The Impact of Cellular Proliferation on the HIV-1 Reservoir


Abstract
1. Introduction
2. Cell Type, Proliferation Potential, and Growth Stimulus
2.1. Introduction to HIV Infection in Patients
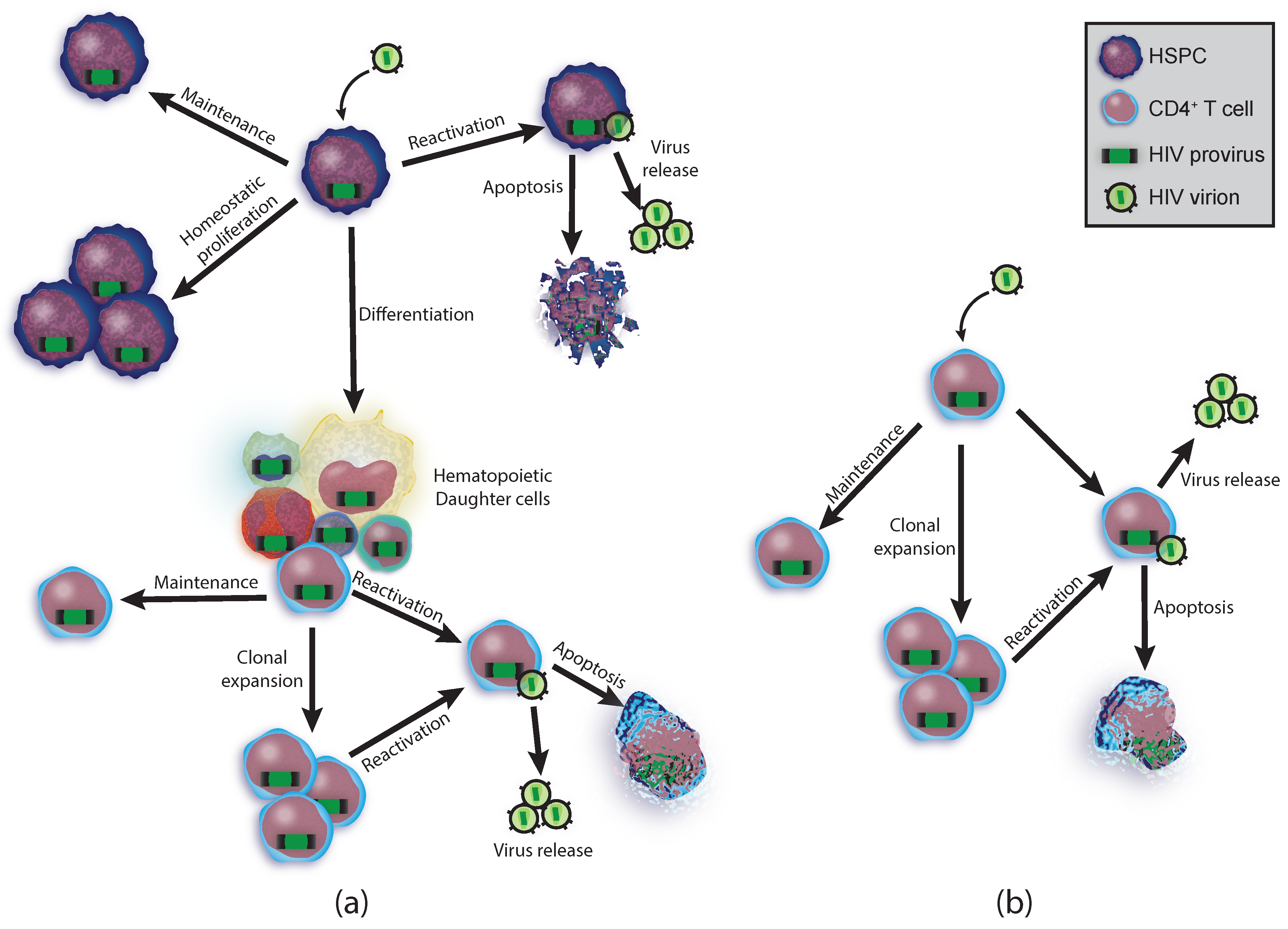
2.2. Hematopoietic Potential
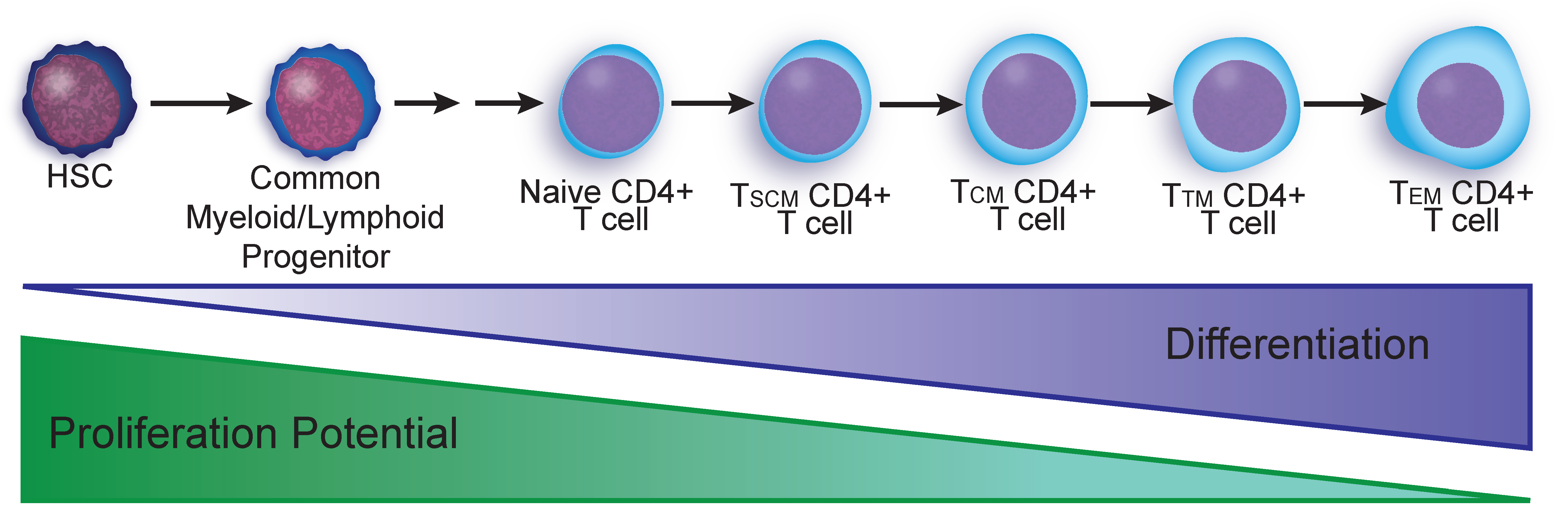
2.3. T Cell Reservoirs
3. HIV Sequence Integrity
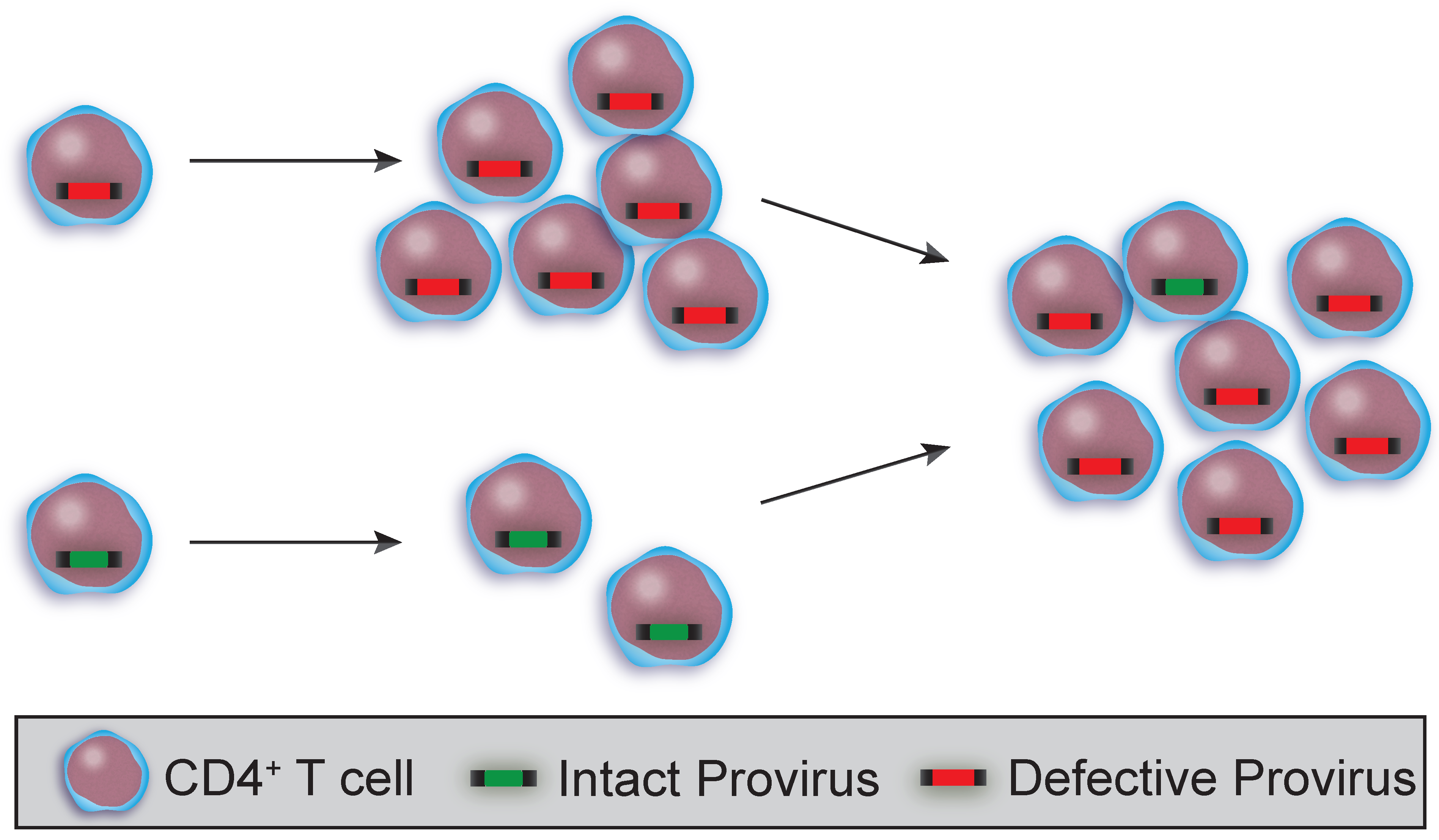
3.1. Clonality and HIV Sequence Integrity in T Cell Subsets and HSPCs
3.2. Using HIV Sequences to Determine Clonality and Decay
4. Methods of Viral Spread
4.1. Cell-to-Cell Viral Spread
4.2. Viral Evolution and Insufficient ART
4.3. Expression from Unintegrated Virus
5. Insertion Site
5.1. Integration Site and Genome Capture Techniques
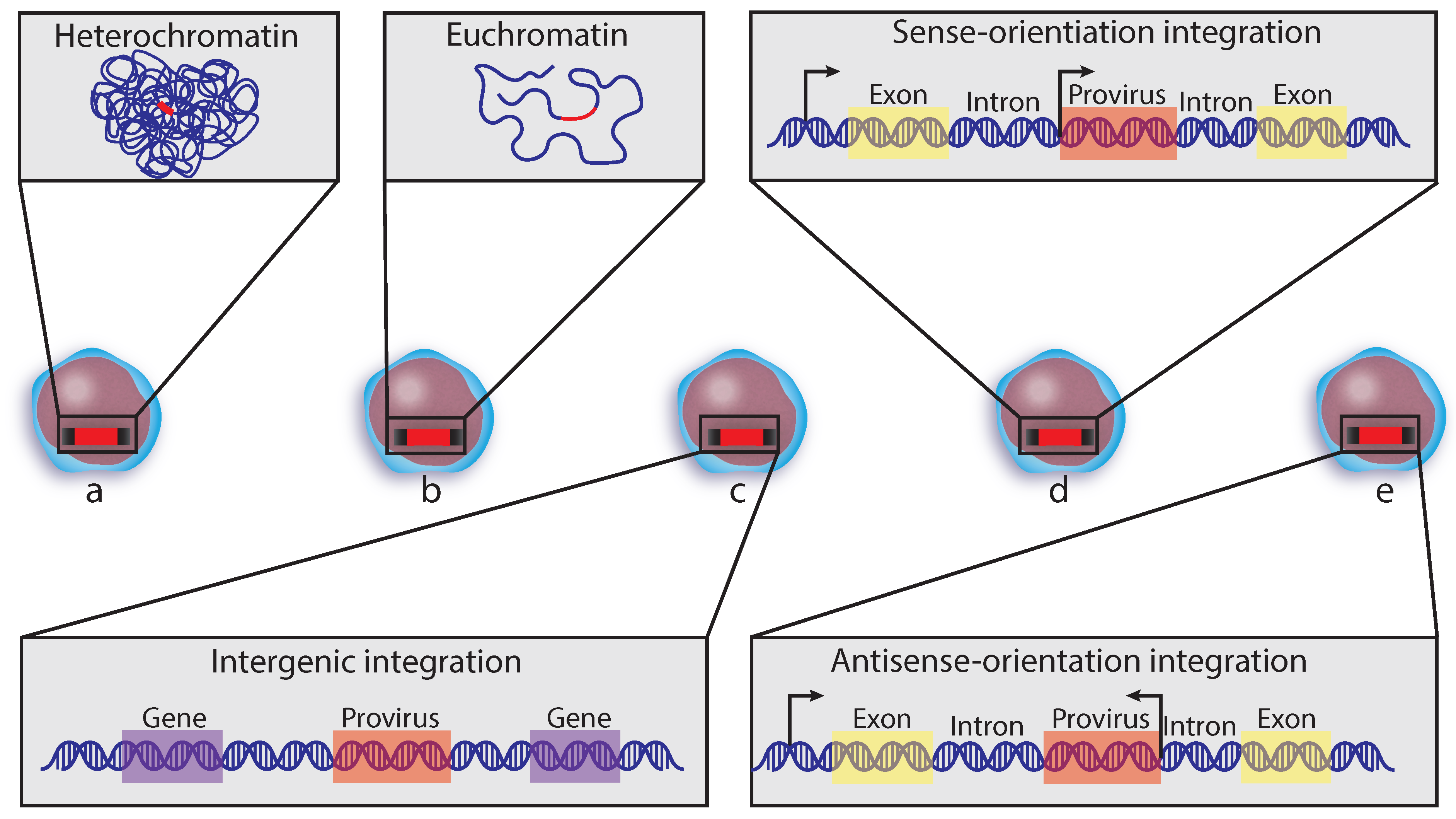
5.2. Integration Site Preferences
5.3. Informing Cure Strategies from Integration Sites
6. Techniques
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PBMC | peripheral blood mononuclear cell |
| BMMC | bone marrow mononuclear cell |
| HSPC | hematopoietic stem and progenitor cell |
| HDI | histone deacetylase inhibitor |
| TCR | T cell antigen receptor |
| RT | reverse transcriptase |
| pMHCII | peptide-bound major histocompatibility complex II |
| APC | antigen presenting cell |
| ORF | open reading frame |
| ELISA | enzyme-linked immunosorbent assay |
