Abstract
Three decades of extensive work in the HIV field have revealed key viral and host cell factors controlling proviral transcription. Various models of transcriptional regulation have emerged based on the collective information from in vitro assays and work in both immortalized and primary cell-based models. Here, we provide a recount of the past and current literature, highlight key regulatory aspects, and further describe potential limitations of previous studies. We particularly delve into critical steps of HIV gene expression including the role of the integration site, nucleosome positioning and epigenomics, and the transition from initiation to pausing and pause release. We also discuss open questions in the field concerning the generality of previous regulatory models to the control of HIV transcription in patients under suppressive therapy, including the role of the heterogeneous integration landscape, clonal expansion, and bottlenecks to eradicate viral persistence. Finally, we propose that building upon previous discoveries and improved or yet-to-be discovered technologies will unravel molecular mechanisms of latency establishment and reactivation in a “new era”.
Keywords:
HIV-1; provirus; persistence; cure; integration; transcription; ART; latency; latent reservoir; epigenomics 1. The Latent Reservoir in the Spotlight
The development of combination anti-retroviral therapy (ART), which targets several steps of the viral life cycle, decreased patient viral titers to below the limit of detection using contemporary methods [1,2]. This remarkable biomedical breakthrough was the tipping point for reducing mortality and extending the lifespan of HIV-infected individuals. However, despite initial hopes of undetectable viremia, the discontinuation of ART quickly led to viral titer rebound [3,4], suggesting the presence of a “latent” yet inducible and replication-competent viral reservoir. Consequently, in efforts to control infection, patients must remain on a lifelong regime of ART to prevent the rebound of plasma viremia.
The best characterized latent reservoir in patients consists of integrated viral DNA in resting memory CD4+ T cells [2,5], which are refractory to immune surveillance and current ART regime. Studies over the past decade have revealed the timeline, size, stability, and composition of the latent reservoir in ART treated patients.
1.1. Timeline, Size, Stability, and Composition of the Latent Reservoir
Kinetically, the latent reservoir is established within days after infection regardless whether the patient has undergone ART [6]. The stability of the latent reservoir in resting memory CD4+ T cells in patients receiving highly suppressive therapy with no detectable viremia for many years has been estimated to have a half-life of ~44 months [7]. Given this long-term stability, it is impossible to cure HIV by waiting for the infected cells to decay over time.
In addition to the stability of the latent reservoir, there have been attempts to estimate its size and composition. Initial studies used PCR-based techniques but were later found to have largely overestimated the reservoir magnitude [8,9,10,11,12], as the majority of latently-infected cells in patients hold replication-incompetent proviruses. This discovery indicated that defective genomes are not the source of the rebound virus upon ceasing ART, although they can otherwise contribute to continued immune activation and exhaustion [10,13].
More recent assays, such as the quantitative viral outgrowth assay (QVOA), measure the latent reservoir by computing the number of latently-infected resting CD4+ T cells that produce replication-competent virus after in vitro stimulation typically with strong T-cell agonists [14,15]. With this assay, it was estimated that about one in a million resting CD4+ T cells in the blood of ART treated patients can be induced to produce replication-competent virus [7,16,17,18]. Nonetheless, in contrast to PCR detection methods, the QVOA largely underestimates the reservoir size as not all intact proviruses reactivate after one round of immune stimulation, i.e., subsequent rounds of treatment can activate a larger (2–3-fold) population of intact proviruses [8,19,20].
In theory, most intact proviruses are capable of producing replication-competent virus. However, the frequency of cells harboring intact proviruses that can, in theory, be stimulated is ~30 times larger than the actual frequency of cells that are induced in the QVOA assay. The collective evidence suggest ~90% of proviral genomes in resting memory CD4+ T cells within individuals on ART are defective, and from the remaining ~10% of intact proviruses [8,19,21] only ~0.1–0.2% can be reactivated, posing the question as to where in the human genome is this small fraction of intact and reactivatable proviruses located, and what are the molecular mechanisms underlying their persistence.
Overall, the scarcity of cells containing intact proviruses has posed limitations for defining the composition and underlying molecular mechanisms regulating these types of proviruses. Recent tools to simultaneously define both integration positions and proviral intactness [22,23] are starting to reveal a clearer picture of the “physiologic” latent reservoir and its heterogeneous mechanism of regulation.
Strikingly, Swanstrom and colleagues reported that the genetic diversity of the latent reservoir largely depends on the timing of initiating ART post-infection (i.e., acute or chronic phase) [24], where ~71% of unique intact proviruses induced from post-ART samples were genetically similar to samples taken shortly before ART initiation. Thus, within the intact latent reservoir, there is a small population of inducible proviruses that can potentially reseed the active viral reservoir if ART is interrupted. Further, to sustain viral lifelong persistence, several recent studies have shown the intact, replication-competent proviruses within populations of resting memory CD4+ T cells are maintained by inconspicuous levels of clonal expansion [20,25,26,27,28] which could influence targetable approaches. Sékaly and colleagues described that proviruses are maintained by either homeostatic proliferation (in memory CD4+ T cells) or antigen stimulation (in resting central memory CD4+ T cells) rather than by viral re-infection [29]. Moreover, it was further suggested that clonal expansion can be driven by cytokine and antigen stimulation without causing antiviral effects or inducing viral production, thereby giving key biological insights suggesting an additional challenge to reverse latency in the clinics [30].
1.2. Routes of Latency Establishment
The previous discoveries on the timeline, size, stability, and composition of the latent reservoir opened a field on itself and led to the long-lasting debate regarding how the reservoir is established. Different proposed routes have been made concerning how latency establishment occurs after HIV integrates into the genome of target immune cells (naïve and effector CD4+ T cell subsets) [4,5,31,32,33,34]. At present, the most widely accepted model of latency establishment is that HIV enters a latent state when effector CD4+ T cells transition to a resting memory state due to cellular relaxation, the so-called effector-memory-transition (EMT) [35,36] (Figure 1A, top panel). We refer to this mechanism as “late” latency because the provirus is initially active and later becomes inactive as a potential consequence of cellular relaxation, thereby suggesting a major contribution of host cell state on viral entry into latency. Alternative, non-mutually exclusive models proposed the direct infection of resting CD4+ T cells [37], and naive or effector CD4+ T cells in which the proviral genome is not efficiently transcribed (Figure 1A, bottom panel). In the latter case, it is likely certain integration sites promote “early” latency, in which the provirus becomes transcriptionally silent immediately after integration into specific, unfavorable genomic areas, despite the beneficial environment for proviral transcription and replication in effector CD4+ T cells (Figure 1A, bottom panel).
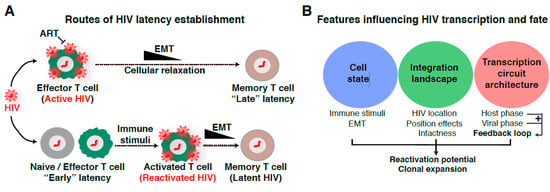
Figure 1.
Routes of latency establishment and features influencing HIV proviral transcription and fate. (A) In “early” latency, HIV integrates into genomic domains not supporting transcription, but is otherwise intact and induced by immune stimuli. Alternatively, HIV infects cells in different states of activation or during effector-memory transition (EMT) [35,36] in which cellular relaxation pushes an initial transcriptionally active provirus into a silent provirus promoting “late” latency. (B) List of features contributing to HIV proviral transcription and fate. The “cell state” denotes CD4+ T cells of any of the major subsets (e.g., Th1, Th2, Th17, Treg, Tcm) in different stages (naïve, effector, resting memory, and effector memory). The “integration landscape” describes the heterogeneous positions of HIV proviruses in the human genome and position effects. The complex “transcription circuit architecture” describes the progressions through the different phases of the HIV transcriptional program (basal, host, and viral) leading to the positive-feedback loop. See text and Figure 4A for details.
In contrast to “late” latency, “early” latency suggests a major contribution of integration landscape, but not cell state, of viral entry into latency. As such, the integration site can influence the provirus in different ways: (1) through a nucleosome position and epigenetic-driven mechanism, and/or (2) through an RNA polymerase II (Pol II) transcription-driven mechanism related to transcriptional interference [38,39]. On this topic, several landmark studies have surveyed the chromatin environment of single-copy integrated proviruses in CD4+ T cell lines such as Jurkat (the J-Lat clones), including nucleosome positioning [40,41], DNA methylation [42], and histone post-translational modifications (PTMs) [41,43,44,45,46] to study their impact on the establishment and/or maintenance of latency, thereby providing an initial view of the potential effect of integration landscape to proviral transcription and fate (active vs. latent; inducible vs. not inducible). In the sections below, we expand on these topics to describe key discoveries, potential limitations, and urgently needed ideas for future investigations to underpin their contributions to disease progression in physiologically relevant models and patient samples.
One major paradigm about “late” latency is whether it is simply attributed to cells dropping immune signaling dynamics during EMT (Figure 1A, top panel), thereby reducing transcription-sustaining host cell factors, or whether the virus actively “pushes” cells into the resting state by rewiring host transcriptional programs [32], or a combination of both. Conversely, other theories have argued in favor of a cell-autonomous model establishing latency where the virus itself shuts-off viral transcription without host cell-intrinsic or cell-extrinsic contributions [47], indicating a “free solo” scenario. However, the potential key viral players involved in the proposed mechanisms remain to be discovered. Together, these studies potentially indicate that both viral and host cell factors influence the establishment and maintenance of proviral latency.
Despite all these possible routes of latency establishment, one outstanding question in the field to address is whether there are several mechanisms operating at different levels due: (1) to the site of proviral integration into the human genome (weighting more on the proposed “early” latency model) and/or (2) to alterations in cell state leading to cellular relaxation (weighting more on the proposed “late” latency model) (Figure 1A).
Because of the multiple routes of latency establishment and features influencing proviral transcription, including immune cell state, integration landscape, and complex transcription circuit architecture (Figure 1B), it has become difficult to ascertain their precise contribution to proviral fate. Indeed, the numerous possible variations on the origin of latency at the single-cell level may account for the rising of a heterogenous proviral population (Figure 2), whereby cells contain proviruses with variable degrees of activity (expression and replication). While ART stops viral replication in cells containing active proviruses, ART is unable to cope with the transcriptionally silent proviruses and those replicating at very low levels. Therefore, these proviruses survive therapy and generate a latent reservoir that upon ART cessation results in a rapid rebound of plasma viremia. We thus propose that a “new era” of research in this field will encompass uncoupling the influence of various regulatory features (immune cell state, integration landscape, and transcription circuit architecture) on proviral transcription (Figure 1B). We envision that building upon previous discoveries and improved technologies with the advent of genomics (e.g., single-cell) and deep learning will facilitate unraveling molecular mechanisms of latency establishment and reactivation. Further, this will enable the identification of novel cell targets, which may guide strategies to eliminate persistent reservoirs to end the HIV epidemic [48].
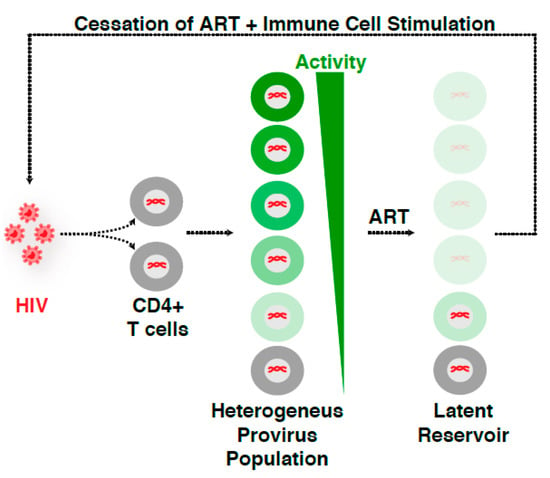
Figure 2.
HIV infection leads to a heterogeneous proviral population. HIV infection of CD4+ T cells yields a “heterogeneous provirus population” with a variable, continuum degree of gene expression and replication. ART stops viral replication and disease progression into the AIDS phase. However, ART is unable to cope with both the transcriptionally silent proviruses and those replicating at very low levels, thereby surviving therapy and generating the so-called “latent reservoir”. As such, ART discontinuation in the presence of immune stimulation results in a rapid rebound of virus, indicating that while ART suppresses viral replication, HIV is able to persist in an infectious state for years.
Given all these previous landmark discoveries, here, we provide a recount of the literature to discuss the evolution of the field, highlighting the use of various latency models and studies with patient isolates. At the same time, we will be presenting potential limitations of previous discoveries and discuss new approaches needed to move the field forward in our race to devise an effective HIV cure strategy in the “new era”. Specifically, we discuss the following: the role of the nuclear architecture in integration site selection, the current knowledge on how the integration landscape may influence HIV transcription dynamics and proviral fate, the role of nucleosomes and chromatin states, the functions of host cell machinery in regulating transcription, and finally combine these basic discoveries to better understand the therapeutic challenges faced.
2. Contribution of Nuclear Topology Shaping the Integration Landscape, Transcriptional Regulation, and Proviral Fate
Studies over the past two decades have defined how HIV integrates into the human genome and how proviral transcription normally operates [49,50,51,52,53]. It has long been known that HIV preferentially integrates into chromatin accessible sites and within or near transcriptionally active regions/genes [54,55,56], which led to the definition of “recurrent integration genes” (RIGs) (Figure 3A) [57,58]. However, integrated proviruses are detected on every human chromosome, in various chromatin landscapes (euchromatic and heterochromatic), and at different locations (intergenic or intragenic) and orientations (sense, divergent or convergent) respective to human genes and regulatory elements (Figure 3B). Despite these landmark discoveries, the underlying molecular basis for this biased integration pattern has long-remained unclear.
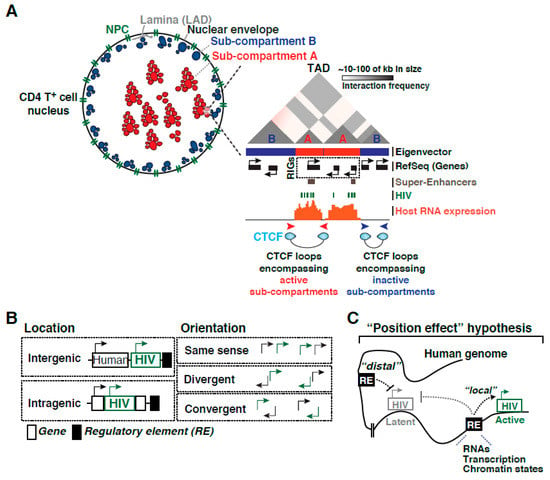
Figure 3.
