
Clinical Manifestations and Epigenetic Regulation of Oral Herpesvirus Infections



{kind=link}
{kind=link}
Abstract
1. Introduction
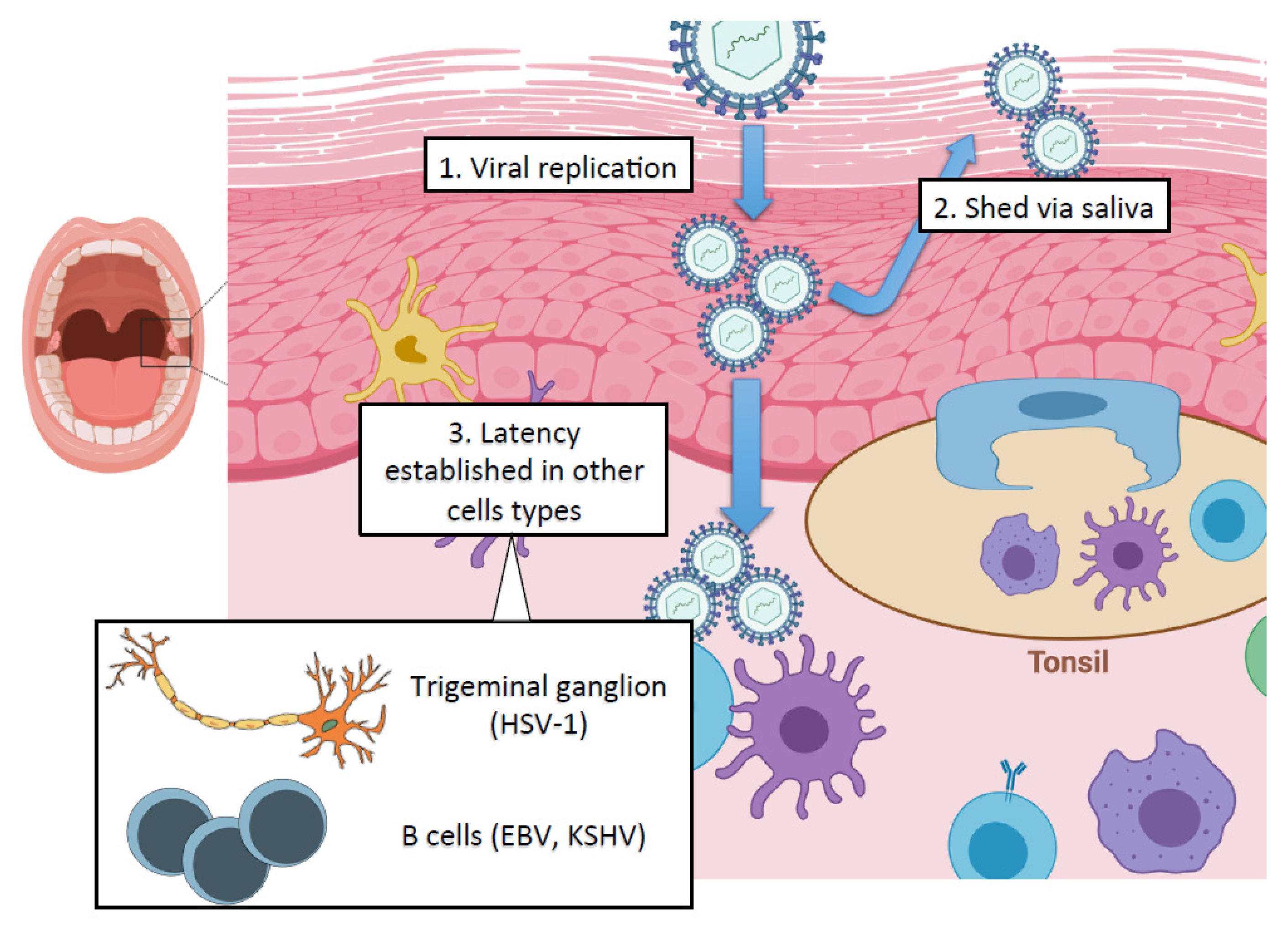
2. Oral Manifestations of Herpesvirus Infections
2.1. Herpes Simplex Virus Type 1 (HSV-1)
2.2. Epstein–Barr Virus (EBV)
2.3. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV)
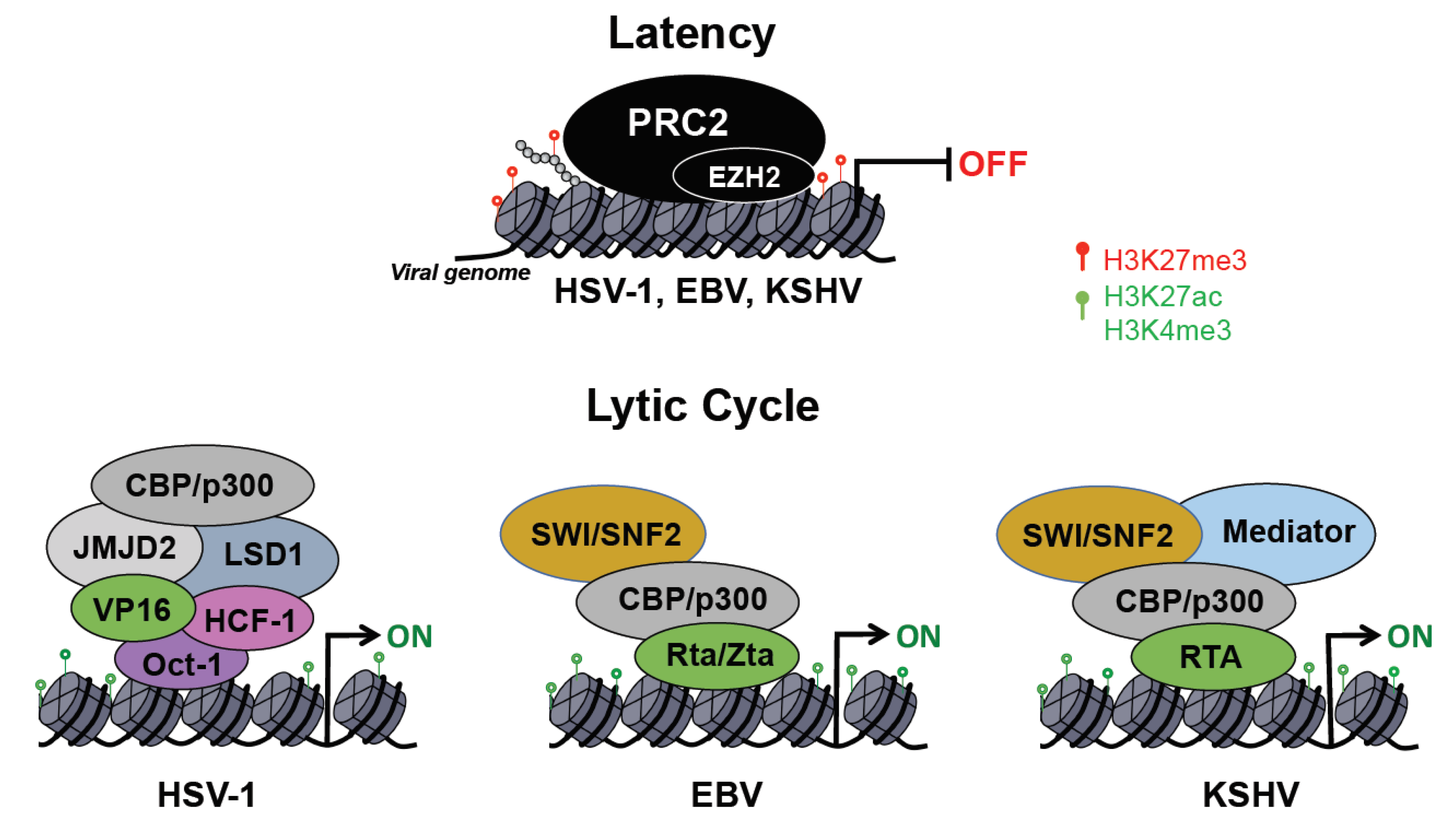
3. Impact of Host Epigenetic Machinery on the Viral Life Cycle
3.1. Transcriptional and Epigenetic Control of HSV-1 Infection
3.2. Epigenetic and Transcriptional Principles Governing EBV Infection
3.3. Epigenetic Regulation of the Biphasic Life Cycle of KSHV
4. Outlook for Epigenetic-Directed Therapeutic Interventions for Viral Infections
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bomsel, M.; Alfsen, A. Entry of viruses through the epithelial barrier: Pathogenic trickery. Nat. Rev. Mol. Cell Biol. 2003, 4, 57–68. [Google Scholar] [CrossRef]
- Groeger, S.E.; Meyle, J. Epithelial barrier and oral bacterial infection. Periodontology 2000 2015, 69, 46–67. [Google Scholar] [CrossRef]
- Groeger, S.; Meyle, J. Oral mucosal epithelial cells. Front. Immunol. 2019, 10, 208. [Google Scholar] [CrossRef]
- Dommisch, H.; Jepsen, S. Diverse functions of defensins and other antimicrobial peptides in periodontal tissues. Periodontol. 2000 2015, 69, 96–110. [Google Scholar] [CrossRef]
- Malamud, D.; Abrams, W.R.; Barber, C.A.; Weissman, D.; Rehtanz, M.; Golub, E. Antiviral activities in human saliva. Adv. Dent. Res. 2011, 23, 34–37. [Google Scholar] [CrossRef]
- Corstjens, P.L.; Abrams, W.R.; Malamud, D. Saliva and viral infections. Periodontology 2000 2016, 70, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef] [PubMed]
- Minhas, V.; Wood, C. Epidemiology and transmission of Kaposi’s sarcoma-associated herpesvirus. Viruses 2014, 6, 4178–4194. [Google Scholar] [CrossRef] [PubMed]
- Sacks, S.L.; Griffiths, P.D.; Corey, L.; Cohen, C.; Cunningham, A.; Dusheiko, G.M.; Self, S.; Spruance, S.; Stanberry, L.R.; Wald, A.; et al. HSV shedding. Antiviral. Res. 2004, 63 (Suppl. 1), S19–S26. [Google Scholar] [CrossRef] [PubMed]
- Grinde, B.; Olsen, I. The role of viruses in oral disease. J. Oral. Microbiol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, E.; Mashkoor, F.; Abdulateef, S. Oral viral infections: Diagnosis and management. Dent. Clin. N. Am. 2017, 61, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Rouse, B.T.; Sehrawat, S. Immunity and immunopathology to viruses: What decides the outcome? Nat. Rev. Immunol. 2010, 10, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Asai, D.; Nakashima, H. Pathogenic viruses commonly present in the oral cavity and relevant antiviral compounds derived from natural products. Medicines (Basel) 2018, 5, 120. [Google Scholar] [CrossRef] [PubMed]
- Tappuni, A.R. The global changing pattern of the oral manifestations of HIV. Oral Dis. 2020, 26 (Suppl. 1), 22–27. [Google Scholar] [CrossRef]
- Heron, S.E.; Elahi, S. HIV infection and compromised mucosal immunity: Oral manifestations and systemic inflammation. Front. Immunol. 2017, 8, 241. [Google Scholar] [CrossRef]
- Li, S.; Kong, L.; Yu, X.; Zheng, Y. Host-virus interactions: From the perspectives of epigenetics. Rev. Med. Virol. 2014, 24, 223–241. [Google Scholar] [CrossRef]
- Nehme, Z.; Pasquereau, S.; Herbein, G. Control of viral infections by epigenetic-targeted therapy. Clin. Epigenet. 2019, 11, 55. [Google Scholar] [CrossRef]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Stoeger, T.; Adler, H. “Novel” triggers of herpesvirus reactivation and their potential health relevance. Front. Microbiol. 2018, 9, 3207. [Google Scholar] [CrossRef]
- El Hayderi, L.; Raty, L.; Failla, V.; Caucanas, M.; Paurobally, D.; Nikkels, A.F. Severe herpes simplex virus type-I infections after dental procedures. Medicina Oral Patología Oral y Cirugia Bucal 2011, 16, e15–e18. [Google Scholar] [CrossRef][Green Version]
- El Hayderi, L.; Delvenne, P.; Rompen, E.; Senterre, J.M.; Nikkels, A.F. Herpes simplex virus reactivation and dental procedures. Clin. Oral Investig. 2013, 17, 1961–1964. [Google Scholar] [CrossRef] [PubMed]
- Grenier, G.; Gagnon, G.; Grenier, D. Detection of herpetic viruses in gingival crevicular fluid of patients suffering from periodontal diseases: Prevalence and effect of treatment. Oral Microbiol. Immunol. 2009, 24, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Victória, J.M.; Guimarães, A.L.; da Silva, L.M.; Kalapothakis, E.; Gomez, R.S. Polymerase chain reaction for identification of herpes simplex virus (HSV-1), cytomegalovirus (CMV) and human herpes virus-type 6 (HHV-6) in oral swabs. Microbiol. Res. 2005, 160, 61–65. [Google Scholar] [CrossRef]