| rCD4 | resting CD4 |
| MOLT-4/CCR5 | cells from the CCR5-expressing T cell line MOLT-4 |
| FACS | Fluorescence Activated Cell Sorting |
| TH | T helper cell |
| TEM | T effector memory |
| TCM | T central memory |
| TTM | T transitional memory |
| TSCM | T stem cell memory |
| ART | antiretroviral therapy |
| PV | peripheral virus |
| MSD | major splice donor site |
| LTR | long terminal repeat |
| NFL | near full length. |
References
- Chun, T.-W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In Vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.; Buck, C.; Chaisson, R.; Quinn, T.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.; Margolick, J.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.; Kajdas, J.; Finzi, D.; Quinn, T.; Chadwick, K.; Margolick, J.; Kovacs, C.; Gange, S.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 2009, 1, 1137–1165. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.; Murray, A.; Pollack, R.; Soliman, M.; Laskey, S.; Capoferri, A.; Lai, J.; Strain, M.; Lada, S.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.; Silva, I.; Oliveira, T.; Rosales, R.; Parrish, E.; Learn, G.; Hahn, B.; Czartoski, J.; McElrath, M.; Lehmann, C.; et al. HIV-1 integration landscape during latent and active infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef]
- Simonetti, F.; Sobolewski, M.; Fyne, E.; Shao, W.; Spindler, J.; Hattori, J.; Anderson, E.; Watters, S.; Hill, S.; Wu, X.; et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 In Vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1883–1888. [Google Scholar] [CrossRef]
- Reeves, D.; Duke, E.; Wagner, T.; Palmer, S.; Spivak, A.; Schiffer, J.T. A majority of HIV persistence during antiretroviral therapy is due to infected cell proliferation. Nat. Commun. 2018, 9, 4811. [Google Scholar] [CrossRef]
- Wagner, T.; McKernan, J.; Tobin, N.; Tapia, K.; Mullins, J.; Frenkel, L.M. An increasing proportion of monotypic HIV-1 dna sequences during antiretroviral treatment suggests proliferation of HIV-Infected Cells. J. Virol. 2013, 87, 1770–1778. [Google Scholar] [CrossRef]
- Zaikos, T.; Terry, V.; Sebastian Kettinger, N.; Lubow, J.; Painter, M.; Virgilio, M.; Neevel, A.; Taschuk, F.; Onafuwa-Nuga, A.; McNamara, L.; et al. Hematopoietic stem and progenitor cells are a distinct hiv reservoir that contributes to persistent viremia in suppressed patients. Cell Rep. 2018, 25, 3759–3773. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, N.; Zaikos, T.; Terry, V.; Taschuk, F.; McNamara, L.; Onafuwa-Nuga, A.; Yucha, R.; Signer, R.A.J.; Riddell, J., IV; Bixby, D.; et al. CD4 is expressed on a heterogeneous subset of hematopoietic progenitors, which persistently harbor CXCR4 and CCR5-tropic HIV proviral genomes In Vivo. PLoS Pathog. 