Contribution of nuclear topology shaping the integration landscape, transcriptional regulation, and proviral fate. (A) Scheme depicting the relationship between nuclear topology, nuclear envelope, nuclear lamina with its associated domains (LADs), topological associated domains (TADs), the position of the eigenvector respective to nuclear sub-compartments A and B, and their genetic composition (genes and regulatory elements like super-enhancers) with their activity (host RNA expression). The position of RIGs in the RefSeq track and other chromatin accessible regions of the human genome “preferred” for HIV integration (such as super-enhancers) are highlighted. (B) Summary of HIV insertion patterns (both location and orientation) relative to genes and regulatory elements in the human genome (e.g., enhancers, mobile elements). (C) Position effect hypothesis highlighting both local and distal effects from human genome regulatory elements and long-range chromatin interactions, thereby influencing proviral fate.
2.1. Importance of CD4+ T Cell Nuclear Topology for HIV Integration
Landmark work by Lusic and colleagues revealed that HIV integration occurs in the outer shell of the nucleus and in close communication with the nuclear pore complex (NPC) [57], thus highlighting a key role for nuclear topology and RIGs strategic positioning for viral integration (Figure 3A). Further evidenced, through electron microscopy, heterochromatin was visualized at the nuclear periphery and overlapping with lamin-associated domains (LADs), whereas euchromatin is typically associated with the NPC resulting in a nonrandom spatial distribution of chromatin (reviewed in [59]). Interestingly, this NPC proximal region contains a subset of host genes characterized by the presence of active transcription chromatin marks in autologous cells. Furthermore, as expected, the heterochromatic regions in the LADs and other transcriptionally active regions located centrally in the nucleus were strongly disfavored [57], consistent with the idea that HIV prefers chromatin accessible regions potentially located in exposed areas of the nucleus and away from LADs (Figure 3A).
Notably, the spatial proximity between the NPC and actively transcribed genes could explain the relationship between HIV integration sites and the first host genomic DNA that HIV would encounter during the translocation of the viral particle from the cytoplasm to the nucleus as a pre-integration complex. Indeed, as the first barrier to access host chromatin, the 30 nucleoporins (Nup) comprising the NPC have been extensively studied in efforts to understand the mechanisms underlying retrovirus integration. By systematically knocking down the expression of each Nup, Bieniasz and colleagues comprehensively explored their contribution to HIV infection and proposed the idea of potential Nup-dependent “pathways” facilitating HIV access to host chromatin [60], implicating nuclear topology with integration site selection. As this review will not focus on the specifics of retroviral integration per se, other key host cell factors facilitating this process are summarized in excellent recent reviews by colleagues in the field [52,61,62].
2.2. CD4+ T Cell Nucleus Sub-Compartments and Regulatory Elements
The three-dimensional (3D) organization of chromosomes enables long-range interactions between enhancers and promoters that are critical for building complex gene regulatory networks [63,64]. Interphase chromosomes occupy separate spaces known as nuclear territories [65] and each chromosome is organized into dynamic, non-random structures containing stretches of transcriptionally active compartments interspersed with sections of transcriptionally inactive compartments [66]. As such, the genome is partitioned into contact domains (Compartments A and B) (Figure 3A) segregating into smaller sub-compartments (A1, A2, B1, B2, B3, and B4) that (1) appear located in different nuclear territories, (2) are associated with distinct patterns of histone marks, and (3) show different expression levels [67].
Interestingly, RIGs are clustered in specific spatial compartments (sub-compartments A) of the CD4+ T cell nucleus [58], which seem to acquire this location during T cell activation, thus explaining their preferred recognition during the integration process (highlighted in green in Figure 3A). Further work to define the genomic features enriched at HIV integration genomic sites and the relevance of their transcriptional activity revealed that RIGs are typically proximal to super-enhancers (referred to as SE). SE were originally defined as locally grouped clusters of enhancers (defined as H3K27ac, H3K4me3, H3K4me1, and active transcription domains within 12.5-kb of each other) driving high levels of transcription of nearby cell-identity genes (Figure 3A) [68]. These results have thus exposed the importance of spatial compartmentalization of the host cell genome for HIV integration during T cell activation, thereby reinforcing the role of nuclear architecture in the infection process as well as in future proviral fate changes.
Recent developments in genomic technologies have led to rapid advances in the study of the 3D genome organization. In particular, Hi-C has been introduced as a method for identifying higher-order chromatin interactions genome-wide [66]. Ren and colleagues have reported megabase-sized local chromatin interaction domains called topologically associated domains (TADs) (Figure 3A) as structural features of the genome organization [69]. TADs boundaries are enriched for the insulator binding protein CCCTC-binding factor (CTCF) forming “CTCF loops” encompassing active sub-compartments A and to block heterochromatin spreading typically present in sub-compartments B (Figure 3A).
Despite these past discoveries, the contributions of nuclear sub-compartments, TADs, and CTCF loops to HIV proviral transcription and fate remains unknown. Since compartments are dynamic in nature, it is likely that their position and chromatin content will change during EMT, thereby potentially converting an active provirus into a resting one during cellular relaxation (Figure 1A). This is consistent with the idea that latency is an unfortunate consequence of infection during a narrow window after CD4+ T cell activation [35].
2.3. Integration Site Effects on HIV Proviral Transcription and Fate
Notwithstanding the key role of the nuclear architecture in the integration process, and since the integration landscape is highly diverse in sequence and chromatin structure [70,71,72], it seems logical to speculate that the integration site could contain information influencing the magnitude of HIV transcription, thereby shaping the heterogeneous proviral fate (Figure 2). Supporting this idea, landmark work by Verdin and colleagues on tens of Jurkat CD4+ T cell clones containing individual HIV integrants revealed that the integration site controls basal and immune stimulation-dependent transcription [71]. This indicates HIV possibly operates in an integration site- and cell state-dependent manner, consistent with theories of “early” and “late” latency establishment (Figure 1A). By analyzing primary patient samples, Nussenzweig and colleagues described most intact proviruses are maintained in silent regions where the cells are in a quiescent state, while the active clonally expanded proviruses are defective, further confirming HIV proviral fate may be cell state- and integration site-dependent (Figure 1B) [21].
Additionally, it is increasingly evident that integration site profiles partially differ in preference for orientation relative to the gene and functional gene classification (i.e., oncogene, housekeeping) between in vitro acutely infected immortalized cell lines (e.g., HeLa, Jurkat) vs. aviremic patient samples under highly suppressive therapy [25,73]. For example, in patient samples, HIV integrated into introns and in the same orientation of proto-oncogenes (e.g., BACH2, MKL2, STAT5B) and also upstream of the transcription start sites (TSS). However, these orientation and location preferences/biases were not observed when comparing to in vitro infected cells which had HIV integrated into both orientations relative to the gene and located throughout the genes [25,73]. These studies suggest potential limitations of using in vitro models of integration establishment for understanding the integration landscape effect on HIV proviral transcription and fate decisions and may also indicate a selective pressure in vivo to keep a certain type of integrants.
Strikingly, Nussenzweig and colleagues described preferred integration in “hotspot” locations at or near Alu sequences/repeats in patient samples [21]. Alu are mobile elements that belong to a class of primate-specific retroelements termed SINEs (short interspersed elements), have a copy number well in excess of 1 million, comprise ~11% of the human genome, and are known to have wide-ranging influences on gene expression (reviewed in [74]). Thus, Alu may function as regulatory elements shaping HIV proviral transcription and fate (Figure 3B) in clonally expanded but deficient proviruses and/or in intact proviruses found in CD4+ T cells that remain in a quiescent state, a critical aspect that remains to be determined.
Despite these past discoveries, it remains unknown what genomic features or combination of features control HIV proviral expression and fate. It has been known for years that the human genome provides the underlying code for the correct transcriptional regulation of most biological processes through precise spatial-temporal interactions between cis-elements present at promoters and enhancers and sequence-specific factors that recognize them, as well as by the position of genes and regulatory elements in the three-dimensional space and in relation to nuclear territories and sub-compartments (Figure 3A). These regulatory elements could provide “local” (related to the processes of transcription and chromatin accessibility) or “distal” (chromatin communication between human and viral genomes) functions that could influence HIV expression, thereby modulating proviral fate (Figure 3C). Depending on the integration site, proviruses might function as constitutively active or silent transcriptional units, and a fraction of the silent proviruses could be reactivated at different levels by immune stimulation. Given these possibilities, we hypothesize that the integration site contains “instructions” or “integration code” (here referred to as “position effect” hypothesis) in which one or more genomic features located proximally and/or distally from the integration site could shape the organization and activity of proviruses in the context of the human genome (Figure 3C).
2.4. Conclusions and Future Directions
Notwithstanding our current knowledge regarding how the HIV transcriptional program operates, we have no clear understanding on the role of the integration landscape on proviral fate. Given the large diversity of the integration landscape as well as the complexity in the regulation of transcription, it is conceivable that a combination of regulatory elements (including the location and orientation of proviruses respective to nearby genes and regulatory elements such as enhancers and mobile elements, chromatin states of the provirus and neighboring genomic domains, and long-range chromatin interactions (Figure 3B)) could define the “integration code.” As such, this major biomedical challenge demands a comprehensive definition of the molecular rules modulating proviral fate before we can even leverage this knowledge in the clinical setting. Studies in the “new era” may require deep, integrated genomic approaches, combined with the interrogation of patient samples and the potential implementation of deep learning models to both predict and test molecular features contributing to proviral expression and persistence [75]. First, human genome codes shaping proviral transcription could be deciphered by combining open-source, large-scale datasets (including epigenetics, transcriptome, and 3D genome architecture) to interrogate the chromatin states, transcription activity landscape, nuclear sub-compartments, TADs, and chromatin loops containing the major regulator CTCF around HIV integration sites in CD4+ T cells (Figure 3A). Second, regulatory features in the human genome can be predicted and experimentally tested by implementing deep learning approaches. Third, interrogating patient samples with methods that can simultaneously assess proviral integration positions, intactness, and activity at the single-cell level may provide physiologic relevance. Together, these efforts could then be compatible with clinical decision-making, for personalized genome editing-mediated elimination of intact proviruses.
3. The HIV Transcriptional Program and Proviral Fate
3.1. Phases of the HIV Transcriptional Program
The HIV transcriptional program is composed of various regulatory phases and is controlled by many host cell and viral activators. First, during normal cell homeostasis, “basal” steady-state transcription keeps a low level of non-productive RNA synthesis, leading to short, immature transcripts (Figure 4A). In this state, the viral encoded activator Tat is not expressed, and HIV does not replicate, thereby promoting latency establishment. In the “host” phase, when cells are exposed to immune stimulation, host transcription factors are activated, leading to an initial low-level boost in proviral transcription. In defective proviruses lacking functional Tat [76] or containing mutations in its binding sites on the trans-activation response (TAR) stem-loop [77], which is formed at the 5′-end of nascent viral pre-mRNAs by Pol II, the host phase leads to a small amount of viral products. However, during productive infections with intact proviruses containing functional Tat-TAR axes, the initial transcriptional boost enables Tat synthesis before the “host” phase turns off. In this case, the “host” phase is rapidly followed by a “viral” phase in which Tat amplifies transcription by more than 100-fold, promoting a positive feedback loop and robust viral replication [78] (Figure 4A).
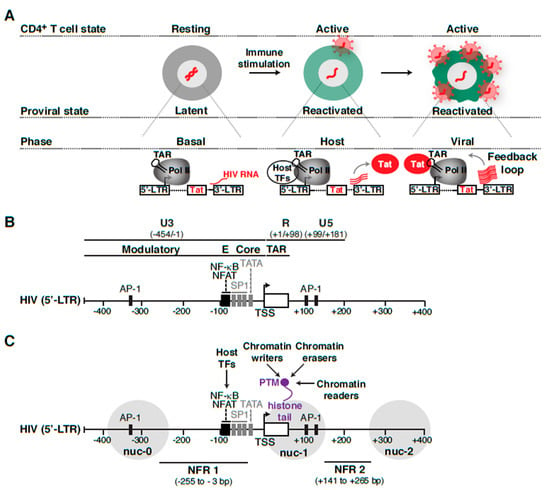
Figure 4.
The multiple layers of control in the HIV transcriptional program. (A) Schematic representation of the progressions through the HIV transcriptional program. See text for details. (B) Schematic representation of the HIV 5′-LTR showing U3, R, and U5 regions. The U3 region is further divided into the modulatory region, enhancer (E), core promoter, and TAR. The U3 and R regions comprise binding sites for SP1, NF-κb/ NFAT, and AP-1 transcription factors. (C) A depiction of chromatin landscape at the HIV promoter showing the three well-positioned nucleosomes (nuc-0, nuc-1, and nuc-2), separated by two large NFRs (NFR-1 and NFR-2) in the basal state (latent proviral fate). The action of chromatin modifiers (Readers, Writers, and Erasers) render the position of nuc-1 downstream of the TSS and regulate the establishment of proviral latency. The position of one histone tail and a general PTM (i.e., acetylation and methylation) is shown as a simple representation. See text for details.
In the basal state of resting CD4+ T cells, most host transcription factors that bind the proviral promoter are in an inhibited state but become activated when infected cells encounter a stimulus from the immune microenvironment. For example, ligands (such as antigens and cytokines) trigger several host cell signaling pathways upon receptor activation, inducing the initial translocation of sequestered, cytoplasmic host factors into the nucleus which subsequently recognize their respective binding element at the viral promoter, driving proviral transcription [79,80]. Conceptually, CD4+ T cell stimulation functions in a broadly similar manner, i.e., through multiple signaling pathways inducing several master cellular activators [79,81].
3.2. Feedback Loop and Theories of Latency Establishment due to Dysfunctions in the HIV Transcriptional Program
While the normal function of the two phases (host and viral) creates a positive feedback loop facilitating viral replication, their interruption promotes latency. However, it has long remained unclear to what extent the host phase influences the outcome of the viral phase. Using experimental and modeling approaches, D’Orso and colleagues recently created a mathematical model recapitulating the complete HIV transcriptional program (“basal-host-viral”). These studies revealed that the host phase is subject to “transcriptional fragility” (due to host co-factor dysfunction), thereby dampening the feedback loop and latency-reversal potential [82] and adding an extra layer of complexity in the mechanisms establishing latency. Concerning patient-derived cells, this model predicts fluctuations in levels of key host co-factors could affect the host phase and, as a consequence, the magnitude of the Tat feedback.
Notably, variations in host co-factor levels alter the outcome of the host phase and impart heterogeneity in the transcriptional responses [82], thereby influencing the reactivation potential of latent HIV. These studies provided a mechanistic explanation on the importance of the host phase to ensure the virus is readily and robustly activated. Interestingly, Weinberger and colleagues have shown that “stochastic” Tat fluctuations drive entry into latency [83], consistent with the idea that the “threshold” of host phase activation influences Tat function [82]. Thus, it is likely that the system stochasticity arises as a consequence of host phase alterations driving Tat synthesis [82]. According to Weinberger, these stochastic fluctuations in Tat drive phenotypic diversity [83], probably originated from transcriptional bursting from the viral promoter [84] and lack of system bistability [85], ultimately influencing the viral latency decision [83]. Thus, the combined transcription heterogeneity and system stochastic variability present a major obstacle for therapeutic strategies to eliminate the latent reservoir [86].
Transcription heterogeneity has been observed in both latently infected clonal CD4+ T cell populations [71,87] and primary resting CD4+ T cells [8,88]. Even after potent immune stimulation, reactivation occurred only in a fraction of cells, while latent proviruses remained inactive in the majority of the cells [8,89]. Recently, a single-cell RNA-seq (scRNA-seq) approach revealed heterogeneous responses during latent HIV reactivation in primary systems (CD4+ T cells) and proposed a model in which cells transition between two alternate states [90], potentially explaining the variable responses.
Given these theories, and to further distinguish whether proviral latency is dependent or independent of cell state, Weinberger and colleagues used an artificially modulated Tat circuit (where the expression of Tat is fused to a controllable-proteolysis tag). They provided evidence that the Tat positive feedback circuit is “hardwired” and sufficient to establish proviral latency, independent of cell activation state [47], thus contradicting theories in which cell state prevails over cell-autonomous, HIV centric mechanisms.
Supporting the importance of cell state, Siliciano and colleagues showed that latency is an unfortunate consequence of infection of CD4+ T cells within a narrow time window after activation in which transcriptional reprogramming during EMT (Figure 1A) renders CD4+ T cells permissive for latency [35]. These discoveries are also consistent with recent studies by Karn and colleagues showing that entry of effector CD4+ T cell subsets into quiescence forces HIV into latency [32], in which the activity of master regulators of the host phase declines over time through their inactivation and sequestration in the cytoplasm (reviewed in [91]). Collectively, while this review does not pretend to address which model is right and if all models are theoretically possible, it is likely that a combination of cell- and HIV-driven forces contribute to the stochastic nature of the HIV transcriptional program.
3.3. Conclusions and Future Directions
Given the multiple routes of latency establishment and features influencing proviral transcription, namely immune cell state, integration landscape, and complex circuit architecture (Figure 1A,B), it has become difficult to ascertain their precise contribution to proviral fate (activation, latency, and reactivation). Collectively, we propose that further studies uncoupling their influence on proviral transcription will unravel molecular mechanisms of latency establishment and reactivation, and allow the identification of novel host targets, which may guide strategies to eliminate the persistent yet inducible latent reservoir.
6. HIV Proviral Nucleosome Positioning and Epigenomics
Following HIV integration into the host genome, a highly ordered chromatin structure is formed encompassing the proviral genome [40,41]. The chromatin structure formed not only governs the establishment and maintenance of proviral latency but also plays a critical role in transcriptional reactivation and latency reversal by serving as the target of chromatin modulators (activators and repressors). The functions of activators/repressors, Pol II, and elongation factors are coordinated by nucleosomes, which are the smallest unit of chromatin. Not merely “beads-on-a-string,” nucleosomes have an active role in the regulation of gene transcription (reviewed in [194]). Given the known functions nucleosomes play in modulating gene activation/repression through chemical modifications and strategic positioning within gene regulatory elements, it is evident that they also exert a layer of HIV transcriptional control, thereby shaping proviral fate. For the purpose of this review, we will mainly focus on chromatin-remodeling complexes regulating nucleosome positioning at the proviral genome in addition to chromatin-modifying complexes (writers, erasers, and readers) regulating the proviral epigenome (Figure 4C).
6.1. Chromatin-Remodeling Complexes and Associated Factors
In the early days, the role of chromatin organization in transcriptional regulation of HIV provirus was studied in immortalized models of latency [ACH-2 (T-cell) and U1 (macrophage)] using Micrococcal nuclease (MNase) digestion followed by enzyme restriction analysis [40], which has low resolution and DNA sequence-cleavage biased. In these models, under “basal” conditions, the 5′ LTR of the integrated provirus appeared to contain an array of five well-positioned nucleosomes (nuc-0 to nuc-4) with two nucleosome-free regions (NFR) spanning −255 to −3 and +141 to +265 bp, respective to TSS (+1 nt) (Figure 4C) [40]. While nuc-0 occupied the beginning of the U3 region and contained several promoter and enhancer elements, nuc-1 was positioned in the R-U5 region and flanked on either side by two NFRs [40]. Further, MNase mapping of the 5′-LTR has shown that nucleosome organization appears to be independent of the integration landscape, which was also confirmed using in vitro nucleosome reconstitution [40,41,123,195]. This nucleosomal arrangement is preserved by virtue of chromatin remodelers [196]. The nuc-1 present downstream of the proviral TSS acts as a barrier for transcription initiation [40] but is rapidly disrupted upon activation in response to a plethora of agonists such as pro-inflammatory cytokines (like TNF, activating NF-κB signaling), phorbol esters (like PMA/TPA, activating protein kinase C (PKC)), and histone deacetylase inhibitors (like trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA)), which appear to induce hyperacetylation of the integrated proviral genome) [40,41,123,196,197]. Interestingly, nucleosome mapping techniques such as indirect end labeling and MNase protection assays revealed nuc-1 position overlaps with the site of Pol II pausing at the proviral genome in immortalized models of latency (U1, ACH-2, and Jurkat), reiterating nuc-1 role in provirus transcription regulation [40,123]. However, experiments of transcription inhibition with α-amanitin, which interacts with the bridge helix in Pol II, thereby interfering with its translocation along the DNA backbone [198], showed that disruption of nuc-1 is independent of transcription [40].
Although the underlying sequence of nuc-1 is thermodynamically unfavorable for nucleosome formation, the position of nuc-1 in the latent proviral state (in Jurkat models of latency) is maintained by the switch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complex [196], which dynamically counteracts the preferred nucleosome over NFR1 onto the energetically suboptimal nuc-1 position. The human analogs of the ATP-dependent SWI/SNF family of chromatin remodelers are composed of two complexes differing in subunit composition: BAF (SWI/SNF-A), which stands for “BRG1/SMARCA4- or BRM/SMARCA2-associated factors”, and PBAF (SWI/SNF-B), which stands for “Polybromo-associated BAF” [199]. Many functions were attributed to these two SWI/SNF complexes including transcriptional activation and repression (reviewed in [200,201]).
Interestingly, both SWI/SNF complexes temporally occupy the proviral promoter with BAF in the basal phase and PBAF during the viral, Tat-dependent activation phase [196]. These observations are consistent with the idea that BAF is crucial for early stages of HIV proviral transcription [202], and that several SWI/SNF subunits interact with Tat [203,204] to facilitate robust proviral activation and sustain the positive feedback loop [196,205,206,207]. In agreement, targeting of BAF by either RNAi-mediated knock down or by pharmacological inhibition with small molecule inhibitors displaced the BAF complex from nuc-1 at the proviral genome [208] which de-repressed basal HIV proviral genome transcription as measured by expression of LTR-driven GFP reporter and viral products. Given this de-repression was in the absence of any CD4+ T cell agonists in both immortalized (Jurkat) and primary models of latency, as well as in aviremic patient samples [196,208], it explains the molecular mechanism underlying latency reversal.
The BAF complex appears to be recruited to the proviral genome to maintain latency by one member of the BET family, specifically the shorter isoform of BRD4 [209]. Upon Tat-dependent transcription activation, the p300 histone acetyltransferase (HAT) acetylates Tat in its Arginine-rich RNA-binding domain (Lys50 and Lys51). This step dislodges Tat from TAR RNA [210,211] to enable recruitment of PBAF (PBAF-specific subunit BAF180) to the HIV proviral promoter [196], dictating a potential switch from BAF to PBAF SWI/SNF complexes regulating the exit from latency. In addition to SWI/SNF, Tat hijacks other chromatin regulatory complexes for either preventing or facilitating nucleosome remodeling for HIV transcription activation to rewire proviral fate decisions [212,213,214,215,216,217,218], some of which will be discussed below.
6.2. Histone Chaperones and Chromatin Reassembly Factors
In addition to the chromatin-modifying and chromatin-remodeling complexes required to switch “on” or “off” provirus transcription, chromatin reassembly factors (CRFs) like SPT6, CHD1, FACT, and histone H3 chaperones (ASF1a and HIRA) are involved in transcription interference [39,219], thus promoting viral latency by maintaining a repressive chromatin environment after HIV integration into the introns of highly active genes [39,219].
The FACT (facilitates chromatin transcription) complex, a heterodimer of SPT16 and SSRP1, removes and then reinstates the histone H2A/H2B dimer after Pol II elongation [220,221]. Two reports have proposed seemingly opposite functions for FACT in HIV transcription with disparate consequences for proviral fate. First, upon activation, FACT was shown to be recruited to nuc-1 to promote viral transcription [222]. However, a more recent study reported that FACT promotes proviral latency in both immortalized (Jurkat) and primary (resting primary CD4+ T) models of latency by interfering with P-TEFb binding to the proviral promoter [223]. These conflicting reports could be due to elongation factors repressing transcription initiation from cryptic HIV promoters integrated within host coding regions [224] and not solely to FACT obstructing the association between P-TEFb and Tat [223], or to unwanted indirect effects due to long-term FACT loss. As such, to fathom FACT functions in the maintenance of, and/or activation from, latency, future studies should prioritize the use of more consistent physiologic models of latency and acute depletion approaches to eliminate unnecessary variables.
6.3. Proviral Chromatin Acetylation/Deacetylation and Associated Factors
Nucleosome positioning is typically concerted with the deposition and/or removal of histone tail PTMs (Figure 4C). For example, the lack of histone tail (primarily H3 and H4) Lys acetylation facilitates interactions between those tails and DNA [225], thus reinforcing the role of DNA sequences in guiding nucleosome positioning.
Histone tails are post-translationally modified to establish regulatory codes that facilitate or prevent factor-chromatin interactions [226]. Specifically, cycles of histone acetylation/deacetylation are required to precisely fine-tune the magnitude and kinetics of transcriptional programs. Given HIV integrates into the host genome, proviral transcription is controlled through enzymes that deposit or remove acetyl groups, namely HATs and histone deacetylases (HDACs), respectively. Thus, recruitment of HDACs to the proviral genome (through transcription factors, like NF-κB/p50) in the basal phase are thought to constantly keep histone (H3 and H4) tails in the unacetylated or hypoacetylated state [46]. Upon transcription activation in the host phase (in response to immune stimulation), HDACs are evicted from the proviral genome and several HATs including CREB-binding protein (CBP), GCN5, and P/CAF are recruited to the proviral genome in immortalized models of latency (U1 and HL3T1 cells) causing the expected increase in histone (H3 and H4) acetylation levels (hyperacetylation) of nucleosomes encompassing the proviral 5′-LTR [213,214,215,216,227]. Nucleosomes upstream and downstream of the TSS remained hypoacetylated in the basal state, but acquired a specific hyperacetylation pattern with increased H3K9ac, H3K14ac, H4K5ac, H4K8ac, and H4K16ac, but not H4K12ac, upon transition to the host-viral phases in response to strong immune stimulation with phorbol esters (PMA) [227].
Since HDACs prevent histone tail acetylation, thereby promoting chromatin compaction, it came to no surprise that HDAC deposition at the proviral genome was linked to, and strongly correlated with, latency maintenance and establishment [41,43]. Thus, expectedly, treatment with HDAC inhibitors (trapoxin (TPX) and TSA, SAHA, romidepsin) triggered a HAT-dependent nuc-1 remodeling, thereby facilitating spontaneous HIV proviral expression in both immortalized (ACH2, OM 10.1, Ul, J49, J-Lat) and primary (resting and memory CD4+ T cells) models of latency [41,46,228,229,230,231]. HDAC activity inhibition promoted Pol II recruitment to the proviral genome but remained non-processive generating only short viral transcripts, consistent with the idea of activation of transcription initiation but not elongation [46].
The HDAC family is composed of five classes (I, IIA, IIB, III, and IV) based on function and sequence similarity (reviewed in [232]). Specifically, HDAC1 within class I co-purified with the yin yang 1–late SV40 factor (YY1-LSF) transcription repressor complex, which binds nuc-1 region on the proviral genome [43,233]. As a result, HDAC1 keeps nuc-1 in a hypoacetylated state, potentially reinforcing nucleosome positioning to prevent spurious activation [43,233] (Figure 4B). Despite strong molecular evidences linking HDAC-mediated control of proviral latency, clinical data later suggested that HDAC inhibition alone was insufficient for latency reversal due to low levels of reactivation and loss of effectiveness after prolonged treatment [234,235]. This may be potentially explained by the generation of read-through transcripts from upstream host promoters [236] indicating a lack of fully-mature and processed HIV transcripts.
6.4. Proviral Chromatin Methylation/Demethylation and Associated Factors
Besides histone acetylation/deacetylation, histone methylation of nucleosomes, by histone methyltransferases (HMTs), surrounding the proviral genome is another epigenetic mechanism contributing to nucleosome positioning, thereby regulating proviral fate. Multiple HMTs (SUV39H1, SUV39H2, and G9a) target distinct chromatin domains with different degrees of histone tail Lys methylation (e.g., mono-, di-, or tri-methylation) [237]. For instance, mono- and di-methylation of H3K9 (H3K9me1/2) are localized within silent domains of euchromatic regions, while tri-methylation of H3K9 (H3K9me3) is enriched at pericentric heterochromatin [237]. In immortalized models of latency (Jurkat), high levels of H3K9me3 were present at the proviral 5′-LTR, which is rapidly lost after immune stimulation (TNF-α) [238]. The SUV39 family of SET-domain containing HMTs, SUV39H1 and G9a, both appear to associate with the proviral 5′-LTR, which correlates with H3K9me2/3 levels to establish and maintain latency [239,240]. First, SUV39H1 binds to the proviral 5′-LTR in immortalized models of latency (HeLa-LTR-luc) and establishes a transcriptionally repressed state by maintaining high levels of H3K9me3 [239]. Second, G9a is responsible for proviral transcription repression in immortalized models of latency (ACH-2) by promoting H3K9me2 of nucleosomes surrounding the 5′-LTR [240]. Third, the SETDB1 HMT appears to regulate proviral transcriptional activity through Tat methylation [241].
Despite previous evidence that these three HMTs regulate proviral transcription, it is still unclear how they target proviruses integrated in diverse chromatin states. Since HMT have varying mechanisms of action at different chromatin domains [237], it is possible that proviruses integrated into different chromatin contexts may be targeted by different, or combination of, HMTs to promote the establishment of latency. This would be achieved through regulation of histone PTMs and nucleosome positioning enforcement. In this context, the role of the transcriptional repressor heterochromatin protein-1 (HP1) is noteworthy. The HP1 family in human cells is composed of three isoforms (HP1-α, HP1-β, and HP1-γ), which bind directly to methylated H3K9 tail but not to unmodified H3 [242]. Although the three HP1 isoforms share sequence similarity and structural organization, their nuclear localization respective to chromatin territories is different [243]. HP1-α and HP1-β are primarily associated with centromeric heterochromatin (like SUV39H1 and SUV39H2), whereas HP1-γ is found in both heterochromatic and euchromatic regions (like G9a) [244]. Interestingly, HP1-γ is present in the transcribed regions of active genes and physically associate with the elongating form of Pol II [245,246]. Remarkably, experiments in Xenopus oocytes showed that HP1 recruitment to chromatin requires not only the presence of H3K9 methylation but also a direct interaction of HP1 with HMTs. Thus, while both SUV39H1 and G9a have H3K9 methyltransferase activity, only SUV39H1 is able to recruit all isoforms of HP1 to chromatin through direct interaction [247].
The multiple HP1 isoforms and their different chromatin distribution profiles opened the question as to which HP1s regulate HIV proviral transcription, and what are the underlying molecular mechanisms. Surprisingly, distinct HP1 isoforms were found to associate with the proviral genome in various models and under different contexts, thus questioning the central mechanism.
In 2007, Benkirane and colleagues ascribed HP1-γ function, but not HP1-α and HP1-β, in maintaining provirus latency in both immortalized (HeLa and Jurkat) models of latency and PBMCs isolated from aviremic patients [239]. In 2008, the Karn lab reported HP1-α occupancy at the proviral promoter in the basal state and its eviction during the transition to the host-viral phases of the program in response to immune stimulation (TNF-α) in immortalized models of latency (Jurkat) [238]. The same year, a third study found HP1-β bound to the proviral genome in the basal state and a switch from HP1-β to HP1-γ during the transition to the host-viral phases of the program in response to immune stimulation (PMA) in an immortalized model of latency (Jurkat A1) [248]. These discrepancies may be because of differences in the chromatin landscape at or surrounding the integration site, including the methylation status of nucleosomes encompassing the proviral 5′-LTR and entire genome. While interesting, the disparate results from these studies urge a careful and comprehensive evaluation of all HP1 isoforms in various models of latency in which the role of cell state and integration site placement is carefully explored. Additionally, a thorough investigation is urgently needed to clarify direct and indirect functions (e.g., through chromatin remodeling complex recruitment) of different HMTs and HP1 isoforms in nucleosome positioning at the proviral genome, as well as in the establishment and/or maintenance of proviral latency. Given the large fraction of defective proviruses over intact proviruses [8] and their potential differential locations in the human genome [23], it is yet completely unclear how the various HMTs and HP1 isoforms target and regulate proviral transcription and fate from these disparate physical and functional proviral groups.
Another histone methylation (H3K27me3) has long been known to have a repressive role (reviewed in [249]) in transcription programs regulating key biological outcomes such as differentiation and development [250,251]. Expectedly, H3K27me3 was detected at the proviral genome in the basal state of immortalized models of latency (Jurkat) and further, H3K27me3 alongside H3K9me3 were lost during the transition to the host-viral phases of the program in response to stimulation (TNF-α) [238].
Consistent with the deposition of H3K27me3 at the proviral genome in immortalized models of latency (Jurkat), EZH2 (the catalytic subunit of the polycomb repressive complex (PRC2) responsible for H3K27 di- and tri-methylation) was also found in the basal state; however, its levels rapidly decreased during the transition to the host-viral phases of the program in response to stimulation (TNF-α) [252], in agreement with its known repressive role. Consistently, silencing or pharmacologic inhibition of EZH2 reactivated latent proviruses [252], presumably due to reduced H3K27me3 levels at the proviral genome. As many other transcriptional and chromatin regulators do, EZH2 cooperates with another HMT (EHMT2: euchromatic histone lysine N-methyltransferase 2) to establish and maintain proviral silencing in primary models of latency (CD4+ T helper (Th17)) and in resting memory T cells isolated from aviremic patients [253].
Together, these initial observations indicate that site-specific H3 Lys methylation (both H3K9me3 and H3K27me3) contributes to proviral latency, potentially by maintaining nucleosomes in a repressed state at the 5′-LTR. Although both H3K9me2/3 and H3K27me3 are involved in gene silencing and can act in a concerted manner for the establishment of heterochromatin loci [249], the functional crosstalk between methylation states in the establishment and maintenance of provirus latency is not fully understood.
More recently, another HMT (SMYD2: SET and MYND domain-containing protein-2) was identified using an RNAi-based screening approach [254]. SMYD2 binds to the proviral 5′-LTR and maintains proviral latency via H4K20me1. Thus, loss or inactivation of SMYD2 function induced latency reversal from both immortalized (Jurkat) and primary (CD4+ T) models of latency [254]. Given the site-specific H4 (H4K20me1) and H3 (H3K9me2/3 and H3K27me3) methylation at the proviral genome, elucidating the potential interplay and crosstalk between their chromatin writers, readers, and erasers seems (Figure 4C) an interesting but poorly studied process.
6.5. Proviral Genome DNA Methylation
Besides histone PTMs, DNA methylation of the proviral promoter is another epigenetic silencing mechanism implicated in transcriptional regulation. Two CpG islands (at positions ‒194 to ‒94 and +180 to +368 respective to TSS) flanking the proviral TSS in immortalized (Jurkat) and primary (CD4+ T) models of latency were proposed [42]. One of those CpG islands was found to be targeted by host methyl–CpG-binding domain protein (MBD2) in both immortalized (Jurkat) and primary (CD4+ T) models of latency, thereby promoting latency establishment [42]. Additionally, hypermethylation of the 5′-LTR in latently infected patient CD4+ T cells under suppressive therapy conferred resistance to provirus reactivation in contrast to hypomethylated HIV 5′-LTR in viremic patients [255], suggesting that the state of DNA methylation of the proviral promoter correlates with its transcriptional activity. As such, methylation of the proviral genome has been proposed as an additional restriction factor for latency stability but not for the initial establishment of proviral silencing. However, in contrast to these two studies, several others demonstrated very low levels of DNA methylation within the 5′-LTR of latently infected patient CD4+ T cells under suppressive therapy [8,256,257], signifying DNA methylation is not the lion’s share in proviral transcription silencing. Collectively, these contrasting results could be due to the fact that a vast majority of latently infected resting CD4+ T cells harbor defective proviruses [8] and may contain hypermethylated HIV 5′-LTRs [256].
6.6. Conclusions and Future Directions
Recent progress made for the identification and isolation of latently infected CD4+ T cells carrying intact proviruses from aviremic patients [8] may clarify and shed light into the role of CpG methylation in promoting and/or maintaining proviral latency. Newer high-throughput sequencing approaches such as NOMe-seq (nucleosome occupancy and methylome sequencing), and more recent single-cell implementations (scNOMe-seq) have been used to obtain locus-specific information of chromatin accessibility, nucleosome phasing, and DNA methylation at the single molecule level [258,259]. Importantly, these newer technologies, especially scNOMe-seq, may provide combinatorial epigenomic signatures of single-cells from heterogeneous populations of patient samples (Figure 2).
Collectively, studies over the past three decades focused on the identification and characterization of host cell factors hijacked by HIV to integrate and transcribe the proviral genome. This was done using either minimalistic models of latency in immortalized clones of CD4+ T cells or population-based studies in ex vivo primary models of latency and aviremic patient samples. These previous studies led to the characterization of a vast number of factors with nucleic-acid binding properties and chromatin-modifying or -remodeling activity, just to name a few. However, we currently have a very little understanding on how these chromatin regulatory factors regulate the most important reservoir in patients, namely intact proviruses that are capable of reseeding the reservoir upon therapy interruption. With the advent of single-cell genomic technologies for ChIP-seq [260], ATAC-seq [261], and Hi-C [262] to analyze individual proviruses (reviewed in [263]), and next-generation tools that could simultaneously assess proviral integration sites, intactness, expression and/or maps of active Pol II (Figure 5), a leap in our future understanding of proviral integration and transcriptional regulation can be made.
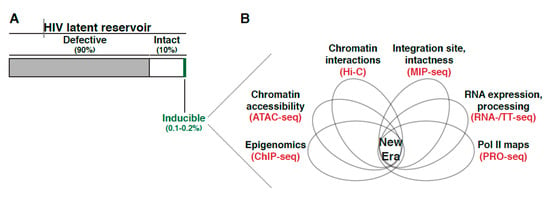
Figure 5.
Studies of the latent reservoir in the “new era”. (A) The latent HIV reservoir is comprised of a large fraction of proviruses with replication-defective genomes (~90%) and a small fraction of proviruses with replication-intact genomes (~10%). From the intact pool, only a tiny (~0.1–0.2%) fraction can be induced after single or multiple rounds of stimulation with potent T cell agonists ex vivo. (B) The “new era” will require use of newer omics tools implemented at the single-cell level allowing for unprecedented opportunities to investigate the multiple facets of the latent reservoir (including integration site, intactness, expression, chromatin accessibility/nucleosome positioning, and mechanistic regulation) for both, patient samples and ex vivo primary models of latency. The wealth of information generated by each of these technologies, integrated using deep learning algorithms, would be akin to a “Swiss army knife” in where any tool or combination of tools could dissect the multifactorial aspects contributing to proviral latency.
Most of our understanding of nucleosome positioning relies on data collected in vitro using clones of proviruses in immortalized models of latency (Jurkat) and low-resolution tools, compared to chemical cleavage techniques, such as MNase digestion followed by foot printing analysis. Despite these pioneer discoveries, it remains unclear how applicable these early models are for intact proviruses persisting in aviremic patients. Is the current dogma in the field concerning nucleosome positioning still valid for intact proviruses in patients and/or for models that can better interrogate and explain disease progression? How does the integration site influence the position of nucleosomes in both defective and intact proviruses? How does the orientation and direction of proviruses respective to nearby regulatory elements in the 1D and 3D dimensions (Figure 3) influence the position of nucleosomes in both defective and intact proviruses? How does the integration landscape in both immortalized and primary models of latency in CD4+ T cells as well as in patient samples regulate nucleosome positioning at the proviral genome? Can the initial results obtained in the above immortalized models be informative of the physiologic mechanism of chromatin control at integrated proviruses? Are those results generalizable? What is/are the role/s of the integration site, nuclear topology, sub-compartments and chromatin states to histone methylation, nucleosome positioning and proviral fate? Does the precise position and magnitude of H3 and H4 site-specific (or combinatorial) methylation matter in terms of restricting, and or allowing future, proviral activation and latency-reversal potential? These are some of the many questions that remain to be answered to fully understand how histone site-specific PTMs shape proviral responses. Certainly, we face an exceeding limitation due to the scarcity of, not only the latent reservoir but importantly, the “intact” latent reservoir (Figure 5A). Together these limitations hamper the need to interrogate the role of nucleosome positioning and epigenomics in physiologically and clinically relevant models of disease.
7. Disease Relevance and Current Therapeutic Challenges
Early on, it has been noted that HIV persistence was due to viral latency and not drug failure citing limited mutation rates [264]. Where latency is maintained by memory CD4+ T cell survival (possibly leading to clonal expansion) and homeostatic proliferation ([29], reviewed in [265]), clinical interventions must include targeting not only viral replication, but the rebound provirus that perpetuates in proliferating memory CD4+ T cells too ([29], reviewed in [265]). As such, many therapeutic efforts have been initiated which fall under different strategic categories: Reducing the reservoir, immunologically control viral rebound, and silencing the reservoir (reviewed in [265]). It is worth noting these efforts are focused to target the intact inducible provirus, which some may argue is not a complete cure as defective provirus may still express viral proteins able to inflict harm [266]. Nonetheless, to highlight a few strategies, one method for reducing the reservoir is “shock and kill” which boosts the provirus out of latency using latency reversing agents (LRA) so that enhanced cytotoxic lymphocytes may completely purge the virus (reviewed in [267,268]). Immunotherapy includes the use of broadly neutralizing antibodies [269,270] and chimeric antigen receptor T (CAR-T) cells [271] to target viral envelope on the cell surface for killing. A “block and lock” strategy could be used for silencing the reservoir in where pharmacologic inhibitors are used to implore HIV into a permanent transcriptionally silent state [272]. Importantly, the efficiency of these strategies and the underlying molecular mechanisms is often biased to singular latency models used, such that when tested in non-homogeneous patient samples (Figure 2), the efficacy in reducing overall reservoir size is often questioned/limited (reviewed in [2]). Therefore, new attempts to capture generalizable model(s) of latency, such as combining cell line data with ex vivo approaches [236], is often at the forefront of identifying and confirming new compounds. It is clear the varying transcription activity within a reservoir is influenced by the heterogeneous integrated nature within a proviral population [75] (Figure 2). Even so, we must also consider the added complexity stemming from the different origins, or genesis, of each unique proviral integrations within the same individual (Figure 1). Distinguishing how the origin of establishing latency, for example an intact provirus arising from clonal expansion vs uniquely integrated intact proviruses, influences viral rebound and reseeding of active infection upon ART cessation may illuminate the strategic development of novel compounds or combination of treatments.
The importance of using primary models of latency is highlighted in the identification of benzotriazoles as a class of LRAs that inhibit STAT5 SUMOylation increasing its presence on the HIV proviral promoter [273]. Yet, as opposed to primary cells and ex vivo models of latency, benzotriazoles fail to reactivate in multiple immortalized models of latency (J-Lat clones) because they lack the IL-2 receptor (CD25), thereby impeding STAT5 activation [273]. Physiologically relevant models are increasingly being used in combination and/or to verify findings derived from immortalized models. This combination of approaches has aided the understanding of the chromatin landscape and offered insights into predicting and validating long-term efficient new compounds. For example, didehydro-cortistatin A (dCA) was shown to inhibit Tat function by preventing TAR binding to reduce viral replication in vitro using immortalized models of latency (HeLa-CD4-LTR-Luc) and later confirmed using patient samples [274,275]. Yet, even now, we are beginning to bypass immortalized models for mechanistic studies all together. For example, to understand the mechanism of dCA, Valente and colleagues assessed how dCA promotes epigenetic silencing by increasing nuc-1 occupancy at the proviral genome (Figure 4), thereby preventing Pol II from binding the promoter using patient samples ex vivo. These results further confirm long term efficacy using the bone marrow-liver-thymus (BLT) mouse model of latency and persistence [272].
Given the importance of chromatin-modifying complexes in silencing proviral transcription, they were the focus of many studies aimed at promoting latency-reversal and reduction of the latent reservoir. The SWI/SNF chromatin modifying complexes have been targets for the development of therapies in the clinical setting. However, given BAF and PBAF complexes share several subunits and have functionally opposed roles in proviral transcription, this has represented a roadblock for “shock and kill” approaches of an HIV cure. Further, several compounds were recently identified to target subunits of the SWI/SNF BAF complex, but were later reported to have toxic off-target effects [208]. Recently, Dykhuizen and colleagues used a high throughput approach to identify non-toxic small molecule inhibitors of the BAF complex, which led to the identification of macrolactam compounds as LRAs that reverse latency both in immortalized and primary models of latency as well as in CD4+ T cells from aviremic patients without causing toxicity or T cell activation [276]. These new compounds target the BAF-specific ARID1A subunit, thereby reducing repressive nucleosome occupancy at the 5′-LTR (Figure 4).
Beyond high-throughput approaches, another tactic others have taken is to provide a better kinetic understanding of the proviral chromatin environment as it transitioned into a latent state in a primary model of latency (CD4+ T cells transduced with the with anti-apoptotic molecule Bcl-2) during EMT (Figure 1) [277]. Expectedly, heterochromatin (named based on H3K9me3 or H3K27me3 densities at the provirus (Figure 4)) gradually stabilized as cells transitioned from the active to the resting state, and thus proviruses became less accessible with reduced activation potential. Such a tool may eventually help specify drug development to accurate chromatin environment profiles.
The strongest indications of LRA’s efficacy are in vivo studies using animal models. Garcia and colleagues showed how treatment with the compound AZD5582 in HIV infected BLT mice and SIV infected rhesus macaques under ART induced HIV and SIV RNA expression in several tissues including lymph node of the macaques and lymph node, thymus, bone marrow, liver, and lung of BLT mice [278]. Though no reduction in reservoir size was detected, these models can lead the way to determine dose, timing and pairing with other compounds to induce the cytotoxic lymphocyte killing of the persistent reservoir. As shown by Silvestri and colleagues though the IL-15 super agonist compound, N-803, is able to reactivate primary human CD4+ T cells latently infected with HIV in vitro, co-culturing with CD8+ T cells inhibited N-803–mediated latent provirus reactivation [279]. This was later confirmed in SIV infected macaques, where N-803 alone had no impact on the reservoir. Yet, combining N-803 with MT807R1, an antibody that depletes CD8+ T cells in vivo resulted in a robust SIV increase. Together, these studies illuminate how animal models can be used not only to test compound efficacy but also to develop latency reversing strategies that take into account physiologic variables.
Overall, the importance of cross-validation using different models of latency, with emphasis on physiologically relevant models, is becoming increasingly apparent. Additionally, a careful analysis of hundreds or even thousands of single proviruses integrated in unique positions and bearing unique genetic variants, and not population-based studies, will hone our understanding on the role of integration landscape and cell state on latency reversal potential and other therapeutic schemes.
8. The Ephemeral Nature of Ideas and Considerations for Future Research
Characterizing the latent reservoir composition was the first core discovery spurring an entire field dedicated to understanding the mechanisms underlying its establishment and maintenance. Though initially capturing the precise reservoir size and composition were low resolution estimates (i.e., either over- or under-estimating the size and intactness), improved and combined methodologies have continuously revised the latent reservoir profile, unveiling that only a miniscule proportion is intact and inducible (Figure 5A). By surveying the genetic environmental landscape in where proviruses have embedded themselves, many were able to define peculiar preferential “behaviors” of this intriguing virus. RIGs, genes where the provirus was found repeatedly integrated in patient samples, began to reveal the integration site could largely influence what would become the position effect phenomena (Figure 3) [75], in where the integration site neighborhood shift proviral activity. Indeed, it is clear the partialities extend beyond the 1D view of simply a proviral insertion into a linear scale, as the importance of 3D nuclear architecture and temporal order of events in respect to cell state have become more apparent. Nonetheless, each new uncovered characteristic adds to understanding the proviral integration code, or molecular rules tempering proviral fate. At the same time, there is a growing consensus that many of these preferences might only be observed in patient samples and lost in immortalized and/or primary models of latency suggesting limitations in applicability of models potentially attributed to in vivo “selective pressures” and fitness of select proviral groups not recapitulated in vitro.
Beyond fixed provirus physical location and compartmentalization, other influences of latency establishment and maintenance are much more fluid. Cell status, as illustrated by large fluctuations of nuclear transcriptional regulators and coregulators, can galvanize transcriptional responses when transitioning from resting to active status. The factor fluctuations may also be small enough only to generate heterogenized threshold within a state (at rest for example) due to stochasticity. Or the fluctuations may be at the local gene level where deposition of epigenetic marks, Pol II pausing, and/or nucleosome positioning can influence the inducibility of an otherwise intact provirus. Again, many conflicting results are often attributed to differences in models used for their respective studies given the multiple layers of variability (Figure 1B).
Because of the multifactorial aspects contributing to latency (establishment, maintenance, and reversal), it is obvious the era of singular cause is over. From a therapeutic perspective, the failure of LRAs has always been attributed to the same reason: inability of a single drug, which often target a single molecular rule of latency (e.g., HDAC inhibitor, BRD4 inhibitor, etc.), to reactivate the entire latent reservoir. This extends to screening for new drug compounds. In the “new era”, it will be imperative to integrate multiple approaches (such as multiple genetic datasets, epigenetics, transcriptomes, proteomics, 3D genome architecture, nuclear topology, chromatin states, transcriptional activity, proviral position, and intactness) followed by deep learning [75] to predict and experiment with emphasis at the single-cell level and across multiple models including patient samples, and interrogate “deep” (Figure 5B). Understanding how each molecular rule fits together (simultaneously or stepwise) and which rules govern individual proviruses (i.e., is it the same set of rules for all proviruses or are there different kits/sets of rules for different proviruses) will aid personalized therapeutic development and eventual eradication of HIV. However, one obvious but extreme challenge is how the dataset integration (Figure 5B) will be achieved if patient samples are composed of a scarce and heterogeneous population in which multiple CD4+ T cells contain proviruses integrated in unique regions in addition to the clonally expanded pool.
As physiological relevance is vital to remove distracting or inapplicable models/mechanisms from the discussion, preference must be given to use patient samples or primary cell systems. Yet, with the limitation of the number of intact proviruses within a patient to collect data from, alternative models may need to be developed. In this context, can primary models of latency be created in the presence of ART, without the ectopic expression of anti-apoptotic factors but allowing their long-term culturing without causing T cell exhaustion? With the advent of genome editing tools [280], can we engineer “anatomically” correct in vitro models of latency (in immortalized but noncancerous cells or naïve healthy donor cells) in where proviruses are directed to the same integration sites as those found in patient samples? Would this approach potentially remove the roadblock of limited number of cells? No doubt, this would raise additional questions as it would uncouple the specific integration site from latency status, i.e., would repeating the same integration site (not by clonal expansion, but by executing into the same insertion site) repeatedly yield the same “inducibility” or not? Will the integration site landscape remain the same? If the results are consistent, then the molecular rules must be stable and therefore targetable.
With our efforts to discuss a surmountable number of key discoveries and situate them in the context of previously published literature, we hope to provoke future work that will advance our understanding of this fascinating biomedical research challenge. New data supporting or refuting the models discussed herein will undoubtedly increase our knowledge and generate discussions to help with future clinical applications rather than engendering unproductive controversy. Even the more skeptical researchers should be attracted to challenge these models and bring their own points of view to fuel progress in the field.
Funding
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the NIH under award numbers R01AI114362 and R33AI116222, and Welch Foundation grant number I-1782 (to I.D.).
Acknowledgments
We thank members of the D’Orso lab (Emily Cruz-Lorenzo, Jinli Wang, and Usman Hyder) and Holly Ruess for critical reading of the manuscript.
Conflicts of Interest
No conflicts to declare
References
- Perelson, A.S.; Essunger, P.; Cao, Y.; Vesanen, M.; Hurley, A.; Saksela, K.; Markowitz, M.; Ho, D.D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 1997, 387, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.T.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef]
- Chun, T.W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4+ T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. XReplication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Imamichia, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel proteincoding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef]
- Hiener, B.; Horsburgh, B.A.; Eden, J.S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4+ T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Q.; Orlova-Fink, N.; Einkauf, K.; Chowdhury, F.Z.; Sun, X.; Harrington, S.; Kuo, H.H.; Hua, S.; Chen, H.R.; Ouyang, Z.; et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J. Clin. Investig. 2017, 127, 2689–2696. [Google Scholar] [CrossRef] [PubMed]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.S.; Capoferri, A.A.; Beg, S.A.; Huang, S.H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol. Biol. 2005, 304, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Rosenbloom, D.I.S.; Lai, J.; Siliciano, R.F.; Siliciano, J.D. Measuring the frequency of latent HIV-1 in resting CD4+ T cells using a limiting dilution coculture assay. Methods Mol. Biol. 2016, 1354, 239–253. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Strain, M.C.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Smith, D.M.; Leigh-Brown, A.J.; Macaranas, T.R.; Lam, R.Y.; Daly, O.A.; Fischer, M.; et al. Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: Intrinsic stability predicts lifelong persistence. Proc. Natl. Acad. Sci. USA 2003, 100, 4819–4824. [Google Scholar] [CrossRef]
- Crooks, A.M.; Bateson, R.; Cope, A.B.; Dahl, N.P.; Griggs, M.K.; Kuruc, J.D.; Gay, C.L.; Eron, J.J.; Margolis, D.M.; Bosch, R.J.; et al. Precise Quantitation of the Latent HIV-1 Reservoir: Implications for Eradication Strategies. J. Infect. Dis. 2015, 212, 1361–1365. [Google Scholar] [CrossRef]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef]
- Hosmane, N.N.; Kwon, K.J.; Bruner, K.M.; Capoferri, A.A.; Beg, S.; Rosenbloom, D.I.S.; Keele, B.F.; Ho, Y.C.; Siliciano, J.D.; Siliciano, R.F. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017, 214, 959–972. [Google Scholar] [CrossRef]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 integration landscape during latent and active infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Broncano, P.; Maddali, S.; Einkauf, K.B.; Jiang, C.; Gao, C.; Chevalier, J.; Chowdhury, F.Z.; Maswabi, K.; Ajibola, G.; Moyo, S.; et al. Early antiretroviral therapy in neonates with HIV-1 infection restricts viral reservoir size and induces a distinct innate immune profile. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Einkauf, K.B.; Lee, G.Q.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.M.; Jiang, C.; Lian, X.; Chowdhury, F.Z.; et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Investig. 2019, 129, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, M.R.; Joseph, S.B.; Garrett, N.; Tyers, L.; Moeser, M.; Archin, N.; Council, O.D.; Matten, D.; Zhou, S.; Doolabh, D.; et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, F.R.; Sobolewski, M.D.; Fyne, E.; Shao, W.; Spindler, J.; Hattori, J.; Anderson, E.M.; Watters, S.A.; Hill, S.; Wu, X.; et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1883–1888. [Google Scholar] [CrossRef]
- Bui, J.K.; Sobolewski, M.D.; Keele, B.F.; Spindler, J.; Musick, A.; Wiegand, A.; Luke, B.T.; Shao, W.; Hughes, S.H.; Coffin, J.M.; et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog. 2017, 13, e1006283. [Google Scholar] [CrossRef]
- Lorenzi, J.C.C.; Cohen, Y.Z.; Cohn, L.B.; Kreider, E.F.; Barton, J.P.; Learn, G.H.; Oliveira, T.; Lavine, C.L.; Horwitz, J.A.; Settler, A.; et al. Paired quantitative and qualitative assessment of the replication-competent HIV-1 reservoir and comparison with integrated proviral DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E7908–E7916. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Wang, Z.; Gurule, E.E.; Brennan, T.P.; Gerold, J.M.; Kwon, K.J.; Hosmane, N.N.; Kumar, M.R.; Beg, S.A.; Capoferri, A.A.; Ray, S.C.; et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc. Natl. Acad. Sci. USA 2018, 115, E2575–E2584. [Google Scholar] [CrossRef]
- Zerbato, J.M.; Serrao, E.; Lenzi, G.; Kim, B.; Ambrose, Z.; Watkins, S.C.; Engelman, A.N.; Sluis-Cremer, N. Establishment and Reversal of HIV-1 Latency in Naive and Central Memory CD4+ T Cells In Vitro. J. Virol. 2016, 90, 8059–8073. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, C.; Valadkhan, S.; Graham, A.C.; Shukla, M.; Ciuffi, A.; Telenti, A.; Karn, J. Entry of Polarized Effector Cells into Quiescence Forces HIV Latency. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Agosto, L.M.; Henderson, A.J. CD4(+) T Cell Subsets and Pathways to HIV Latency. Aids. Res. Hum. Retrovir. 2018, 34, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Agosto, L.M.; Herring, M.B.; Mothes, W.; Henderson, A.J. HIV-1-Infected CD4+ T Cells Facilitate Latent Infection of Resting CD4+ T Cells through Cell-Cell Contact. Cell Rep. 2018, 24, 2088–2100. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Deng, K.; Gao, H.; Xing, S.; Capoferri, A.A.; Durand, C.M.; Rabi, S.A.; Laird, G.M.; Kim, M.; Hosmane, N.N.; et al. Transcriptional Reprogramming during Effector-to-Memory Transition Renders CD4(+) T Cells Permissive for Latent HIV-1 Infection. Immunity 2017, 47, 766–775.e3. [Google Scholar] [CrossRef] [PubMed]
- Julg, B.; Barouch, D.H. HIV-1 Latency by Transition. Immunity 2017, 47, 611–612. [Google Scholar] [CrossRef][Green Version]
- Chavez, L.; Calvanese, V.; Verdin, E. HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells. PLoS Pathog. 2015, 11, e1004955. [Google Scholar] [CrossRef]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef]
- Gallastegui, E.; Millan-Zambrano, G.; Terme, J.-M.; Chavez, S.; Jordan, A. Chromatin Reassembly Factors Are Involved in Transcriptional Interference Promoting HIV Latency. J. Virol. 2011, 85, 3187–3202. [Google Scholar] [CrossRef]
- Verdin, E.; Paras, P.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef] [PubMed]
- Coull, J.J.; Romerio, F.; Sun, J.-M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The Human Factors YY1 and LSF Repress the Human Immunodeficiency Virus Type 1 Long Terminal Repeat via Recruitment of Histone Deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef] [PubMed]
- Hsia, S.-C.V.; Shi, Y.-B. Chromatin Disruption and Histone Acetylation in Regulation of the Human Immunodeficiency Virus Type 1 Long Terminal Repeat by Thyroid Hormone Receptor. Mol. Cell. Biol. 2002, 22, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Greene, W.C. NF-kappaB/Rel: Agonist and antagonist roles in HIV-1 latency. Curr. Opin. HIV Aids 2011, 6, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef]
- Razooky, B.S.; Pai, A.; Aull, K.; Rouzine, I.M.; Weinberger, L.S. A hardwired HIV latency program. Cell 2015, 160, 990–1001. [Google Scholar] [CrossRef]
- Fauci, A.S.; Redfield, R.R.; Sigounas, G.; Weahkee, M.D.; Giroir, B.P. Ending the HIV Epidemic: A Plan for the United States. JAMA 2019, 321, 844–845. [Google Scholar] [CrossRef]
- Mbonye, U.; Karn, J. Control of HIV latency by epigenetic and non-epigenetic mechanisms. Curr. Hiv Res. 2011, 9, 554–567. [Google Scholar]
- Transcriptional control of HIV latency: Cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 2014, 454–455, 328–339. [CrossRef]
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.H.; Coffin, J.M. What Integration Sites Tell Us about HIV Persistence. Cell Host Microbe 2016, 19, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Michieletto, D.; Lusic, M.; Marenduzzo, D.; Orlandini, E. Physical principles of retroviral integration in the human genome. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.C.; O’Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, R.F. Orientation-Dependent Regulation of Integrated HIV-1 Expression by Host Gene Transcriptional Readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Schröder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Han, Y.; Lassen, K.; Monie, D.; Sedaghat, A.R.; Shimoji, S.; Liu, X.; Pierson, T.C.; Margolick, J.B.; Siliciano, R.F.; Siliciano, J.D. Resting CD4+ T Cells from Human Immunodeficiency Virus Type 1 (HIV-1)-Infected Individuals Carry Integrated HIV-1 Genomes within Actively Transcribed Host Genes. J. Virol. 2004, 78, 6122–6133. [Google Scholar] [CrossRef]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef]
- Lucic, B.; Chen, H.C.; Kuzman, M.; Zorita, E.; Wegner, J.; Minneker, V.; Wang, W.; Fronza, R.; Laufs, S.; Schmidt, M.; et al. Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Wong, R.W.; Mamede, J.I.; Hope, T.J. Impact of Nucleoporin-Mediated Chromatin Localization and Nuclear Architecture on HIV Integration Site Selection. J. Virol. 2015, 89, 9702–9705. [Google Scholar] [CrossRef][Green Version]
- Kane, M.; Rebensburg, S.V.; Takata, M.A.; Zang, T.M.; Yamashita, M.; Kvaratskhelia, M.; Bieniasz, P.D. Nuclear pore heterogeneity influences HIV-1 infection and the antiviral activity of MX2. eLlife 2018, 7. [Google Scholar] [CrossRef]
- Lusic, M.; Siliciano, R.F. Nuclear landscape of HIV-1 infection and integration. Nat. Rev. Microbiol. 2017, 15, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.M.; Maldarelli, F. The role of integration and clonal expansion in HIV infection: Live long and prosper. Retrovirology 2018, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ren, B. The Three-Dimensional Organization of Mammalian Genomes. Annu. Rev. Cell Dev. Biol. 2017, 33, 265–289. [Google Scholar] [CrossRef] [PubMed]
- Meaburn, K.J.; Misteli, T. Cell biology: Chromosome territories. Nature 2007, 445, 379–781. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Stanwick, T.L.; Dempsey, M.P.; Stevenson, M. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 1991, 254, 423–427. [Google Scholar] [CrossRef]
- Jordan, A.; Defechereux, P.; Verdin, E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 2001, 20, 1726–1738. [Google Scholar] [CrossRef] [PubMed]
- Sherrill-Mix, S.; Lewinski, M.K.; Famiglietti, M.; Bosque, A.; Malani, N.; Ocwieja, K.E.; Berry, C.C.; Looney, D.; Shan, L.; Agosto, L.M.; et al. HIV latency and integration site placement in five cell-based models. Retrovirology 2013, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.K.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef]
- Ruess, H.; Lee, J.; Guzman, C.; Malladi, V.; D’Orso, I. An integrated genomics approach towards deciphering human genome codes shaping HIV-1 proviral transcription and fate. bioRxiv 2020. [Google Scholar] [CrossRef]
- Emiliani, S.; Fischle, W.; Ott, M.; Van Lint, C.; Amella, C.A.; Verdin, E. Mutations in the tat Gene Are Responsible for Human Immunodeficiency Virus Type 1 Postintegration Latency in the U1 Cell Line. J. Virol. 1998, 72, 1666–1670. [Google Scholar] [CrossRef]
- Emiliani, S.; Van Lint, C.; Fischle, W.; Paras, P.; Ott, M.; Brady, J.; Verdin, E. A point mutation in the HIV-1 Tat responsive element is associated with postintegration latency. Proc. Natl. Acad. Sci. USA 1996, 93, 6377–6381. [Google Scholar] [CrossRef]
- Feinberg, M.B.; Baltimore, D.; Frankel, A.D. The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. USA 1991, 88, 4045–4049. [Google Scholar] [CrossRef]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef]
- Morton, E.L.; Forst, C.V.; Zheng, Y.; DePaula-Silva, A.B.; Ramirez, N.G.P.; Planelles, V.; D’Orso, I. Transcriptional Circuit Fragility Influences HIV Proviral Fate. Cell Rep. 2019, 27, 154–171.e9. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D.V. Stochastic Gene Expression in a Lentiviral Positive-Feedback Loop: HIV-1 Tat Fluctuations Drive Phenotypic Diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Razooky, B.; Cox, C.D.; Simpson, M.L.; Weinberger, L.S. Transcriptional Bursting from the HIV-1 Promoter Is a Significant Source of Stochastic Noise in HIV-1 Gene Expression. Biophys. J. 2010, 98, L32–L34. [Google Scholar] [CrossRef] [PubMed]
- Razooky, B.S.; Weinberger, L.S. Mapping the architecture of the HIV-1 Tat circuit: A decision-making circuit that lacks bistability and exploits stochastic noise. Methods 2011, 53, 68–77. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rouzine, I.M.; Razooky, B.S.; Weinberger, L.S. Stochastic variability in HIV affects viral eradication. Proc. Natl. Acad. Sci. USA 2014, 111, 13251–13252. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef]
- Lassen, K.G.; Hebbeler, A.M.; Bhattacharyya, D.; Lobritz, M.A.; Greene, W.C. A Flexible Model of HIV-1 Latency Permitting Evaluation of Many Primary CD4 T-Cell Reservoirs. PLoS ONE 2012, 7, e30176. [Google Scholar] [CrossRef]
- Cillo, A.R.; Sobolewski, M.D.; Bosch, R.J.; Fyne, E.; Piatak, M.; Coffin, J.M.; Mellors, J.W. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 7078–7083. [Google Scholar] [CrossRef]
- Golumbeanu, M.; Cristinelli, S.; Rato, S.; Munoz, M.; Cavassini, M.; Beerenwinkel, N.; Ciuffi, A. Single-Cell RNA-Seq Reveals Transcriptional Heterogeneity in Latent and Reactivated HIV-Infected Cells. Cell Rep. 2018, 23, 942–950. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Klaver, B.; Berkhout, B. Comparison of 5′ and 3′ long terminal repeat promoter function in human immunodeficiency virus. J. Virol. 1994, 68, 3830–3840. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R.; Lomedico, P.T.; Ju, G. Transcriptional interference in avian retroviruses—implications for the promoter insertion model of leukaemogenesis. Nature 1984, 307, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. HIV-1 gene expression: Lessons from provirus and non-integrated DNA. Retrovirology 2004, 1, 13. [Google Scholar] [CrossRef]
- Delannoy, A.; Poirier, M.; Bell, B. Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses 2019, 11, 269. [Google Scholar] [CrossRef]
- Taube, R.; Peterlin, M. Lost in transcription: Molecular mechanisms that control HIV latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef]
- Pereira, L.A. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000, 28, 663–668. [Google Scholar] [CrossRef]
- Mousseau, G.; Valente, S.T. Role of Host Factors on the Regulation of Tat-Mediated HIV-1 Transcription. Curr. Pharm. Des. 2017, 23, 4079–4090. [Google Scholar] [CrossRef]
- Jones, K.; Kadonaga, J.; Luciw, P.; Tjian, R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science 1986, 232, 755–759. [Google Scholar] [CrossRef]
- Garcia, J.A.; Harrich, D.; Soultanakis, E.; Wu, F.; Mitsuyasu, R.; Gaynor, R.B. Human immunodeficiency virus type 1 LTR TATA and TAR region sequences required for transcriptional regulation. EMBO J. 1989, 8, 765–778. [Google Scholar] [CrossRef]
- Rittner, K.; Churcher, M.J.; Gait, M.J.; Karn, J. The Human Immunodeficiency Virus Long Terminal Repeat Includes a Specialised Initiator Element which is Required for Tat-responsive Transcription. J. Mol. Biol. 1995, 248, 562–580. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Lis, J.T.; Jeang, K.T. Promoter activity of Tat at steps subsequent to TATA-binding protein recruitment. Mol. Cell. Biol. 1997, 17, 6898–6905. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Edwards, N.L.; Duckett, C.S.; Agranoff, A.B.; Schmid, R.M.; Nabel, G.J. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993, 12, 3551–3558. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Agranoff, A.B.; Pascal, E.; Nabel, G.J. An interaction between the DNA-binding domains of RelA(p65) and Sp1 mediates human immunodeficiency virus gene activation. Mol. Cell. Biol. 1994, 14, 6570–6583. [Google Scholar] [CrossRef] [PubMed]
- Sif, S.; Gilmore, T.D. Interaction of the v-Rel oncoprotein with cellular transcription factor Sp1. J. Virol. 1994, 68, 7131–7138. [Google Scholar] [CrossRef]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef]
- Cron, R.Q.; Bartz, S.R.; Clausell, A.; Bort, S.J.; Klebanoff, S.J.; Lewis, D.B. NFAT1 Enhances HIV-1 Gene Expression in Primary Human CD4 T Cells. Clin. Immunol. 2000, 94, 179–191. [Google Scholar] [CrossRef]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-Cell Receptor Signaling Enhances Transcriptional Elongation from Latent HIV Proviruses by Activating P-TEFb through an ERK-Dependent Pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef]
- Chen, B.K.; Feinberg, M.B.; Baltimore, D. The kappaB sites in the human immunodeficiency virus type 1 long terminal repeat enhance virus replication yet are not absolutely required for viral growth. J. Virol. 1997, 71, 5495–5504. [Google Scholar] [CrossRef]
- Alcamí, J.; Laín de Lera, T.; Folgueira, L.; Pedraza, M.A.; Jacqué, J.M.; Bachelerie, F.; Noriega, A.R.; Hay, R.T.; Harrich, D.; Gaynor, R.B. Absolute dependence on kappa B responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4 T lymphocytes. EMBO J. 1995, 14, 1552–1560. [Google Scholar] [CrossRef]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV Latency in Primary CD4+ Cells Is due to Epigenetic Transcriptional Silencing and P-TEFb Restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.G.; Arlen, P.A.; Gao, L.; Kitchen, C.M.R.; Zack, J.A. Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc. Natl. Acad. Sci. USA 2003, 100, 12955–12960. [Google Scholar] [CrossRef] [PubMed]
- Coiras, M.; López-Huertas, M.; Rullas, J.; Mittelbrunn, M.; Alcamí, J. Basal shuttle of NF-κB/IκBα in resting T lymphocytes regulates HIV-1 LTR dependent expression. Retrovirology 2007, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Ne, E.; Palstra, R.-J.; Mahmoudi, T. Transcription: Insights From the HIV-1 Promoter. Int. Rev. Cell Mol. Biol. 2018, 335, 191–243. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An Integrated Overview of HIV-1 Latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.; Gabuzda, D. ERK MAP Kinase Links Cytokine Signals to Activation of Latent HIV-1 Infection by Stimulating a Cooperative Interaction of AP-1 and NF-κB. J. Biol. Chem. 1999, 274, 27981–27988. [Google Scholar] [CrossRef]
- Duverger, A.; Wolschendorf, F.; Zhang, M.; Wagner, F.; Hatcher, B.; Jones, J.; Cron, R.Q.; van der Sluis, R.M.; Jeeninga, R.E.; Berkhout, B.; et al. An AP-1 Binding Site in the Enhancer/Core Element of the HIV-1 Promoter Controls the Ability of HIV-1 To Establish Latent Infection. J. Virol. 2013, 87, 2264–2277. [Google Scholar] [CrossRef]
- El Kharroubi, A.; Piras, G.; Zensen, R.; Martin, M.A. Transcriptional Activation of the Integrated Chromatin-Associated Human Immunodeficiency Virus Type 1 Promoter. Mol. Cell. Biol. 1998, 18, 2535–2544. [Google Scholar] [CrossRef]
- Olson, A.; Basukala, B.; Wong, W.W.; Henderson, A.J. Targeting HIV-1 proviral transcription. Curr. Opin. Virol. 2019, 38, 89–96. [Google Scholar] [CrossRef]
- Muse, G.W.; Gilchrist, D.A.; Nechaev, S.; Shah, R.; Parker, J.S.; Grissom, S.F.; Zeitlinger, J.; Adelman, K. RNA polymerase is poised for activation across the genome. Nat. Genet. 2007, 39, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative Elongation Factor Is Required for the Maintenance of Proviral Latency but Does Not Induce Promoter-Proximal Pausing of RNA Polymerase II on the HIV Long Terminal Repeat. Mol. Cell. Biol. 2014, 34, 1911–1928. [Google Scholar] [CrossRef]
- Zhang, Z.; Klatt, A.; Gilmour, D.S.; Henderson, A.J. Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J. Biol. Chem. 2007, 282, 16981–16988. [Google Scholar] [CrossRef]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Wada, T.; Watanabe, D.; Takagi, T.; Hasegawa, J.; Handa, H. Structure and Function of the Human Transcription Elongation Factor DSIF. J. Biol. Chem. 1999, 274, 8085–8092. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a Multisubunit Complex Containing RD, Cooperates with DSIF to Repress RNA Polymerase II Elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Rao, J.N.; Neumann, L.; Wenzel, S.; Schweimer, K.; Rösch, P.; Wöhrl, B.M. Structural studies on the RNA-recognition motif of NELF E, a cellular negative transcription elongation factor involved in the regulation of HIV transcription. Biochem. J. 2006, 400, 449–456. [Google Scholar] [CrossRef]
- Pagano, J.M.; Kwak, H.; Waters, C.T.; Sprouse, R.O.; White, B.S.; Ozer, A.; Szeto, K.; Shalloway, D.; Craighead, H.G.; Lis, J.T. Defining NELF-E RNA Binding in HIV-1 and Promoter-Proximal Pause Regions. PLoS Genet. 2014, 10, e1004090. [Google Scholar] [CrossRef]
- Narita, T.; Yamaguchi, Y.; Yano, K.; Sugimoto, S.; Chanarat, S.; Wada, T.; Kim, D.-K.; Hasegawa, J.; Omori, M.; Inukai, N.; et al. Human Transcription Elongation Factor NELF: Identification of Novel Subunits and Reconstitution of the Functionally Active Complex. Mol. Cell. Biol. 2003, 23, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Gressel, S.; Schwalb, B.; Decker, T.M.; Qin, W.; Leonhardt, H.; Eick, D.; Cramer, P. CDK9-dependent RNA polymerase II pausing controls transcription initiation. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Zeitlinger, J. Paused RNA polymerase II inhibits new transcriptional initiation. Nat. Genet. 2017, 49, 1045–1051. [Google Scholar] [CrossRef]
- Sathyan, K.M.; McKenna, B.D.; Anderson, W.D.; Duarte, F.M.; Core, L.; Guertin, M.J. An improved auxin-inducible degron system preserves native protein levels and enables rapid and specific protein depletion. Genes Dev. 2019, 33, 1441–1455. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A.; et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Aoi, Y.; Smith, E.R.; Shah, A.P.; Rendleman, E.J.; Marshall, S.A.; Woodfin, A.R.; Chen, F.X.; Shiekhattar, R.; Shilatifard, A. NELF Regulates a Promoter-Proximal Step Distinct from RNA Pol II Pause-Release. Mol. Cell 2020, 78, 261–274.e5. [Google Scholar] [CrossRef]
- Hartzog, G.A.; Basrai, M.A.; Ricupero-Hovasse, S.L.; Hieter, P.; Winston, F. Identification and analysis of a functional human homolog of the SPT4 gene of Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 2848–2856. [Google Scholar] [CrossRef][Green Version]
- Glover-Cutter, K.; Kim, S.; Espinosa, J.; Bentley, D.L. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat. Struct. Mol. Biol. 2008, 15, 71–78. [Google Scholar] [CrossRef]
- Gilchrist, D.A.; Dos Santos, G.; Fargo, D.C.; Xie, B.; Gao, Y.; Li, L.; Adelman, K. Pausing of RNA Polymerase II Disrupts DNA-Specified Nucleosome Organization to Enable Precise Gene Regulation. Cell 2010, 143, 540–551. [Google Scholar] [CrossRef]
- Missra, A.; Gilmour, D.S. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc. Natl. Acad. Sci. USA 2010, 107, 11301–11306. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Shatkin, A.J. Transcription elongation factor hSPT5 stimulates mRNA capping. Genes Dev. 1999, 13, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, R.A.; Sharp, P.A. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991, 10, 4189–4196. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV Aids 2011, 6, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Mancebo, H.S.Y.; Lee, G.; Flygare, J.; Tomassini, J.; Luu, P.; Zhu, Y.; Peng, J.; Blau, C.; Hazuda, D.; Price, D.; et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997, 11, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pe’ery, T.; Peng, J.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendt, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of Human Immunodeficiency Virus Transcription: P-TEFb Phosphorylates RD and Dissociates Negative Effectors from the Transactivation Response Element. Mol. Cell. Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef]
- Yamada, T.; Yamaguchi, Y.; Inukai, N.; Okamoto, S.; Mura, T.; Handa, H. P-TEFb-Mediated Phosphorylation of hSpt5 C-Terminal Repeats Is Critical for Processive Transcription Elongation. Mol. Cell 2006, 21, 227–237. [Google Scholar] [CrossRef]
- Bourgeois, C.F.; Kim, Y.K.; Churcher, M.J.; West, M.J.; Karn, J. Spt5 Cooperates with Human Immunodeficiency Virus Type 1 Tat by Preventing Premature RNA Release at Terminator Sequences. Mol. Cell. Biol. 2002, 22, 1079–1093. [Google Scholar] [CrossRef]
- Marshall, N.F.; Peng, J.; Xie, Z.; Price, D.H. Control of RNA Polymerase II Elongation Potential by a Novel Carboxyl-terminal Domain Kinase. J. Biol. Chem. 1996, 271, 27176–27183. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, Y.; Rajpara, S.M.; Reza, S.M.; Lees, E.; Shuman, S.; Mathews, M.B.; Pe’ery, T. Three RNA Polymerase II Carboxyl-terminal Domain Kinases Display Distinct Substrate Preferences. J. Biol. Chem. 2001, 276, 10913–10920. [Google Scholar] [CrossRef] [PubMed]
- Komarnitsky, P.; Cho, E.J.; Buratowski, S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000, 14, 2452–2460. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, B.M.; Price, D.H. Controlling the Elongation Phase of Transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, T.; Price, D.H. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012, 81, 119–143. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef]
- Chen, R.; Yang, Z.; Zhou, Q. Phosphorylated Positive Transcription Elongation Factor b (P-TEFb) Is Tagged for Inhibition through Association with 7SK snRNA. J. Biol. Chem. 2004, 279, 4153–4160. [Google Scholar] [CrossRef]
- Li, Q.; Price, J.P.; Byers, S.A.; Cheng, D.; Peng, J.; Price, D.H. Analysis of the Large Inactive P-TEFb Complex Indicates That It Contains One 7SK Molecule, a Dimer of HEXIM1 or HEXIM2, and Two P-TEFb Molecules Containing Cdk9 Phosphorylated at Threonine 186. J. Biol. Chem. 2005, 280, 28819–28826. [Google Scholar] [CrossRef]
- Xue, Y.; Yang, Z.; Chen, R.; Zhou, Q. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res. 2010, 38, 360–369. [Google Scholar] [CrossRef]
- Krueger, B.J.; Jeronimo, C.; Roy, B.B.; Bouchard, A.; Barrandon, C.; Byers, S.A.; Searcey, C.E.; Cooper, J.J.; Bensaude, O.; Cohen, E.A.; et al. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008, 36, 2219–2229. [Google Scholar] [CrossRef]
- Jeronimo, C.; Forget, D.; Bouchard, A.; Li, Q.; Chua, G.; Poitras, C.; Thérien, C.; Bergeron, D.; Bourassa, S.; Greenblatt, J.; et al. Systematic Analysis of the Protein Interaction Network for the Human Transcription Machinery Reveals the Identity of the 7SK Capping Enzyme. Mol. Cell 2007, 27, 262–274. [Google Scholar] [CrossRef]
- He, N.; Jahchan, N.S.; Hong, E.; Li, Q.; Bayfield, M.A.; Maraia, R.J.; Luo, K.; Zhou, Q. A La-Related Protein Modulates 7SK snRNP Integrity to Suppress P-TEFb-Dependent Transcriptional Elongation and Tumorigenesis. Mol. Cell 2008, 29, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Bacon, C.W.; D’Orso, I. CDK9: A signaling hub for transcriptional control. Transcription 2019, 10, 57–75. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.P.; McCann, J.L.; Gudipaty, S.A.; D’Orso, I. Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 2013, 5, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Faust, T.B.; Li, Y.; Bacon, C.W.; Jang, G.M.; Weiss, A.; Jayaraman, B.; Newton, B.W.; Krogan, N.J.; D’Orso, I.; Frankel, A.D. The HIV-1 Tat protein recruits a ubiquitin ligase to reorganize the 7SK snRNP for transcriptional activation. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- D’Orso, I.; Frankel, A.D. RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat. Struct. Mol. Biol. 2010, 17, 815–821. [Google Scholar] [CrossRef]
- D’Orso, I.; Jang, G.M.; Pastuszak, A.W.; Faust, T.B.; Quezada, E.; Booth, D.S.; Frankel, A.D. Transition Step during Assembly of HIV Tat:P-TEFb Transcription Complexes and Transfer to TAR RNA. Mol. Cell. Biol. 2012, 32, 4780–4793. [Google Scholar] [CrossRef] [PubMed]
- Barboric, M.; Yik, J.H.N.; Czudnochowski, N.; Yang, Z.; Chen, R.; Contreras, X.; Geyer, M.; Matija Peterlin, B.; Zhou, Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007, 35, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Sedore, S.C.; Byers, S.A.; Biglione, S.; Price, J.P.; Maury, W.J.; Price, D.H. Manipulation of P-TEFb control machinery by HIV: Recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007, 35, 4347–4358. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.E.; Mayall, T.P.; Suess, E.M.; Meisenhelder, J.; Thompson, N.E.; Jones, K.A. CDK9 Autophosphorylation Regulates High-Affinity Binding of the Human Immunodeficiency Virus Type 1 Tat-P-TEFb Complex to TAR RNA. Mol. Cell. Biol. 2000, 20, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Moon, K.J.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and Host AFF4 Recruit Two Transcription Elongation Factors into a Bifunctional Complex for Coordinated Activation of HIV-1 Transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat Assembles a Multifunctional Transcription Elongation Complex and Stably Associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef]
- McNamara, R.P.; Reeder, J.E.; McMillan, E.A.; Bacon, C.W.; McCann, J.L.; D’Orso, I. KAP1 Recruitment of the 7SK snRNP Complex to Promoters Enables Transcription Elongation by RNA Polymerase II. Mol. Cell 2016, 61, 39–53. [Google Scholar] [CrossRef]
- Lin, C.; Smith, E.R.; Takahashi, H.; Lai, K.C.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C.; Shilatifard, A. AFF4, a Component of the ELL/P-TEFb Elongation Complex and a Shared Subunit of MLL Chimeras, Can Link Transcription Elongation to Leukemia. Mol. Cell 2010, 37, 429–437. [Google Scholar] [CrossRef]
- Chou, S.; Upton, H.; Bao, K.; Schulze-Gahmen, U.; Samelson, A.J.; He, N.; Nowak, A.; Lu, H.; Krogan, N.J.; Zhou, Q.; et al. HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc. Natl. Acad. Sci. USA 2013, 110, E123–E131. [Google Scholar] [CrossRef]
- Lu, H.; Li, Z.; Xue, Y.; Schulze-Gahmen, U.; Johnson, J.R.; Krogan, N.J.; Alber, T.; Zhou, Q. AFF1 is a ubiquitous P-TEFb partner to enable Tat extraction of P-TEFb from 7SK snRNP and formation of SECs for HIV transactivation. Proc. Natl. Acad. Sci. USA 2014, 111, E15–E24. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Guest, E.; Garrett, A.S.; Mohaghegh, N.; Swanson, S.; Marshall, S.; Florens, L.; Washburn, M.P.; Shilatifard, A. The super elongation complex family of RNA polymerase II elongation factors: Gene target specificity and transcriptional output. Mol. Cell Biol. 2012, 32, 2608–2617. [Google Scholar] [CrossRef]
- Li, Z.; Lu, H.; Zhou, Q. A Minor Subset of Super Elongation Complexes Plays a Predominant Role in Reversing HIV-1 Latency. Mol. Cell. Biol. 2016, 36, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.-C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.-W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J.; Singer, D.S. BRD4 is an atypical kinase that phosphorylates Serine2 of the RNA Polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Brd4 and JMJD6-Associated Anti-Pause Enhancers in Regulation of Transcriptional Pause Release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef]
- D’Orso, I. 7SKiing on chromatin: Move globally, act locally. RNA Biol. 2016, 13, 545–553. [Google Scholar] [CrossRef]
- Wolf, D.; Goff, S.P. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 2007, 131, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Bacon, C.W.; Challa, A.; Hyder, U.; Shukla, A.; Borkar, A.N.; Bayo, J.; Liu, J.; Wu, S.Y.; Chiang, C.-M.; Kutateladze, T.G.; et al. A chromatin reader couples steps RNA polymerase II transcription to sustain oncogenic programs. Mol. Cell 2020, 78, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Pugh, B.F. Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161–172. [Google Scholar] [CrossRef]
- Steger, D.J.; Workman, J.L. Stable co-occupancy of transcription factors and histones at the HIV-1 enhancer. EMBO J. 1997, 16, 2463–2472. [Google Scholar] [CrossRef]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR Nucleosome Positioning by the BAF Complex Is Required for HIV Latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef]
- Verdin, E.; Becker, N.; Bex, F.; Droogmans, L.; Burny, A. Identification and characterization of an enhancer in the coding region of the genome of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1990, 87, 4874–4878. [Google Scholar] [CrossRef]
- Bushnell, D.A.; Cramer, P.; Kornberg, R.D. Structural basis of transcription: -Amanitin-RNA polymerase II cocrystal at 2.8 A resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 1218–1222. [Google Scholar] [CrossRef]
- Wang, W.; Xue, Y.; Zhou, S.; Kuo, A.; Cairns, B.R.; Crabtree, G.R. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996, 10, 2117–2130. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv. 2015, 1, e1500447. [Google Scholar] [CrossRef]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef]
- Mizutani, T.; Ishizaka, A.; Tomizawa, M.; Okazaki, T.; Yamamichi, N.; Kawana-Tachikawa, A.; Iwamoto, A.; Iba, H. Loss of the Brm-Type SWI/SNF Chromatin Remodeling Complex Is a Strong Barrier to the Tat-Independent Transcriptional Elongation of Human Immunodeficiency Virus Type 1 Transcripts. J. Virol. 2009, 83, 11569–11580. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sodroski, J.; Rosen, C.; Wong-Staal, F.; Salahuddin, S.; Popovic, M.; Arya, S.; Gallo, R.; Haseltine, W. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science 1985, 227, 171–173. [Google Scholar] [CrossRef]
- Sodroski, J.; Rosen, C.; Haseltine, W. Trans-acting transcriptional activation of the long terminal repeat of human T lymphotropic viruses in infected cells. Science 1984, 225, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Tréand, C.; Du Chéné, I.; Brès, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [CrossRef]
- Mahmoudi, T.; Parra, M.; Vries, R.G.J.; Kauder, S.E.; Verrijzer, C.P.; Ott, M.; Verdin, E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 2006, 281, 19960–19968. [Google Scholar] [CrossRef] [PubMed]
- Agbottah, E.; Deng, L.; Dannenberg, L.O.; Pumfery, A.; Kashanchi, F. Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription. Retrovirology 2006, 3, 48. [Google Scholar] [CrossRef]
- Stoszko, M.; De Crignis, E.; Rokx, C.; Khalid, M.M.; Lungu, C.; Palstra, R.-J.; Kan, T.W.; Boucher, C.; Verbon, A.; Dykhuizen, E.C.; et al. Small Molecule Inhibitors of BAF.; A Promising Family of Compounds in HIV-1 Latency Reversal. EBioMedicine 2016, 3, 108–121. [Google Scholar] [CrossRef]
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.-M.; Ott, M. The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol. Cell 2017, 67, 1001–1012.e6. [Google Scholar] [CrossRef]
- Ott, M.; Schnölzer, M.; Garnica, J.; Fischle, W.; Emiliani, S.; Rackwitz, H.-R.; Verdin, E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol. 1999, 9, 1489–1493. [Google Scholar] [CrossRef]
- Kiernan, R.E. HIV-1 Tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV–human protein complexes. Nature 2012, 481, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Benkirane, M.; Chun, R.F.; Xiao, H.; Ogryzko, V.V.; Howard, B.H.; Nakatani, Y.; Jeang, K.-T. Activation of integrated provirus requires histone acetyl- transferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 1998, 273, 24898–24905. [Google Scholar] [CrossRef] [PubMed]
- Col, E.; Caron, C.; Seigneurin-Berny, D.; Gracia, J.; Favier, A.; Khochbin, S. The Histone Acetyltransferase, hGCN5, Interacts with and Acetylates the HIV Transactivator, Tat. J. Biol. Chem. 2001, 276, 28179–28184. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Nabel, G.J. Interaction of human immunodeficiency virus type 1 Tat with the transcriptional coactivators p300 and CREB binding protein. J. Virol. 1998, 72, 8252–8256. [Google Scholar] [CrossRef]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 Tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef]
- Vardabasso, C.; Manganaro, L.; Lusic, M.; Marcello, A.; Giacca, M. The histone chaperone protein Nucleosome Assembly Protein-1 (hNAP-1) binds HIV-1 Tat and promotes viral transcription. Retrovirology 2008, 5, 8. [Google Scholar] [CrossRef]
- Gautier, V.W.; Gu, L.; O’Donoghue, N.; Pennington, S.; Sheehy, N.; Hall, W.W. In vitro nuclear interactome of the HIV-1 Tat protein. Retrovirology 2009, 6, 47. [Google Scholar] [CrossRef]
- Vanti, M.; Gallastegui, E.; Respaldiza, I.; Rodríguez-Gil, A.; Gómez-Herreros, F.; Jimeno-González, S.; Jordan, A.; Chávez, S. Yeast Genetic Analysis Reveals the Involvement of Chromatin Reassembly Factors in Repressing HIV-1 Basal Transcription. PLoS Genet. 2009, 5, e1000339. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. FACT facilitates transcription-dependent nucleosome alteration. Science 2003, 301, 1090–1093. [Google Scholar] [CrossRef]
- Orphanides, G.; LeRoy, G.; Chang, C.-H.; Luse, D.S.; Reinberg, D. FACT, a Factor that Facilitates Transcript Elongation through Nucleosomes. Cell 1998, 92, 105–116. [Google Scholar] [CrossRef]
- Easley, R.; Carpio, L.; Dannenberg, L.; Choi, S.; Alani, D.; Van Duyne, R.; Guendel, I.; Klase, Z.; Agbottah, E.; Kehn-Hall, K.; et al. Transcription through the HIV-1 nucleosomes: Effects of the PBAF complex in Tat activated transcription. Virology 2010, 405, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Santoso, N.; Power, D.; Simpson, S.; Dieringer, M.; Miao, H.; Gurova, K.; Giam, C.-Z.; Elledge, S.J.; Zhu, J. FACT Proteins, SUPT16H and SSRP1, Are Transcriptional Suppressors of HIV-1 and HTLV-1 That Facilitate Viral Latency. J. Biol. Chem. 2015, 290, 27297–27310. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, C.D. Transcription Elongation Factors Repress Transcription Initiation from Cryptic Sites. Science 2003, 301, 1096–1099. [Google Scholar] [CrossRef]
- Anderson, J.; Lowary, P.; Widom, J. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2001, 307, 977–985. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Lusic, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef]
- Ylisastigui, L.; Archin, N.M.; Lehrman, G.; Bosch, R.J.; Margolis, D.M. Coaxing HIV-1 from resting CD4 T cells: Histone deacetylase inhibition allows latent viral expression. AIDS 2004, 18, 1101–1108. [Google Scholar] [CrossRef]
- Archin, N.M.; Bateson, R.; Tripathy, M.K.; Crooks, A.M.; Yang, K.-H.; Dahl, N.P.; Kearney, M.F.; Anderson, E.M.; Coffin, J.M.; Strain, M.C.; et al. HIV-1 Expression Within Resting CD4+ T Cells After Multiple Doses of Vorinostat. J. Infect. Dis. 2014, 210, 728–735. [Google Scholar] [CrossRef]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone Deacetylase Inhibitor Romidepsin Induces HIV Expression in CD4 T Cells from Patients on Suppressive Antiretroviral Therapy at Concentrations Achieved by Clinical Dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef]
- Shirakawa, K.; Chavez, L.; Hakre, S.; Calvanese, V.; Verdin, E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013, 21, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Margolis, D.M. Counterregulation of Chromatin Deacetylation and Histone Deacetylase Occupancy at the Integrated Promoter of Human Immunodeficiency Virus Type 1 (HIV-1) by the HIV-1 Repressor YY1 and HIV-1 Activator Tat. Mol. Cell. Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.M.; Garcia, J.V.; Hazuda, D.J.; Haynes, B.F. Latency reversal and viral clearance to cure HIV-1. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol. Cell 2003, 12, 1591–1598. [Google Scholar] [CrossRef]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic Silencing of Human Immunodeficiency Virus (HIV) Transcription by Formation of Restrictive Chromatin Structures at the Viral Long Terminal Repeat Drives the Progressive Entry of HIV into Latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef]
- Chéné, I.D.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of Histone H3 Lysine 9 (H3K9) Methyltransferase G9a in the Maintenance of HIV-1 Latency and Its Reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef]
- Van Duyne, R.; Easley, R.; Wu, W.; Berro, R.; Pedati, C.; Klase, Z.; Kehn-Hall, K.; Flynn, E.K.; Symer, D.E.; Kashanchi, F. Lysine methylation of HIV-1 Tat regulates transcriptional activity of the viral LTR. Retrovirology 2008, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Maison, C.; Almouzni, G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004, 5, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Minc, E.; Courvalin, J.-C.; Buendia, B. HP1γ associates with euchromatin and heterochromatin in mammalian nuclei and chromosomes. Cytogenet. Genome Res. 2000, 90, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Mandat, S.A.; Olenchock, B.A.; Blobel, G.A. Histone H3 Lysine 9 Methylation and HP1γ Are Associated with Transcription Elongation through Mammalian Chromatin. Mol. Cell 2005, 19, 381–391. [Google Scholar] [CrossRef]
- Kwon, S.H.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Abmayr, S.M.; Workman, J.L. Heterochromatin protein 1 (HP1) connects the FACT histone chaperone complex to the phosphorylated CTD of RNA polymerase II. Genes Dev. 2010, 24, 2133–2145. [Google Scholar] [CrossRef]
- Stewart, M.D.; Li, J.; Wong, J. Relationship between Histone H3 Lysine 9 Methylation, Transcription Repression, and Heterochromatin Protein 1 Recruitment. Mol. Cell. Biol. 2005, 25, 2525–2538. [Google Scholar] [CrossRef]
- Mateescu, B.; Bourachot, B.; Rachez, C.; Ogryzko, V.; Muchardt, C. Regulation of an inducible promoter by an HP1β-HP1γ switch. EMBO Rep. 2008, 9, 267–272. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications – writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of Developmental Regulators by Polycomb in Human Embryonic Stem Cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Cho, W.-K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic Silencing of HIV-1 by the Histone H3 Lysine 27 Methyltransferase Enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Das, B.; Dobrowolski, C.; Karn, J. Multiple Histone Lysine Methyltransferases Are Required for the Establishment and Maintenance of HIV-1 Latency. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Jeng, M.; Camus, G.; Gramatica, A.; Schwarzer, R.; Johnson, J.R.; Hull, P.A.; Montano, M.; Sakane, N.; Pagans, S.; et al. SMYD2-Mediated Histone Methylation Contributes to HIV-1 Latency. Cell Host Microbe 2017, 21, 569–579.e6. [Google Scholar] [CrossRef]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG Methylation Controls Reactivation of HIV from Latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef]
- Blazkova, J.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.K.; Moir, S.; Chun, T.-W.; Fauci, A.S. Paucity of HIV DNA Methylation in Latently Infected, Resting CD4+ T Cells from Infected Individuals Receiving Antiretroviral Therapy. J. Virol. 2012, 86, 5390–5392. [Google Scholar] [CrossRef]
- Weber, S.; Weiser, B.; Kemal, K.S.; Burger, H.; Ramirez, C.M.; Korn, K.; Anastos, K.; Kaul, R.; Kovacs, C.; Doerfler, W. Epigenetic analysis of HIV-1 proviral genomes from infected individuals: Predominance of unmethylated CpG’s. Virology 2014, 449, 181–189. [Google Scholar] [CrossRef][Green Version]
- Pott, S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife 2017, 6. [Google Scholar] [CrossRef]
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [CrossRef]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef]
- Mezger, A.; Klemm, S.; Mann, I.; Brower, K.; Mir, A.; Bostick, M.; Farmer, A.; Fordyce, P.; Linnarsson, S.; Greenleaf, W. High-throughput chromatin accessibility profiling at single-cell resolution. Nat. Commun. 2018, 9, 3647. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Kok, Y.L.; Ciuffi, A.; Metzner, K.J. Unravelling HIV-1 Latency, One Cell at a Time. Trends Microbiol. 2017, 25, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.C. HIV/AIDS Research for the Future. Cell Host Microbe 2020, 27, 499–501. [Google Scholar] [CrossRef]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef]
- Bar, K.J.; Sneller, M.C.; Harrison, L.J.; Justement, J.S.; Overton, E.T.; Petrone, M.E.; Salantes, D.B.; Seamon, C.A.; Scheinfeld, B.; Kwan, R.W.; et al. Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. N. Engl. J. Med. 2016, 375, 2037–2050. [Google Scholar] [CrossRef]
- Mendoza, P.; Gruell, H.; Nogueira, L.; Pai, J.A.; Butler, A.L.; Millard, K.; Lehmann, C.; Suárez, I.; Oliveira, T.Y.; Lorenzi, J.C.C.; et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 2018, 561, 479–484. [Google Scholar] [CrossRef]
- Herzig, E.; Kim, K.C.; Packard, T.A.; Vardi, N.; Schwarzer, R.; Gramatica, A.; Deeks, S.G.; Williams, S.R.; Landgraf, K.; Killeen, N.; et al. Attacking Latent HIV with convertibleCAR-T Cells, a Highly Adaptable Killing Platform. Cell 2019, 179, 880–894.e10. [Google Scholar] [CrossRef] [PubMed]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep 2017, 21, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Bosque, A.; Nilson, K.A.; Macedo, A.B.; Spivak, A.M.; Archin, N.M.; Van Wagoner, R.M.; Martins, L.J.; Novis, C.L.; Szaniawski, M.A.; Ireland, C.M.; et al. Benzotriazoles Reactivate Latent HIV-1 through Inactivation of STAT5 SUMOylation. Cell Rep. 2017, 18, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An Analog of the Natural Steroidal Alkaloid Cortistatin A Potently Suppresses Tat-Dependent HIV Transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Jablonski, J.; Paris, J.; Clementz, M.; Thenin-Houssier, S.; McLaughlin, J.; Valente, S. Didehydro-Cortistatin A Inhibits HIV-1 Tat Mediated Neuroinflammation and Prevents Potentiation of Cocaine Reward in Tat Transgenic Mice. Curr. HIV Res. 2015, 13, 64–79. [Google Scholar] [CrossRef]
- Marian, C.A.; Stoszko, M.; Wang, L.; Leighty, M.W.; de Crignis, E.; Maschinot, C.A.; Gatchalian, J.; Carter, B.C.; Chowdhury, B.; Hargreaves, D.C.; et al. Small Molecule Targeting of Specific BAF (mSWI/SNF) Complexes for HIV Latency Reversal. Cell Chem. Biol. 2018, 25, 1443–1455.e14. [Google Scholar] [CrossRef]
- Lindqvist, B.; Svensson Akusjärvi, S.; Sönnerborg, A.; Dimitriou, M.; Svensson, J.P. Chromatin maturation of the HIV-1 provirus in primary resting CD4+ T cells. PLoS Pathog. 2020, 16, e1008264. [Google Scholar] [CrossRef]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- McBrien, J.B.; Mavigner, M.; Franchitti, L.; Smith, S.A.; White, E.; Tharp, G.K.; Walum, H.; Busman-Sahay, K.; Aguilera-Sandoval, C.R.; Thayer, W.O.; et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 2020, 578, 154–159. [Google Scholar] [CrossRef]
- Bialek, J.K.; Walther, T.; Hauber, J.; Lange, U.C. CRISPR-Cas9-based Genome Engineering to Generate Jurkat Reporter Models for HIV-1 Infection with Selected Proviral Integration Sites. J. Vis. Exp. 2018, 141, e58572. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).