- Casper, C.; Krantz, E.; Selke, S.; Kuntz, S.R.; Wang, J.; Huang, M.L.; Pauk, J.S.; Corey, L.; Wald, A. Frequent and asymptomatic oropharyngeal shedding of human herpesvirus 8 among immunocompetent men. J. Infect. Dis. 2007, 195, 30–36. [Google Scholar] [CrossRef]
- Slots, J. Herpesviral-bacterial synergy in the pathogenesis of human periodontitis. Curr. Opin. Infect. Dis. 2007, 20, 278–283. [Google Scholar] [CrossRef]
- Zhu, C.; Li, F.; Wong, M.C.; Feng, X.P.; Lu, H.X.; Xu, W. Association between herpesviruses and chronic periodontitis: A meta-analysis based on case-control studies. PLoS ONE 2015, 10, e0144319. [Google Scholar] [CrossRef]
- Emecen-Huja, P.; Danaher, R.J.; Dawson, D.R.; Wang, C.; Kryscio, R.J.; Ebersole, J.L.; Miller, C.S. Relationship between herpesviruses and periodontal disease progression. J. Clin. Periodontol. 2020, 47, 442–450. [Google Scholar] [CrossRef]
- Doumas, S.; Vladikas, A.; Papagianni, M.; Kolokotronis, A. Human cytomegalovirus-associated oral and maxillo-facial disease. Clin. Microbiol. Infect. 2007, 13, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Sällberg, M. Oral viral infections of children. Periodontol. 2000 2009, 49, 87–95. [Google Scholar] [CrossRef]
- Petti, S.; Lodi, G. The controversial natural history of oral herpes simplex virus type 1 infection. Oral Dis. 2019, 25, 1850–1865. [Google Scholar] [CrossRef]
- Arduino, P.G.; Porter, S.R. Herpes Simplex Virus Type 1 infection: Overview on relevant clinico-pathological features. J. Oral Pathol. Med. 2008, 37, 107–121. [Google Scholar] [CrossRef]
- Kumar, S.; Chandy, M.; Shanavas, M.; Khan, S.; Suresh, K. Pathogenesis and life cycle of herpes simplex virus infection-stages of primary, latency and recurrence. J. Oral Maxillofac. Surg. Med. Pathol. 2016, 28, 350–353. [Google Scholar] [CrossRef]
- Yan, C.; Luo, Z.; Li, W.; Li, X.; Dallmann, R.; Kurihara, H.; Li, Y.F.; He, R.R. Disturbed Yin-Yang balance: Stress increases the susceptibility to primary and recurrent infections of herpes simplex virus type 1. Acta Pharm. Sin. B 2020, 10, 383–398. [Google Scholar] [CrossRef]
- Tovaru, S.; Parlatescu, I.; Tovaru, M.; Cionca, L.; Arduino, P.G. Recurrent intraoral HSV-1 infection: A retrospective study of 58 immunocompetent patients from Eastern Europe. Medicina Oral Patología Oral y Cirugia Bucal 2011, 16, e163–e169. [Google Scholar] [CrossRef]
- Lerch, M.; Mainetti, C.; Beretta-Piccoli, B.; Harr, T. Current perspectives on erythema multiforme. Clin. Rev. Allergy Immunol. 2018, 54, 177–184. [Google Scholar] [CrossRef]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 encephalitis in adults: Pathophysiology, diagnosis, and management. Neurotherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef]
- Farooq, A.V.; Shukla, D. Herpes simplex epithelial and stromal keratitis: An epidemiologic update. Surv. Ophthalmol. 2012, 57, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Birkmann, A.; Zimmermann, H. HSV antivirals—Current and future treatment options. Curr. Opin. Virol. 2016, 18, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Longnecker, R. Epithelial cell infection by Epstein-Barr virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Webster-Cyriaque, J.; Duus, K.; Cooper, C.; Duncan, M. Oral EBV and KSHV infection in HIV. Adv. Dent. Res. 2006, 19, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Inoue, H.; Miyazaki, Y.; Ide, F.; Kojima, M.; Kusama, K. Epstein-Barr virus (EBV)-associated epithelial and non-epithelial lesions of the oral cavity. Jpn. Dent. Sci. Rev. 2017, 53, 95–109. [Google Scholar] [CrossRef]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein-Barr virus epidemiology, serology, and genetic variability of LMP-1 oncogene among healthy population: An update. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Walling, D.M.; Flaitz, C.M.; Nichols, C.M.; Hudnall, S.D.; Adler-Storthz, K. Persistent productive Epstein-Barr virus replication in normal epithelial cells in vivo. J. Infect. Dis. 2001, 184, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.D.; Neuhierl, B.; Baldwin, G.; Rickinson, A.B.; Delecluse, H.J. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Sixbey, J.W.; Nedrud, J.G.; Raab-Traub, N.; Hanes, R.A.; Pagano, J.S. Epstein-Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 1984, 310, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Tonoyan, L.; Vincent-Bugnas, S.; Olivieri, C.V.; Doglio, A. New viral facets in oral diseases: The EBV paradox. Int. J. Mol. Sci. 2019, 20, 5861. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Greenspan, D.; Lennette, E.T.; Abrams, D.I.; Conant, M.A.; Petersen, V.; Freese, U.K. Replication of Epstein-Barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 1985, 313, 1564–1571. [Google Scholar] [CrossRef]
- Henle, W.; Henle, G. Evidence for a relation of Epstein-Barr virus to Burkitt’s lymphoma and nasopharyngeal carcinoma. Bibl. Haematol. 1970, 706–713. [Google Scholar] [CrossRef]
- Guidry, J.T.; Birdwell, C.E.; Scott, R.S. Epstein-Barr virus in the pathogenesis of oral cancers. Oral Dis. 2018, 24, 497–508. [Google Scholar] [CrossRef]
- Silva, T.D.; Ferreira, C.B.; Leite, G.B.; de Menezes Pontes, J.R.; Antunes, H.S. Oral manifestations of lymphoma: A systematic review. Ecancermedicalscience 2016, 10, 665. [Google Scholar] [CrossRef]
- Jiang, R.; Ekshyyan, O.; Moore-Medlin, T.; Rong, X.; Nathan, S.; Gu, X.; Abreo, F.; Rosenthal, E.L.; Shi, M.; Guidry, J.T.; et al. Association between human papilloma virus/Epstein-Barr virus coinfection and oral carcinogenesis. J. Oral Pathol. Med. 2015, 44, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.F.; Kenney, S.C.; Lambert, P.F. Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef]
- Etemad, S.A.; Dewan, A.K. Kaposi sarcoma updates. Dermatol. Clin. 2019, 37, 505–517. [Google Scholar] [CrossRef]
- Pantanowitz, L.; Khammissa, R.A.; Lemmer, J.; Feller, L. Oral HIV-associated Kaposi sarcoma. J. Oral Pathol. Med. 2013, 42, 201–207. [Google Scholar] [CrossRef]
- Kalpidis, C.D.; Lysitsa, S.N.; Lombardi, T.; Kolokotronis, A.E.; Antoniades, D.Z.; Samson, J. Gingival involvement in a case series of patients with acquired immunodeficiency syndrome-related Kaposi sarcoma. J. Periodontol. 2006, 77, 523–533. [Google Scholar] [CrossRef]
- Fatahzadeh, M.; Schwartz, R.A. Oral Kaposi’s sarcoma: A review and update. Int. J. Dermatol. 2013, 52, 666–672. [Google Scholar] [CrossRef]
- Rohrmus, B.; Thoma-Greber, E.M.; Bogner, J.R.; Rocken, M. Outlook in oral and cutaneous Kaposi’s sarcoma. Lancet 2000, 356, 2160. [Google Scholar] [CrossRef]
- Vieira, J.; Huang, M.L.; Koelle, D.M.; Corey, L. Transmissible Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in saliva of men with a history of Kaposi’s sarcoma. J. Virol. 1997, 71, 7083–7087. [Google Scholar] [CrossRef]
- Pauk, J.; Huang, M.L.; Brodie, S.J.; Wald, A.; Koelle, D.M.; Schacker, T.; Celum, C.; Selke, S.; Corey, L. Mucosal shedding of human herpesvirus 8 in men. N. Engl. J. Med. 2000, 343, 1369–1377. [Google Scholar] [CrossRef]
- Duus, K.M.; Lentchitsky, V.; Wagenaar, T.; Grose, C.; Webster-Cyriaque, J. Wild-type Kaposi’s sarcoma-associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J. Virol. 2004, 78, 4074–4084. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.S.; Maronian, N.; Vieira, J. Activation of Kaposi’s sarcoma-associated herpesvirus lytic gene expression during epithelial differentiation. J. Virol. 2005, 79, 13769–13777. [Google Scholar] [CrossRef]
- Hassman, L.M.; Ellison, T.J.; Kedes, D.H. KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. J. Clin. Investig. 2011, 121, 752–768. [Google Scholar] [CrossRef]
- Myoung, J.; Ganem, D. Infection of lymphoblastoid cell lines by Kaposi’s sarcoma-associated herpesvirus: Critical role of cell-associated virus. J. Virol. 2011, 85, 9767–9777. [Google Scholar] [CrossRef]
- Dai, L.; Qin, Z.Q.; Defee, M.; Toole, B.P.; Kirkwood, K.L.; Parsons, C. Kaposi sarcoma-associated herpesvirus (KSHV) induces a functional tumor-associated phenotype for oral fibroblasts. Cancer Lett. 2012, 318, 214–220. [Google Scholar] [CrossRef]
- Dai, L.; Bai, L.H.; Lin, Z.; Qiao, J.; Yang, L.; Flemington, E.K.; Zabaleta, J.; Qin, Z.Q. Transcriptomic analysis of KSHV-infected primary oral fibroblasts: The role of interferon-induced genes in the latency of oncogenic virus. Oncotarget 2016, 7, 47052–47060. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Barrett, L.; Plaisance-Bonstaff, K.; Post, S.R.; Qin, Z.Q. Porphyromonas gingivalis coinfects with KSHV in oral cavities of HIV plus patients and induces viral lytic reactivation. J. Med. Virol. 2020, 6. [Google Scholar] [CrossRef]
- Dai, L.; Qiao, J.; Yin, J.; Goldstein, A.; Lin, H.-Y.; Post, S.R.; Qin, Z. Kaposi sarcoma–associated herpesvirus and Staphylococcus aureus coinfection in oral cavities of HIV-positive patients: A unique niche for oncogenic virus lytic reactivation. J. Infect. Dis. 2019, 221, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.L.; Shahir, A.M.; Sha, J.F.; Feng, Z.M.; Eapen, B.; Nithianantham, S.; Das, B.; Karn, J.; Weinberg, A.; Bissada, N.F.; et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi’s sarcoma-associated herpesvirus replication. J. Virol. 2014, 88, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Milavetz, B.I.; Balakrishnan, L. Viral epigenetics. Methods Mol. Biol. 2015, 1238, 569–596. [Google Scholar] [CrossRef] [PubMed]
- Crimi, E.; Benincasa, G.; Figueroa-Marrero, N.; Galdiero, M.; Napoli, C. Epigenetic susceptibility to severe respiratory viral infections: Pathogenic and therapeutic implications: A narrative review. Br. J. Anaesth. 2020. [Google Scholar] [CrossRef]
- Strang, B.L.; Stow, N.D. Circularization of the herpes simplex virus type 1 genome upon lytic infection. J. Virol. 2005, 79, 12487–12494. [Google Scholar] [CrossRef]
- Lieberman, P.M. Epigenetics and genetics of viral latency. Cell Host Microbe 2016, 19, 619–628. [Google Scholar] [CrossRef]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; et al. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef]
- Kent, J.R.; Zeng, P.Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar] [CrossRef]
- Huang, J.; Kent, J.R.; Placek, B.; Whelan, K.A.; Hollow, C.A.; Zeng, P.Y.; Fraser, N.W.; Berger, S.L. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J. Virol. 2006, 80, 5740–5746. [Google Scholar] [CrossRef] [PubMed]
- Kristie, T.M.; Sharp, P.A. Interactions of the Oct-1 POU subdomains with specific DNA sequences and with the HSV alpha-trans-activator protein. Genes Dev. 1990, 4, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Gerster, T.; Roeder, R.G. A herpesvirus t rans-activating protein interacts with transcription factor OTF-1 and other cellular proteins. Proc. Natl. Acad. Sci. USA 1988, 85, 6347–6351. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kristie, T.M.; Vogel, J.L.; Sears, A.E. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc. Natl. Acad. Sci. USA 1999, 96, 1229–1233. [Google Scholar] [CrossRef] [PubMed]
- Kristie, T.M. Dynamic modulation of HSV chromatin drives initiation of infection and provides targets for epigenetic therapies. Virology 2015, 479–480, 555–561. [Google Scholar] [CrossRef]
- Hill, J.M.; Quenelle, D.C.; Cardin, R.D.; Vogel, J.L.; Clement, C.; Bravo, F.J.; Foster, T.P.; Bosch-Marce, M.; Raja, P.; Lee, J.S.; et al. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Liang, Y.; Vogel, J.L.; Arbuckle, J.H.; Rai, G.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; Kristie, T.M. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Tavalai, N.; Stamminger, T. Interplay between herpesvirus infection and host defense by PML nuclear bodies. Viruses 2009, 1, 1240–1264. [Google Scholar] [CrossRef]
- Gu, H.; Zheng, Y. Role of ND10 nuclear bodies in the chromatin repression of HSV-1. Virol. J. 2016, 13, 62. [Google Scholar] [CrossRef]
- Gu, H.D.; Liang, Y.; Mandel, G.; Roizman, B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 7571–7576. [Google Scholar] [CrossRef]
- Gibeault, R.L.; Conn, K.L.; Bildersheim, M.D.; Schang, L.M. An essential viral transcription activator modulates chromatin dynamics. PLoS Pathog. 2016, 12, e1005842. [Google Scholar] [CrossRef]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 restricts HSV-1 replication by accumulating on the HSV-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J. Virol. 2016, 90, 8351–8359. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology 2015, 479, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Watson, Z.; Dhummakupt, A.; Messer, H.; Phelan, D.; Bloom, D. Role of polycomb proteins in regulating HSV-1 latency. Viruses 2013, 5, 1740–1757. [Google Scholar] [CrossRef]
- Arbuckle, J.H.; Gardina, P.J.; Gordon, D.N.; Hickman, H.D.; Yewdell, J.W.; Pierson, T.C.; Myers, T.G.; Kristie, T.M. Inhibitors of the histone methyltransferases EZH2/1 induce a potent antiviral state and suppress infection by diverse viral pathogens. mBio 2017, 8. [Google Scholar] [CrossRef]
- Edwards, T.G.; Bloom, D.C. Lund human mesencephalic (LUHMES) neuronal cell line supports herpes simplex virus 1 latency in vitro. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Washington, S.D.; Edenfield, S.I.; Lieux, C.; Watson, Z.L.; Taasan, S.M.; Dhummakupt, A.; Bloom, D.C.; Neumann, D.M. Depletion of the insulator protein CTCF results in herpes simplex virus 1 reactivation. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Watson, Z.L.; Washington, S.D.; Phelan, D.M.; Lewin, A.S.; Tuli, S.S.; Schultz, G.S.; Neumann, D.M.; Bloom, D.C. In vivo knockdown of the herpes simplex virus 1 latency-associated transcript reduces reactivation from Latency. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef]
- Day, L.; Chau, C.M.; Nebozhyn, M.; Rennekamp, A.J.; Showe, M.; Lieberman, P.M. Chromatin profiling of Epstein-Barr virus latency control region. J. Virol. 2007, 81, 6389–6401. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Chromatin structure of Epstein-Barr virus latent episomes. Curr. Top. Microbiol. Immunol. 2015, 390, 71–102. [Google Scholar] [CrossRef]
- Arvey, A.; Tempera, I.; Tsai, K.; Chen, H.S.; Tikhmyanova, N.; Klichinsky, M.; Leslie, C.; Lieberman, P.M. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 2012, 12, 233–245. [Google Scholar] [CrossRef]
- Scott, R.S. Epstein-Barr virus: A master epigenetic manipulator. Curr. Opin. Virol. 2017, 26, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Tempera, I.; Lieberman, P.M. Epigenetic regulation of EBV persistence and oncogenesis. Semin. Cancer Biol. 2014, 26, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Keeping it quiet: Chromatin control of gammaherpesvirus latency. Nat. Rev. Microbiol. 2013, 11, 863–875. [Google Scholar] [CrossRef]
- Holdorf, M.M.; Cooper, S.B.; Yamamoto, K.R.; Miranda, J.J. Occupancy of chromatin organizers in the Epstein-Barr virus genome. Virology 2011, 415, 1–5. [Google Scholar] [CrossRef]
- Ramasubramanyan, S.; Osborn, K.; Flower, K.; Sinclair, A.J. Dynamic chromatin environment of key lytic cycle regulatory regions of the Epstein-Barr virus genome. J. Virol. 2012, 86, 1809–1819. [Google Scholar] [CrossRef][Green Version]
- Tempera, I.; Wiedmer, A.; Dheekollu, J.; Lieberman, P.M. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 2010, 6, e1001048. [Google Scholar] [CrossRef] [PubMed]
- Ambinder, R.F.; Robertson, K.D.; Tao, Q. DNA methylation and the Epstein-Barr virus. Semin. Cancer Biol. 1999, 9, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Parker, G.A.; Watanatanasup, E.; White, R.E.; Allday, M.J. BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res. 2012, 40, 7233–7246. [Google Scholar] [CrossRef] [PubMed]
- Leong, M.M.L.; Cheung, A.K.L.; Dai, W.; Tsao, S.W.; Tsang, C.M.; Dawson, C.W.; Mun Yee Ko, J.; Lung, M.L. EBV infection is associated with histone bivalent switch modifications in squamous epithelial cells. Proc. Natl. Acad. Sci. USA 2019, 116, 14144–14153. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; El-Guindy, A.; Countryman, J.; Ye, J.; Gradoville, L. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv. Cancer Res. 2007, 97, 81–109. [Google Scholar] [CrossRef]
- Kalla, M.; Schmeinck, A.; Bergbauer, M.; Pich, D.; Hammerschmidt, W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. USA 2010, 107, 850–855. [Google Scholar] [CrossRef]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog. 2012, 8, e1002902. [Google Scholar] [CrossRef]
- Kalla, M.; Göbel, C.; Hammerschmidt, W. The lytic phase of epstein-barr virus requires a viral genome with 5-methylcytosine residues in CpG sites. J. Virol. 2012, 86, 447–458. [Google Scholar] [CrossRef]
- Zerby, D.; Chen, C.J.; Poon, E.; Lee, D.; Shiekhattar, R.; Lieberman, P.M. The amino-terminal C/H1 domain of CREB binding protein mediates zta transcriptional activation of latent Epstein-Barr virus. Mol. Cell. Biol. 1999, 19, 1617–1626. [Google Scholar] [CrossRef][Green Version]
- Young, L.S.; Lau, R.; Rowe, M.; Niedobitek, G.; Packham, G.; Shanahan, F.; Rowe, D.T.; Greenspan, D.; Greenspan, J.S.; Rickinson, A.B. Differentiation-associated expression of the Epstein-Barr virus BZLF1 transactivator protein in oral hairy leukoplakia. J. Virol. 1991, 65, 2868–2874. [Google Scholar] [CrossRef]
- Becker, J.; Leser, U.; Marschall, M.; Langford, A.; Jilg, W.; Gelderblom, H.; Reichart, P.; Wolf, H. Expression of proteins encoded by Epstein-Barr virus trans-activator genes depends on the differentiation of epithelial cells in oral hairy leukoplakia. Proc. Natl. Acad. Sci. USA 1991, 88, 8332–8336. [Google Scholar] [CrossRef] [PubMed]
- Niedobitek, G.; Young, L.S.; Lau, R.; Brooks, L.; Greenspan, D.; Greenspan, J.S.; Rickinson, A.B. Epstein-Barr virus infection in oral hairy leukoplakia: Virus replication in the absence of a detectable latent phase. J. Gen. Virol. 1991, 72 Pt 12, 3035–3046. [Google Scholar] [CrossRef]
- Feederle, R.; Neuhierl, B.; Bannert, H.; Geletneky, K.; Shannon-Lowe, C.; Delecluse, H.J. Epstein-Barr virus B95.8 produced in 293 cells shows marked tropism for differentiated primary epithelial cells and reveals interindividual variation in susceptibility to viral infection. Int. J. Cancer 2007, 121, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Sandvej, K.; Krenács, L.; Hamilton-Dutoit, S.J.; Rindum, J.L.; Pindborg, J.J.; Pallesen, G. Epstein-Barr virus latent and replicative gene expression in oral hairy leukoplakia. Histopathology 1992, 20, 387–395. [Google Scholar] [CrossRef]
- Kikuchi, K.; Noguchi, Y.; de Rivera, M.W.; Hoshino, M.; Sakashita, H.; Yamada, T.; Inoue, H.; Miyazaki, Y.; Nozaki, T.; González-López, B.S.; et al. Detection of Epstein-Barr virus genome and latent infection gene expression in normal epithelia, epithelial dysplasia, and squamous cell carcinoma of the oral cavity. Tumour Biol. 2016, 37, 3389–3404. [Google Scholar] [CrossRef] [PubMed]
- Eichelberg, M.R.; Welch, R.; Guidry, J.T.; Ali, A.; Ohashi, M.; Makielski, K.R.; McChesney, K.; Van Sciver, N.; Lambert, P.F.; Keleș, S.; et al. Epstein-Barr virus infection promotes epithelial cell growth by attenuating differentiation-dependent exit from the cell cycle. mBio 2019, 10. [Google Scholar] [CrossRef]
- Birdwell, C.E.; Queen, K.J.; Kilgore, P.; Rollyson, P.; Trutschl, M.; Cvek, U.; Scott, R.S. Genome-wide DNA methylation as an epigenetic consequence of Epstein-Barr virus infection of immortalized keratinocytes. J. Virol. 2014, 88, 11442–11458. [Google Scholar] [CrossRef]
- Wille, C.K.; Nawandar, D.M.; Panfil, A.R.; Ko, M.M.; Hagemeier, S.R.; Kenney, S.C. Viral genome methylation differentially affects the ability of BZLF1 versus BRLF1 to activate Epstein-Barr virus lytic gene expression and viral replication. J. Virol. 2013, 87, 935–950. [Google Scholar] [CrossRef]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-dependent KLF4 expression promotes lytic Epstein-Barr virus infection in epithelial cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef]
- Nawandar, D.M.; Ohashi, M.; Djavadian, R.; Barlow, E.; Makielski, K.; Ali, A.; Lee, D.; Lambert, P.F.; Johannsen, E.; Kenney, S.C. Differentiation-dependent LMP1 expression is required for efficient lytic Epstein-Barr virus reactivation in epithelial cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [CrossRef]
- Birdwell, C.E.; Prasai, K.; Dykes, S.; Jia, Y.; Munroe, T.G.C.; Bienkowska-Haba, M.; Scott, R.S. Epstein-Barr virus stably confers an invasive phenotype to epithelial cells through reprogramming of the WNT pathway. Oncotarget 2018, 9, 10417–10435. [Google Scholar] [CrossRef]
- Chang, H.; Wachtman, L.M.; Pearson, C.B.; Lee, J.S.; Lee, H.R.; Lee, S.H.; Vieira, J.; Mansfield, K.G.; Jung, J.U. Non-human primate model of Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog. 2009, 5, e1000606. [Google Scholar] [CrossRef]
- Bechtel, J.T.; Liang, Y.; Hvidding, J.; Ganem, D. Host range of Kaposi’s sarcoma-associated herpesvirus in cultured cells. J. Virol. 2003, 77, 6474–6481. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Ganem, D. A unique herpesviral transcriptional program in KSHV-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 2013, 13, 429–440. [Google Scholar] [CrossRef]
- Golas, G.; Alonso, J.D.; Toth, Z. Characterization of de novo lytic infection of dermal lymphatic microvascular endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Virology 2019, 536, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Wu, N.C.; Xie, Y.F.; Feng, J.; Tong, L.M.; Brulois, K.F.; Luan, H.D.; Du, Y.S.; Jung, J.U.; Wang, C.Y.; et al. Kaposi’s sarcoma-associated herpesvirus ORF18 and ORF30 are essential for late gene expression during lytic replication. J. Virol. 2014, 88, 11369–11382. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.X.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef]
- Toth, Z.; Brulois, K.; Lee, H.R.; Izumiya, Y.; Tepper, C.; Kung, H.J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef]
- Gwack, Y.; Baek, H.J.; Nakamura, H.; Lee, S.H.; Meisterernst, M.; Roeder, R.G.; Jung, J.U. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi’s sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell. Biol. 2003, 23, 2055–2067. [Google Scholar] [CrossRef]
- Papp, B.; Motlagh, N.; Smindak, R.J.; Jang, S.J.; Sharma, A.; Alonso, J.D.; Toth, Z. Genome-wide identification of direct RTA targets reveals key host factors for Kaposi’s sarcoma-associated herpesvirus lytic reactivation. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, O.; DeCotiis, J.; Goyeneche, C.; Mello, H.; Vicente-Ortiz, B.A.; Shin, H.J.; Driscoll, K.E.; Du, P.C.; Palmeri, D.; Lukac, D.M. A herpesvirus transactivator and cellular POU proteins extensively regulate DNA binding of the host Notch signaling protein RBP-J kappa to the virus genome. J. Biol. Chem. 2019, 294, 13073–13092. [Google Scholar] [CrossRef] [PubMed]
- Aneja, K.K.; Yuan, Y. Reactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus: An update. Front. Microbiol. 2017, 8, 613. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.X.; Wang, S.Z.E.; Hayward, G.S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 2005, 22, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Gould, F.; Harrison, S.M.; Hewitt, E.W.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 2009, 83, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Kobayashi, K.; Kim, K.Y.; Pochampalli, M.; Izumiya, C.; Shevchenko, B.; Wang, D.H.; Huerta, S.B.; Martinez, A.; Campbell, M.; et al. Kaposi’s sarcoma-associated herpesvirus K-Rta exhibits SUMO-targeting ubiquitin ligase (STUbL) like activity and is essential for viral reactivation. PLoS Pathog. 2013, 9, e1003506. [Google Scholar] [CrossRef]
- Gunther, T.; Grundhoff, A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2010, 6, e1000935. [Google Scholar] [CrossRef]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef]
- Sun, R.; Tan, X.H.; Wang, X.; Wang, X.D.; Yang, L.; Robertson, E.S.; Lan, K. Epigenetic landscape of Kaposi’s sarcoma-associated herpesvirus genome in classic Kaposi’s sarcoma tissues. PLoS Pathog. 2017, 13, e1006167. [Google Scholar] [CrossRef]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [CrossRef]
- Gunther, T.; Schreiner, S.; Dobner, T.; Tessmer, U.; Grundhoff, A. Influence of ND10 components on epigenetic determinants of early KSHV latency establishment. PLoS Pathog. 2014, 10, e1004274. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Papp, B.; Brulois, K.; Choi, Y.J.; Gao, S.J.; Jung, J.U. LANA-mediated recruitment of host Polycomb Repressive Complexes onto the KSHV genome during denovo Infection. PLoS Pathog. 2016, 12, e1005878. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; Hsia, D.A.; Izumiya, Y.; Wu, C.Y.; Hsieh, W.P.; Lin, S.F.; Campbell, M.; Lam, K.S.; Luciw, P.A.; et al. Histone demethylase JMJD2A regulates Kaposi’s sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J. Virol. 2011, 85, 3283–3293. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 2012, 8, e1002680. [Google Scholar] [CrossRef]
- Gunther, T.; Frohlich, J.; Herrde, C.; Ohno, S.; Burkhardt, L.; Adler, H.; Grundhoff, A. A comparative epigenome analysis of gammaherpesviruses suggests cis-acting sequence features as critical mediators of rapid polycomb recruitment. PLoS Pathog. 2019, 15, e1007838. [Google Scholar] [CrossRef] [PubMed]
- Naik, N.G.; Nguyen, T.H.; Roberts, L.; Fischer, L.T.; Glickman, K.; Golas, G.; Papp, B.; Toth, Z. Epigenetic factor siRNA screen during primary KSHV infection identifies novel host restriction factors for the lytic cycle of KSHV. PLoS Pathog. 2020, 16, e1008268. [Google Scholar] [CrossRef]
- Janzer, A.; Stamm, K.; Becker, A.; Zimmer, A.; Buettner, R.; Kirfel, J. The H3K4me3 histone demethylase Fbxl10 is a regulator of chemokine expression, cellular morphology, and the metabolome of fibroblasts. J. Biol. Chem. 2012, 287, 30984–30992. [Google Scholar] [CrossRef]
- He, J.; Anh, T.N.; Zhang, Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 2011, 117, 3869–3880. [Google Scholar] [CrossRef]
- Kang, J.Y.; Kim, J.Y.; Kim, K.B.; Park, J.W.; Cho, H.; Hahm, J.Y.; Chae, Y.C.; Kim, D.; Kook, H.; Rhee, S.; et al. KDM2B is a histone H3K79 demethylase and induces transcriptional repression via sirtuin-1-mediated chromatin silencing. FASEB J. 2018, 32, 5737–5750. [Google Scholar] [CrossRef]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife 2012, 1. [Google Scholar] [CrossRef]
- Batie, M.; Druker, J.; D’Ignazio, L.; Rocha, S. KDM2 family members are regulated by HIF-1 in hypoxia. Cells 2017, 6, 8. [Google Scholar] [CrossRef]
- Naik, N.G.; Lee, S.C.; Alonso, J.D.; Toth, Z. KDM2B overexpression facilitates lytic de novo KSHV infection by inducing AP-1 activity through interaction with the SCF E3 ubiquitin ligase complex. J. Virol. 2021. [Google Scholar] [CrossRef]
- Aubert, Y.; Egolf, S.; Capell, B.C. The unexpected noncatalytic roles of histone modifiers in development and disease. Trends Genet. 2019, 35, 645–657. [Google Scholar] [CrossRef]
- Lu, L.; Gao, Y.; Zhang, Z.; Cao, Q.; Zhang, X.N.; Zou, J.H.; Cao, Y. Kdm2a/b lysine demethylases regulate canonical wnt signaling by modulating the stability of nuclear beta-catenin. Dev. Cell 2015, 33, 660–674. [Google Scholar] [CrossRef]
- Han, X.R.; Zha, Z.; Yuan, H.X.; Feng, X.; Xia, Y.K.; Lei, Q.Y.; Guan, K.L.; Xiong, Y. KDM2B/FBXL10 targets c-Fos for ubiquitylation and degradation in response to mitogenic stimulation. Oncogene 2016, 35, 4179–4190. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y.; Tan, M.J.; Zhang, Q.; Yang, F.; Wang, S.Y.; Li, H.; Xiong, X.F.; Sun, Y. LSD1 destabilizes FBXW7 and abrogates FBXW7 functions independent of its demethylase activity. Proc. Natl. Acad. Sci. USA 2019, 116, 12311–12320. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yin, X.T.; Yang, H.R.; Xu, Y.H. Histone demethylase LSD2 acts as an E3 biquitin ligase and inhibits cancer cell growth through promoting proteasomal degradation of OGT. Mol. Cell 2015, 58, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.L.; Arnold, R.R.; Webster-Cyriaque, J. Signaling cascades triggered by bacterial metabolic end products during reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 6032–6042. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Feng, Z.M.; Yuan, G.X.; Emerson, C.C.; Stewart, P.L.; Ye, F.C.; Jin, G. Human immunodeficiency virus-associated exosomes promote Kaposi’s sarcoma-associated herpesvirus infection via the epidermal growth factor receptor. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Slots, J. Oral viral infections of adults. Periodontology 2000 2009, 49, 60–86. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hao, D.; Wang, L.; Wang, H.; Wang, Y.; Zhao, Z.; Li, P.; Deng, C.; Di, L.J. Epigenetic targeting drugs potentiate chemotherapeutic effects in solid tumor therapy. Sci. Rep. 2017, 7, 4035. [Google Scholar] [CrossRef]
- Archin, N.M.; Margolis, D.M. Emerging strategies to deplete the HIV reservoir. Curr. Opin. Infect. Dis. 2014, 27, 29–35. [Google Scholar] [CrossRef]
- Lichterfeld, M. Reactivation of latent HIV moves shock-and-kill treatments forward. Nature 2020, 578, 42–43. [Google Scholar] [CrossRef]
- Ahlenstiel, C.L.; Symonds, G.; Kent, S.J.; Kelleher, A.D. Block and lock HIV cure strategies to control the latent reservoir. Front. Cell. Infect. Microbiol. 2020, 10, 424. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Balogh, K.K.; Brendle, S.A.; Campbell, C.A.; Chen, M.X.; Furze, R.C.; Harada, I.L.; Holyer, I.D.; Kumar, U.; Lee, K.; et al. BET bromodomain inhibitors show anti-papillomavirus activity in vitro and block CRPV wart growth in vivo. Antiviral. Res. 2018, 154, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Sinclair, J.H.; Wills, M.R. Bromodomain inhibitors as therapeutics for herpesvirus-related disease: All BETs are off? Front. Cell. Infect. Microbiol. 2020, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Quenelle, D.; Vogel, J.L.; Mascaro, C.; Ortega, A.; Kristie, T.M. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. mBio 2013, 4. [Google Scholar] [CrossRef]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atyeo, N.; Rodriguez, M.D.; Papp, B.; Toth, Z. Clinical Manifestations and Epigenetic Regulation of Oral Herpesvirus Infections. Viruses 2021, 13, 681. https://doi.org/10.3390/v13040681
Atyeo N, Rodriguez MD, Papp B, Toth Z. Clinical Manifestations and Epigenetic Regulation of Oral Herpesvirus Infections. Viruses. 2021; 13(4):681. https://doi.org/10.3390/v13040681
Chicago/Turabian StyleAtyeo, Natalie, Michelle D. Rodriguez, Bernadett Papp, and Zsolt Toth. 2021. "Clinical Manifestations and Epigenetic Regulation of Oral Herpesvirus Infections" Viruses 13, no. 4: 681. https://doi.org/10.3390/v13040681
APA StyleAtyeo, N., Rodriguez, M. D., Papp, B., & Toth, Z. (2021). Clinical Manifestations and Epigenetic Regulation of Oral Herpesvirus Infections. Viruses, 13(4), 681. https://doi.org/10.3390/v13040681