2017, 13, e1006509. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.; Sedaghat, A.; Kieffer, T.; Brennan, T.; Lee, P.; Wind-Rotolo, M.; Haggerty, C.; Kamireddi, A.; Liu, Y.; Lee, J.; et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol. 2006, 80, 6441–6457. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Orlova-Fink, N.; Einkauf, K.; Chowdhury, F.; Sun, X.; Harrington, S.; Kuo, H.-H.; Hua, S.; Chen, H.-R.; Ouyang, Z.; et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J. Clin. Invest. 2017, 127, 2689–2696. [Google Scholar] [CrossRef]
- Hiener, B.; Horsburgh, B.; Eden, J.-S.; Barton, K.; Schlub, T.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.; Liegler, T.; et al. Identification of genetically intact HIV-1 proviruses in specific CD4+ T cells from effectively treated participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef]
- Carter, C.; Onafuwa-Nuga, A.; McNamara, L.; Riddell, J.; Bixby, D.; Savona, M.; Collins, K.L. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat. Med. 2010, 16, 446–451. [Google Scholar] [CrossRef]
- Perelson, A.; Essunger, P.; Cao, Y.; Vesanen, M.; Hurley, A.; Saksela, K.; Markowitz, M.; Ho, D.D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 1997, 387, 188–191. [Google Scholar] [CrossRef]
- Ho, D.; Neumann, U.; Perelson, A.; Chen, W.; Leonardt, J.; Markowitz, M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [Google Scholar] [CrossRef]
- Chun, T.W.; Davey, R.T., Jr.; Ostrowski, M.; Justement, J.S.; Engel, D.; Mullins, J.I.; Fauci, A.S. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 2000, 6, 757–761. [Google Scholar] [CrossRef]
- Chun, T.-W.; Stuyver, L.; Mizell, S.; Ehler, L.; Mican, J.A.M.; Baseler, M.; Lloyd, A.; Nowak, M.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef]
- Buzon, M.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.; Li, J.; et al. HIV-1 persistence in CD4+ T cells with stem cell–like properties. Nat. Med. 2014, 20, 139–142. [Google Scholar] [CrossRef]
- McNamara, L.; Ganesh, J.; Collins, K.L. Latent HIV-1 infection occurs in multiple subsets of hematopoietic progenitor cells and is reversed by NF-KB activation. J. Virol. 2012, 86, 9337–9350. [Google Scholar] [CrossRef]
- Berger, E.; Murphy, P.; Farber, J.M. Chemokine receptors AS HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef]
- Alkhatib, G. The biology of CCR5 and CXCR4. Curr. Opin. HIV AIDS 2009, 4, 96. [Google Scholar] [CrossRef]
- Scarlatti, G.; Tresoldi, E.; Björndal, Å.; Fredriksson, R.; Colognesi, C.; Kui Deng, H.; Malnati, M.; Plebani, A.; