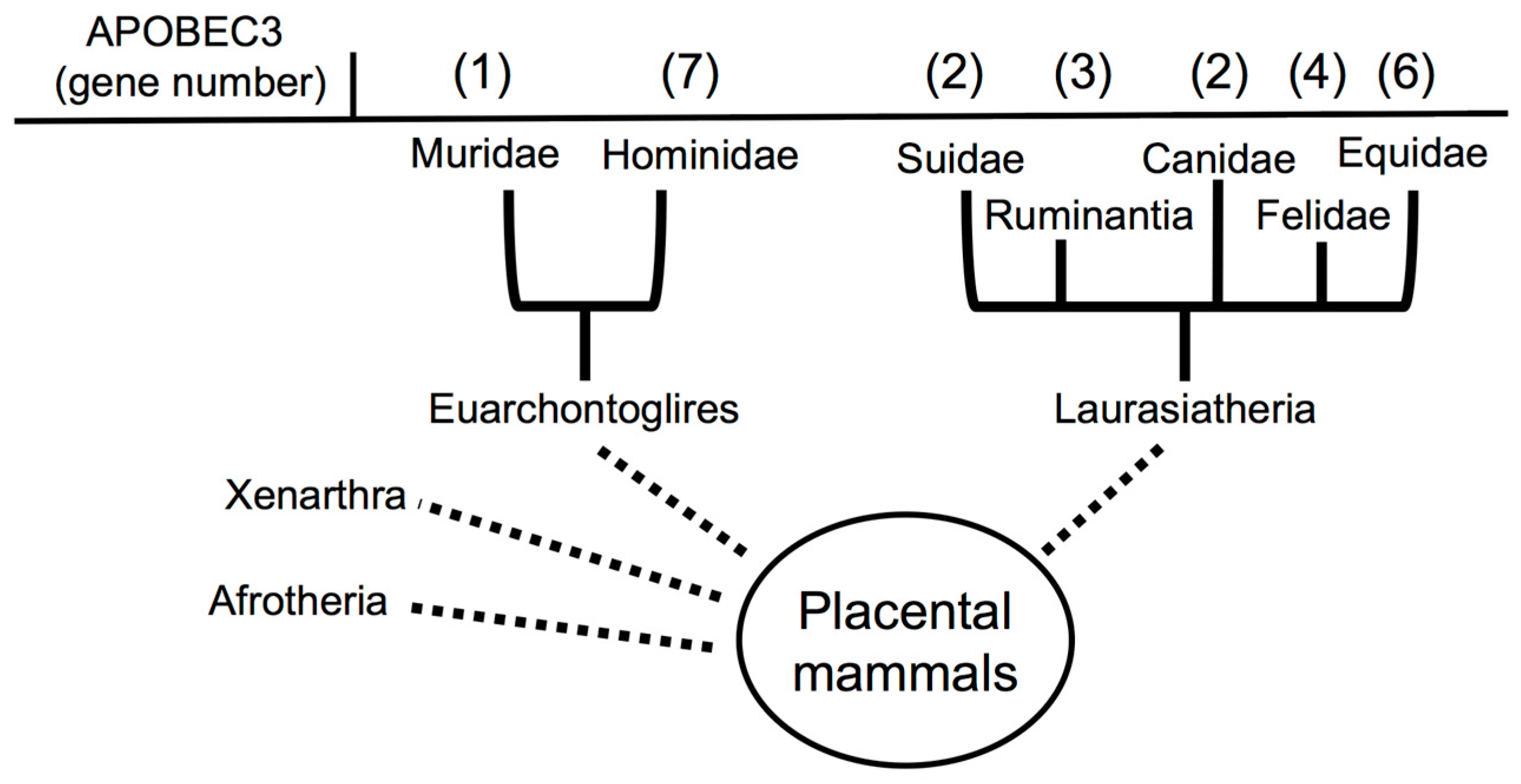
2.2. Evolution of APOBEC3s
2.3. Target Specificity of APOBEC3s
2.4. RNA Editing by APOBEC3A
2.5. Transcriptional Regulation of APOBEC3A and APOBEC3B
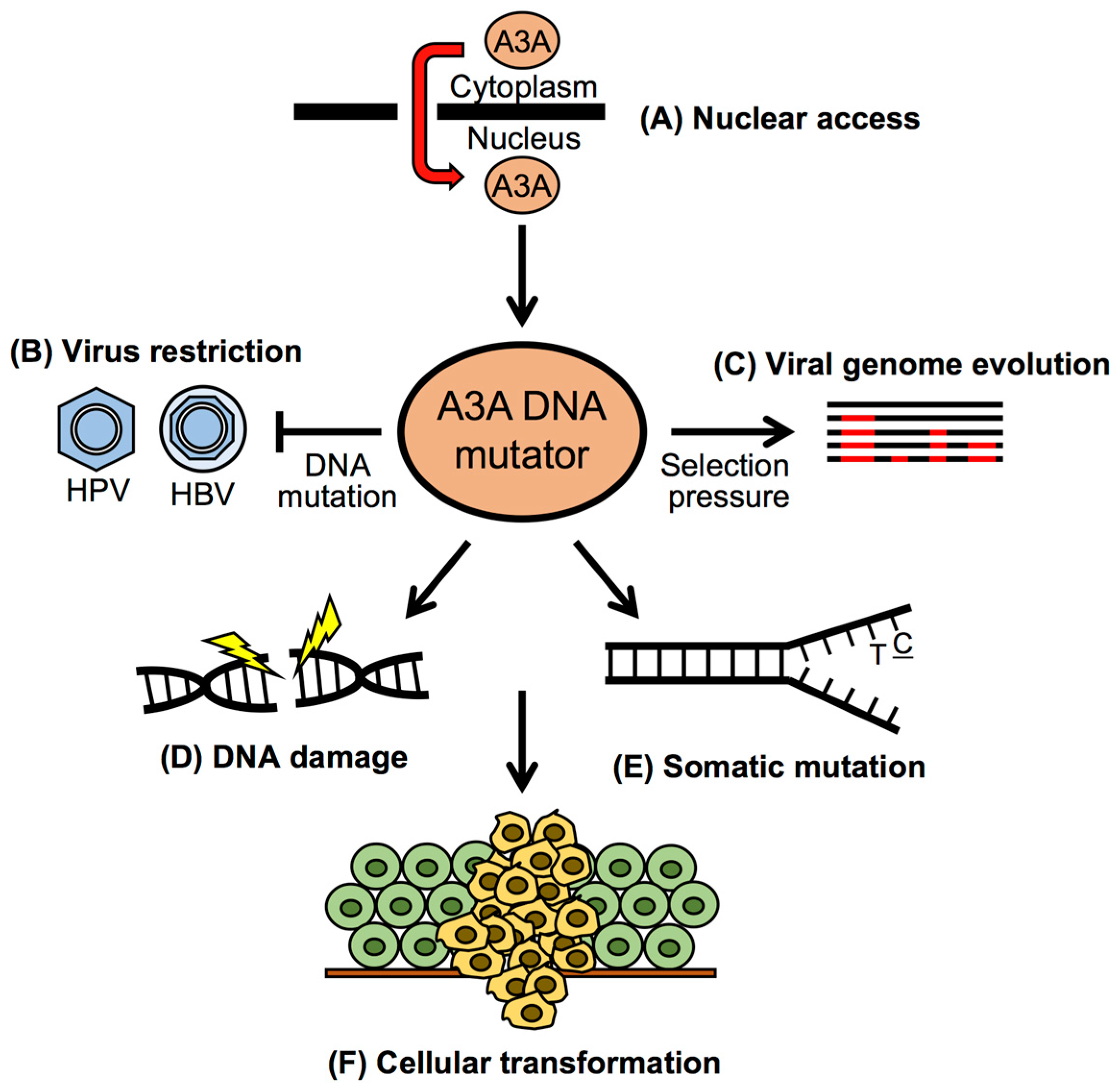
3. Restriction of DNA Viruses by APOBEC3A
3.1. APOBEC3A Restriction of HPV Infection
3.2. APOBEC3A-Mediated Clearance of HPV DNA during Persistent Infection
3.3. Viral Evasion of APOBEC3A-Mediated Restriction
4. Cancer Mutagenesis by APOBEC3A
4.1. Sources of APOBEC3 Mutational Signatures in HPV-Positive Cancer
4.2. The Relative Contributions of APOBEC3A and APOBEC3B to Cancer Mutagenesis
4.3. Source of APOBEC3 Signature in Other Virus-Associated Cancers
4.4. APOBEC3-Mediated Somatic Mutations and Clinical Outcomes of HPV-Positive Cancers
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Wiegand, H.L.; Doehle, B.P.; Lueders, K.K.; Cullen, B.R. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006, 34, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Wiegand, H.L.; Hulme, A.E.; Garcia-Perez, J.L.; O’Shea, K.S.; Moran, J.V.; Cullen, B.R. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 8780–8785. [Google Scholar] [CrossRef] [PubMed]
- Muckenfuss, H.; Hamdorf, M.; Held, U.; Perkovic, M.; Löwer, J.; Cichutek, K.; Flory, E.; Schumann, G.G.; Münk, C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 2006, 281, 22161–22172. [Google Scholar] [CrossRef] [PubMed]
- Stenglein, M.D.; Harris, R.S. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 2006, 281, 16837–16841. [Google Scholar] [CrossRef] [PubMed]
- Doehle, B.P.; Sch fer, A.; Cullen, B.R. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 2005, 339, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Wiegand, H.L.; Doehle, B.P.; Cullen, B.R. The intrinsic antiretroviral factor APOBEC3B contains two enzymatically active cytidine deaminase domains. Virology 2007, 364, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Durand, S.; Fargier, G.; Nguyen, X.-N.; Cordeil, S.; Bouaziz, S.; Muriaux, D.; Darlix, J.-L.; Cimarelli, A. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog. 2011, 7, e1002221. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lilley, C.E.; Yu, Q.; Lee, D.V.; Chou, J.; Narvaiza, I.; Landau, N.R.; Weitzman, M.D. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 2006, 16, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Narvaiza, I.; Linfesty, D.C.; Greener, B.N.; Hakata, Y.; Pintel, D.J.; Logue, E.; Landau, N.R.; Weitzman, M.D. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 2009, 5, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, Y.; Stavrou, S.; Blouch, K.; Tattersall, P.; Ross, S.R. In vivo examination of mouse APOBEC3- and human APOBEC3A- and APOBEC3G-mediated restriction of parvovirus and herpesvirus infection in mouse models. J. Virol. 2016, 90, 8005–8012. [Google Scholar] [CrossRef] [PubMed]
- Minkah, N.; Chavez, K.; Shah, P.; Maccarthy, T.; Chen, H.; Landau, N.; Krug, L.T. Host restriction of murine gammaherpesvirus 68 replication by human APOBEC3 cytidine deaminases but not murine APOBEC3. Virology 2014, 454-455, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Suspène, R.; Aynaud, M.-M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.-P.; Meyerhans, A.; Wain-Hobson, S. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.; Guétard, D.; Suspène, R.; Rusniok, C.; Wain-Hobson, S.; Vartanian, J.-P. Genetic editing of HBV DNA by monodomain human APOBEC3 cytidine deaminases and the recombinant nature of APOBEC3G. PLoS ONE 2009, 4, e4277. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L. Pyeon, APOBEC3A functions as a restriction factor of human papillomavir. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Ahasan, M.M.; Wakae, K.; Wang, Z.; Kitamura, K.; Liu, G.; Koura, M.; Imayasu, M.; Sakamoto, N.; Hanaoka, K.; Nakamura, M.; et al. APOBEC3A and 3C decrease human papillomavirus 16 pseudovirion infectivity. Biochem. Biophys. Res. Commun. 2015, 457, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.-P.; Guétard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Kostrzak, A.; Henry, M.; Demoyen, P.L.; Wain-Hobson, S.; Vartanian, J.P. APOBEC3A catabolism of electroporated plasmid DNA in mouse muscle. Gene Ther. 2015, 22, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Stenglein, M.D.; Burns, M.B.; Li, M.; Lengyel, J.; Harris, R.S. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010, 17, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Patnaik, S.K.; Kemer, Z.; Baysal, B.E. Transient overexpression of exogenous APOBEC3A causes C-to-U RNA editing of thousands of genes. RNA Biol. 2016, 14, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Niavarani, A.; Currie, E.; Reyal, Y.; Anjos-Afonso, F.; Horswell, S.; Griessinger, E.; Sardina, J.L.; Bonnet, D. APOBEC3A is implicated in a novel class of G-to-A mRNA editing in WT1 transcripts. PLoS ONE 2015, 10, e0120089. [Google Scholar] [CrossRef] [PubMed]
- Landry, S.; Narvaiza, I.; Linfesty, D.C.; Weitzman, M.D. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011, 12, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Kotler, M. APOBEC3 Cytidine Deaminases in Double-Strand DNA Break Repair and Cancer Promotion. Cancer Res. 2013, 73, 3494–3498. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Landry, S.; Budagyan, K.; Avgousti, D.C.; Shalhout, S.; Bhagwat, A.S.; Weitzman, M.D. APOBEC3A damages the cellular genome during DNA replication. Cell Cycle 2016, 15, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Roberts, S.A.; Klimczak, L.J.; Sterling, J.F.; Saini, N.; Malc, E.P.; Kim, J.; Kwiatkowski, D.J.; Fargo, D.C.; Mieczkowski, P.A.; et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 2015, 47, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, Å.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Kanu, N.; Cerone, M.A.; Goh, G.; Zalmas, L.-P.; Bartkova, J.; Dietzen, M.; McGranahan, N.; Rogers, R.; Law, E.K.; Gromova, I.; et al. DNA replication stress mediates APOBEC3 family mutagenesis in breast cancer. Genome Biol. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Delahanty, R.J.; Li, G.; Gao, Y.-T.; Lu, W.; Cai, Q.; Xiang, Y.-B.; Li, C.; Ji, B.-T.; Zheng, Y.; et al. A Common Deletion in the APOBEC3 Genes and Breast Cancer Risk. JNCI J. Natl. Cancer Inst. 2013, 105, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Wedge, D.C.; Alexandrov, L.B.; Petljak, M.; Butler, A.P.; Bolli, N.; Davies, H.R.; Knappskog, S.; Martin, S.; Papaemmanuil, E.; et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 2014, 46, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Bulliard, Y.; Narvaiza, I.; Bertero, A.; Peddi, S.; Röhrig, U.F.; Ortiz, M.; Zoete, V.; Castro-Díaz, N.; Turelli, P.; Telenti, A.; et al. Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J. Virol. 2011, 85, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Suspène, R.; Guétard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.-P. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8321–8326. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, M.; Achermann, F.; Greeve, I.; Stroka, D.; Keogh, A.; Inderbitzin, D.; Candinas, D.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.-P.; et al. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 2006, 43, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Rösler, C.; Malim, M.H.; von Weizsäcker, F. Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3C. Hepatology 2007, 46, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Mussil, B.; Suspène, R.; Aynaud, M.-M.; Gauvrit, A.; Vartanian, J.-P.; Wain-Hobson, S. Human APOBEC3A Isoforms Translocate to the Nucleus and Induce DNA Double Strand Breaks Leading to Cell Stress and Death. PLoS ONE 2013, 8, e73641. [Google Scholar] [CrossRef] [PubMed]
- Lackey, L.; Law, E.K.; Brown, W.L.; Harris, R.S. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 2013, 12, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Lackey, L.; Demorest, Z.L.; Land, A.M.; Hultquist, J.F.; Brown, W.L.; Harris, R.S. APOBEC3B and AID have similar nuclear import mechanisms. J. Mol. Biol. 2012, 419, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Io, K.; Shindo, K.; Matsui, M.; Sakamoto, T.; Tada, K.; Kobayashi, M.; Kadowaki, N.; Takaori-Kondo, A. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci. Rep. 2012, 2, 806. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshall, J.; Ramakrishna, M.; et al. Breast Cancer Working Group of the International Cancer Genome Consortium The life history of 21 breast cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Kuong, K.J.; Loeb, L.A. APOBEC3B mutagenesis in cancer. Nat. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.A.; Lambert, P.F.; Howley, P.M.; et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 2014, 5, e02234-14. [Google Scholar] [CrossRef] [PubMed]
- Jarmuz, A.; Chester, A.; Bayliss, J.; Gisbourne, J.; Dunham, I.; Scott, J.; Navaratnam, N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar] [CrossRef] [PubMed]
- LaRue, R.S.; Andrésdóttir, V.; Blanchard, Y.; Conticello, S.G.; Derse, D.; Emerman, M.; Greene, W.C.; Jónsson, S.R.; Landau, N.R.; Löchelt, M.; et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009, 83, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Bransteitter, R.; Prochnow, C.; Chen, X.S. The current structural and functional understanding of APOBEC deaminases. Cell. Mol. Life Sci. 2009, 66, 3137–3147. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.; Bollman, B.; Chen, H.; König, R.; Yu, Q.; Chiles, K.; Landau, N.R. Complementary function of the two catalytic domains of APOBEC3G. Virology 2005, 333, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.N.C.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral Function of APOBEC3G Can Be Dissociated from Cytidine Deaminase Activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, S.R.; Haché, G.; Stenglein, M.D.; Fahrenkrug, S.C.; Andrésdóttir, V.; Harris, R.S. Evolutionarily conserved and non-conserved retrovirus restriction activities of artiodactyl APOBEC3F proteins. Nucleic Acids Res. 2006, 34, 5683–5694. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.K.; Koning, F.A.; Bishop, K.N.; Malim, M.H. APOBEC3F Can Inhibit the Accumulation of HIV-1 Reverse Transcription Products in the Absence of Hypermutation COMPARISONS WITH APOBEC3G. J. Biol. Chem. 2007, 282, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, M.; Greeve, J. Effects of point mutations in the cytidine deaminase domains of APOBEC3B on replication and hypermutation of hepatitis B virus in vitro. J. Gen. Virol. 2007, 88, 3270–3274. [Google Scholar] [CrossRef] [PubMed]
- Logue, E.C.; Bloch, N.; Dhuey, E.; Zhang, R.; Cao, P.; Herate, C.; Chauveau, L.; Hubbard, S.R.; Landau, N.R. A DNA Sequence Recognition Loop on APOBEC3A Controls Substrate Specificity. PLoS ONE 2014, 9, e97062. [Google Scholar] [CrossRef] [PubMed]
- Shlyakhtenko, L.S.; Lushnikov, A.J.; Li, M.; Harris, R.S.; Lyubchenko, Y.L. Interaction of APOBEC3A with DNA assessed by atomic force microscopy. PLoS ONE 2014, 9, e99354. [Google Scholar] [CrossRef] [PubMed]
- Bohn, M.-F.; Shandilya, S.M.D.; Silvas, T.V.; Nalivaika, E.A.; Kouno, T.; Kelch, B.A.; Ryder, S.P.; Kurt-Yilmaz, N.; Somasundaran, M.; Schiffer, C.A. The ssDNA mutator APOBEC3A is regulated by cooperative dimerization. Structure 2015, 23, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Byeon, I.-J.L.; Ahn, J.; Mitra, M.; Byeon, C.-H.; Hercík, K.; Hritz, J.; Charlton, L.M.; Levin, J.G.; Gronenborn, A.M. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 2013, 4, 1890. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.; Hercík, K.; Byeon, I.-J.L.; Ahn, J.; Hill, S.; Hinchee-Rodriguez, K.; Singer, D.; Byeon, C.-H.; Charlton, L.M.; Nam, G.; et al. Structural determinants of human APOBEC3A enzymatic and nucleic acid binding properties. Nucleic Acids Res. 2014, 42, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Amrine-Madsen, H.; Koepfli, K.-P.; Wayne, R.K.; Springer, M.S. A new phylogenetic marker, apolipoprotein B, provides compelling evidence for eutherian relationships. Mol. Phylogenet. Evol. 2003, 28, 225–240. [Google Scholar] [CrossRef]
- Murphy, W.J.; Eizirik, E.; Johnson, W.E.; Zhang, Y.P.; Ryder, O.A.; O’Brien, S.J. Molecular phylogenetics and the origins of placental mammals. Nature 2001, 409, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Madsen, O.; Scally, M.; Douady, C.J.; Kao, D.J.; DeBry, R.W.; Adkins, R.; Amrine, H.M.; Stanhope, M.J.; de Jong, W.W.; Springer, M.S. Parallel adaptive radiations in two major clades of placental mammals. Nature 2001, 409, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Liddament, M.T. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004, 4, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Mikl, M.C.; Watt, I.N.; Lu, M.; Reik, W.; Davies, S.L.; Neuberger, M.S.; Rada, C. Mice deficient in APOBEC2 and APOBEC3. Mol. Cell Biol. 2005, 25, 7270–7277. [Google Scholar] [CrossRef] [PubMed]
- Münk, C.; Willemsen, A.; Bravo, I.G. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol. Biol. 2012, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Münk, C.; Beck, T.; Zielonka, J.; Hotz-Wagenblatt, A.; Chareza, S.; Battenberg, M.; Thielebein, J.; Cichutek, K.; Bravo, I.G.; O’Brien, S.J.; et al. Functions, structure, and read-through alternative splicing of feline APOBEC3 genes. Genome Biol. 2008, 9, R48. [Google Scholar] [CrossRef] [PubMed]
- LaRue, R.S.; Jónsson, S.R.; Silverstein, K.A.T.; Lajoie, M.; Bertrand, D.; El-Mabrouk, N.; Hötzel, I.; Andrésdóttir, V.; Smith, T.P.L.; Harris, R.S. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 2008, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Tallmadge, R.L.; Oaks, J.L.; Carpenter, S.; Cullen, B.R. Equine infectious anemia virus resists the antiretroviral activity of equine APOBEC3 proteins through a packaging-independent mechanism. J. Virol. 2008, 82, 11889–11901. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004, 2, E275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Webb, D.M. Rapid evolution of primate antiviral enzyme APOBEC3G. Hum. Mol. Genet. 2004, 13, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.N.; Gable, J.T.; Wittkopp, C.J.; Emerman, M.; Malik, H.S. Conservation and Innovation of APOBEC3A Restriction Functions during Primate Evolution. Mol. Biol. Evol. 2016, 33, 1889–1901. [Google Scholar] [CrossRef] [PubMed]
- Duggal, N.K.; Malik, H.S.; Emerman, M. The breadth of antiviral activity of Apobec3DE in chimpanzees has been driven by positive selection. J. Virol. 2011, 85, 11361–11371. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, N.R.; Sawyer, S.L. Two-stepping through time: mammals and viruses. Trends Microbiol. 2011, 19, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Shi, K.; Carpenter, M.A.; Banerjee, S.; Shaban, N.M.; Kurahashi, K.; Salamango, D.J.; McCann, J.L.; Starrett, G.J.; Duffy, J.V.; Demir, Ö.; et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kouno, T.; Silvas, T.V.; Hilbert, B.J.; Shandilya, S.M.D.; Bohn, M.F.; Kelch, B.A.; Royer, W.E.; Somasundaran, M.; Kurt-Yilmaz, N.; Matsuo, H.; et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017, 8, 15024. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.-P.; Henry, M.; Marchio, A.; Suspène, R.; Aynaud, M.-M.; Guétard, D.; Cervantes-Gonzalez, M.; Battiston, C.; Mazzaferro, V.; Pineau, P.; et al. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 2010, 6, e1000928. [Google Scholar] [CrossRef] [PubMed]
- Suspène, R.; Aynaud, M.-M.; Guétard, D.; Henry, M.; Eckhoff, G.; Marchio, A.; Pineau, P.; Dejean, A.; Vartanian, J.-P.; Wain-Hobson, S. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc. Natl. Acad. Sci. USA 2011, 108, 4858–4863. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Hoopes, J.I.; Cortez, L.M.; Mertz, T.M.; Malc, E.P.; Mieczkowski, P.A.; Roberts, S.A. APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication. Cell Rep. 2016, 14, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Seplyarskiy, V.B.; Soldatov, R.A.; Popadin, K.Y.; Antonarakis, S.E.; Bazykin, G.A.; Nikolaev, S.I. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016, 26, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; De Jong, K.; Liu, B.; Wang, J.; Patnaik, S.K.; Wallace, P.K.; Taggart, R.T. Hypoxia-inducible C-to-U coding RNA editing downregulates SDHB in monocytes. PeerJ 2013, 1, e152. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, S.; Crawford, D.; Blouch, K.; Browne, E.P.; Kohli, R.M.; Ross, S.R. Different modes of retrovirus restriction by human APOBEC3A and APOBEC3G in vivo. PLoS Pathog. 2014, 10, e1004145. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 675–687. [Google Scholar] [CrossRef] [PubMed]
- García-Sastre, A.; Biron, C.A. Type 1 Interferons and the Virus-Host Relationship: A Lesson in Détente. Science 2006, 312, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Lei, K.J.; Jin, W.; Greenwell-Wild, T.; Wahl, S.M. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 2006, 203, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Mohanram, V.; Sköld, A.E.; Bächle, S.M.; Pathak, S.K.; Spetz, A.-L. IFN-α induces APOBEC3G, F, and A in immature dendritic cells and limits HIV-1 spread to CD4+ T cells. J. Immunol. 2013, 190, 3346–3353. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-X.; Huang, J.; Zhang, H.; Ma, X.; Zhang, H. APOBEC3G upregulation by alpha interferon restricts human immunodeficiency virus type 1 infection in human peripheral plasmacytoid dendritic cells. J. Gen. Virol. 2008, 89, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.; McCann, J.L.; Starrett, G.J.; Kosyakovsky, L.; Luengas, E.M.; Molan, A.M.; Burns, M.B.; McDougle, R.M.; Parker, P.J.; Brown, W.L.; et al. The PKC/NF-κB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015, 75, 4538–4547. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, W.; Shirakawa, K.; Matsui, H.; Matsumoto, T.; Yamazaki, H.; Sarca, A.D.; Kazuma, Y.; Kobayashi, M.; Shindo, K.; Takaori-Kondo, A. Classical NF-κB pathway is responsible for APOBEC3B expression in cancer cells. Biochem. Biophys. Res. Commun. 2016, 478, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Wakae, K.; Wakisaka, N.; Nakanishi, Y.; Ishikawa, K.; Komori, T.; Moriyama-Kita, M.; Endo, K.; Murono, S.; Wang, Z.; et al. APOBEC3A associates with human papillomavirus genome integration in oropharyngeal cancers. Oncogene 2017, 36, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Kukimoto, I. Identification of APOBEC3B promoter elements responsible for activation by human papillomavirus type 16 E6. Biochem. Biophys. Res. Commun. 2015, 460, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Yugawa, T.; Kiyono, T.; Nishina, H.; Kukimoto, I. Human papillomavirus 16 E6 upregulates APOBEC3B via the TEAD transcription factor. J. Virol. 2017, 91, e02413-16. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Nguyen, T.-A.; Snipe, J.; Resnick, M.A. The cytidine deaminase APOBEC3 family is subject to transcriptional regulation by p53. Mol. Cancer Res. 2017, 15, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Gasco, M.; Shami, S.; Crook, T. The p53 pathway in breast cancer. Breast Cancer Res. 2002, 4, 70. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Greenwell-Wild, T.; Nares, S.; Jin, W.; Lei, K.J.; Rangel, Z.G.; Munson, P.J.; Wahl, S.M. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 2007, 110, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Koning, F.A.; Newman, E.N.C.; Kim, E.-Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Van Doorslaer, K.; Pandey, A.; Espinosa, J.M.; Pyeon, D. Role of the host restriction factor APOBEC3 on papillomavirus evolution. Virus Evol. 2015, 1, vev015. [Google Scholar] [CrossRef] [PubMed]
- Love, R.P.; Xu, H.; Chelico, L. Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem. 2012, 287, 30812–30822. [Google Scholar] [CrossRef] [PubMed]
- Beggel, B.; Münk, C.; Däumer, M.; Hauck, K.; Häussinger, D.; Lengauer, T.; Erhardt, A. Full genome ultra-deep pyrosequencing associates G-to-A hypermutation of the hepatitis B virus genome with the natural progression of hepatitis B. J. Viral Hepat. 2013, 20, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Wakae, K.; Aoyama, S.; Wang, Z.; Kitamura, K.; Liu, G.; Monjurul, A.M.; Koura, M.; Imayasu, M.; Sakamoto, N.; Nakamura, M.; et al. Detection of hypermutated human papillomavirus type 16 genome by Next-Generation Sequencing. Virology 2015, 485, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Kukimoto, I.; Mori, S.; Aoyama, S.; Wakae, K.; Muramatsu, M.; Kondo, K. Hypermutation in the E2 gene of human papillomavirus type 16 in cervical intraepithelial neoplasia. J. Med. Virol. 2015, 87, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Herdman, M.T.; Pett, M.R.; Roberts, I.; Alazawi, W.O.F.; Teschendorff, A.E.; Zhang, X.-Y.; Stanley, M.A.; Coleman, N. Interferon-β treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis 2006, 27, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.E.; Pena, L.; Sen, G.C.; Park, J.K.; Laimins, L.A. Long-term effect of interferon on keratinocytes that maintain human papillomavirus type 31. J. Virol. 2002, 76, 8864–8874. [Google Scholar] [CrossRef] [PubMed]
- Terenzi, F.; Saikia, P.; Sen, G.C. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO 2008, 27, 3311–3321. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Griffin, L.M.; Little, A.S.; Huang, I.-C.; Farzan, M.; Pyeon, D. The Antiviral Restriction Factors IFITM1, 2 and 3 Do Not Inhibit Infection of Human Papillomavirus, Cytomegalovirus and Adenovirus. PLoS ONE 2014, 9, e96579. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Lambert, P.F.; Ahlquist, P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 9311–9316. [Google Scholar] [CrossRef] [PubMed]
- Pett, M.R.; Herdman, M.T.; Palmer, R.D.; Yeo, G.S.H.; Shivji, M.K.; Stanley, M.A.; Coleman, N. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc. Natl. Acad. Sci. USA 2006, 103, 3822–3827. [Google Scholar] [CrossRef] [PubMed]
- Alazawi, W.; Pett, M.; Arch, B.; Scott, L.; Freeman, T.; Stanley, M.A.; Coleman, N. Changes in cervical keratinocyte gene expression associated with integration of human papillomavirus 16. Cancer Res. 2002, 62, 6959–6965. [Google Scholar] [PubMed]
- Lace, M.J.; Anson, J.R.; Haugen, T.H.; Dierdorff, J.M.; Turek, L.P. Interferon treatment of human keratinocytes harboring extrachromosomal, persistent HPV-16 plasmid genomes induces de novo viral integration. Carcinogenesis 2015, 36, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Turek, L.P.; Byrne, J.C.; Lowy, D.R.; Dvoretzky, I.; Friedman, R.M.; Howley, P.M. Interferon induces morphologic reversion with elimination of extrachromosomal viral genomes in bovine papillomavirus-transformed mouse cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7914–7918. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.-F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, Z.; Ehrlich, E.S.; Yu, X.; Yu, X.-F. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004, 18, 2867–2872. [Google Scholar] [CrossRef] [PubMed]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.-Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Münger, K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Jones, D.L.; Thompson, D.A.; Münger, K. Destabilization of the RB Tumor Suppressor Protein and Stabilization of p53 Contribute to HPV Type 16 E7-Induced Apoptosis. Virology 1997, 239, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Westrich, J.A.; Warren, C.J.; Klausner, M.J.; Vermeer, D.W.; Guo, K.; Lee, J.H.; Liu, C.; Santiago, M.L.; Pyeon, D. High-risk Human Papillomavirus E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-dependent Protein Degradation. J. Virol. 2017. Under revision. [Google Scholar]
- Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J. Mol. Evol. 2006, 62, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R. Presence and role of cytosine methylation in DNA viruses of animals. Nucleic Acids Res. 2008, 36, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, M.; Samal, J.; Kandpal, M.; Vasaikar, S.; Biswas, B.; Gomes, J.; Vivekanandan, P. CpG dinucleotide frequencies reveal the role of host methylation capabilities in parvovirus evolution. J. Virol. 2013, 87, 13816–13824. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, M.; Vivekanandan, P. Depletion of CpG Dinucleotides in Papillomaviruses and Polyomaviruses: A Role for Divergent Evolutionary Pressures. PLoS ONE 2015, 10, e0142368. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.-M.; Fauquet, C.; Broker, T.R.; Bernard, H.-U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Pyeon, D. APOBEC3 in papillomavirus restriction, evolution and cancer progression. Oncotarget 2015, 6, 39385–39386. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Koneva, L.A.; Virani, S.; Arthur, A.E.; Virani, A.; Hall, P.B.; Warden, C.D.; Carey, T.E.; Chepeha, D.B.; Prince, M.E.; et al. Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clin. Cancer Res. 2016, 22, 4735–4745. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of Human Papillomavirus-Induced Oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Cescon, D.W.; Haibe-Kains, B.; Mak, T.W. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc. Natl. Acad. Sci. USA 2015, 112, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.; Hart, S.N.; Burns, M.B.; Carpenter, M.A.; Temiz, N.A.; Rathore, A.; Vogel, R.I.; Nikas, J.B.; Law, E.K.; Brown, W.L.; et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013, 73, 7222–7231. [Google Scholar] [CrossRef] [PubMed]
- Middlebrooks, C.D.; Banday, A.R.; Matsuda, K.; Udquim, K.-I.; Onabajo, O.O.; Paquin, A.; Figueroa, J.D.; Zhu, B.; Koutros, S.; Kubo, M.; et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat. Genet. 2016, 48, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Marouf, C.; Göhler, S.; Filho, M.I.D.S.; Hajji, O.; Hemminki, K.; Nadifi, S.; Försti, A. Analysis of functional germline variants in APOBEC3 and driver genes on breast cancer risk in Moroccan study population. BMC Cancer 2016, 16, 165. [Google Scholar] [CrossRef] [PubMed]
- Revathidevi, S.; Manikandan, M.; Rao, A.K.D.M.; Vinothkumar, V.; Arunkumar, G.; Rajkumar, K.S.; Ramani, R.; Rajaraman, R.; Ajay, C.; Munirajan, A.K. Analysis of APOBEC3A/3B germline deletion polymorphism in breast, cervical and oral cancers from South India and its impact on miRNA regulation. Tumour Biol. 2016, 37, 11983–11990. [Google Scholar] [CrossRef] [PubMed]
- Caval, V.; Suspène, R.; Shapira, M.; Vartanian, J.-P.; Wain-Hobson, S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3'UTR enhances chromosomal DNA damage. Nat. Commun. 2014, 5, 5129. [Google Scholar] [CrossRef] [PubMed]
- Winder, D.M.; Pett, M.R.; Foster, N.; Shivji, M.K.K.; Herdman, M.T.; Stanley, M.A.; Venkitaraman, A.R.; Coleman, N. An increase in DNA double-strand breaks, induced by Ku70 depletion, is associated with human papillomavirus 16 episome loss and de novo viral integration events. J. Pathol. 2007, 213, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Verhalen, B.; Starrett, G.J.; Harris, R.S.; Jiang, M. Functional upregulation of the DNA cytosine deaminase APOBEC3B by polyomaviruses. J. Virol. 2016, 90, 6379–6386. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.C.; Palma, D.A.; Chow, W.; Tan, S.; Rajakumar, C.; Rizzo, G.; Fung, K.; Kwan, K.; Wehrli, B.; Winquist, E.; et al. High frequency of activating PIK3CA mutations in human papillomavirus-positive oropharyngeal cancer. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Stevanović, S.; Pasetto, A.; Helman, S.R.; Gartner, J.J.; Prickett, T.D.; Howie, B.; Robins, H.S.; Robbins, P.F.; Klebanoff, C.A.; Rosenberg, S.A.; et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017, 356, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Jabbar, S.F.; Park, S.; Schweizer, J.; Berard-Bergery, M.; Pitot, H.C.; Lee, D.; Lambert, P.F. Cervical cancers require the continuous expression of the human papillomavirus type 16 E7 oncoprotein even in the presence of the viral E6 oncoprotein. Cancer Res. 2012, 72, 4008–4016. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Kornepati, A.V.R.; Goldstein, M.; Bogerd, H.P.; Poling, B.C.; Whisnant, A.W.; Kastan, M.B.; Cullen, B.R. Inactivation of the human papillomavirus E6 or E7 gene in cervical carcinoma cells by using a bacterial CRISPR/Cas RNA-guided endonuclease. J. Virol. 2014, 88, 11965–11972. [Google Scholar] [CrossRef] [PubMed]
- Hanning, J.E.; Saini, H.K.; Murray, M.J.; Caffarel, M.M.; van Dongen, S.; Ward, D.; Barker, E.M.; Scarpini, C.G.; Groves, I.J.; Stanley, M.A.; et al. Depletion of HPV16 early genes induces autophagy and senescence in a cervical carcinogenesis model, regardless of viral physical state. J. Pathol. 2013, 231, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Sima, N.; Wang, S.; Wang, W.; Kong, D.; Xu, Q.; Tian, X.; Luo, A.; Zhou, J.; Xu, G.; Meng, L.; et al. Antisense targeting human papillomavirus type 16 E6 and E7 genes contributes to apoptosis and senescence in SiHa cervical carcinoma cells. Gynecol. Oncol. 2007, 106, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Johung, K.; Goodwin, E.C.; DiMaio, D. Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J. Virol. 2007, 81, 2102–2116. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Putral, L.; Hengst, K.; Minto, K.; Saunders, N.A.; Leggatt, G.; McMillan, N.A.J. Inhibition of cervical cancer cell growth in vitro and in vivo with lentiviral-vector delivered short hairpin RNA targeting human papillomavirus E6 and E7 oncogenes. Cancer Gene Ther. 2006, 13, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; DeFilippis, R.A.; Manuelidis, L.; DiMaio, D. Repression of the human papillomavirus E6 gene initiates p53-dependent, telomerase-independent senescence and apoptosis in HeLa cervical carcinoma cells. J. Virol. 2004, 78, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Boonden, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | APOBEC3 Family Members | Functions on Viral Genome | References |
|---|---|---|---|
| Parvovirus | A3A | Unknown | [11,12,13,35] |
| Herpesvirus | A3A, A3G | DNA editing, unknown | [14,15] |
| Papillomavirus | A3A, A3C (?) | DNA editing, unknown | [18,20,36] |
| Hepadnavirus | A3A, A3B, A3C, A3F, A3G, A3H | DNA editing, deamination, and degradation | [16,17,37,38,39,40] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warren, C.J.; Westrich, J.A.; Doorslaer, K.V.; Pyeon, D. Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression. Viruses 2017, 9, 233. https://doi.org/10.3390/v9080233
Warren CJ, Westrich JA, Doorslaer KV, Pyeon D. Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression. Viruses. 2017; 9(8):233. https://doi.org/10.3390/v9080233
Chicago/Turabian StyleWarren, Cody J., Joseph A. Westrich, Koenraad Van Doorslaer, and Dohun Pyeon. 2017. "Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression" Viruses 9, no. 8: 233. https://doi.org/10.3390/v9080233
APA StyleWarren, C. J., Westrich, J. A., Doorslaer, K. V., & Pyeon, D. (2017). Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression. Viruses, 9(8), 233. https://doi.org/10.3390/v9080233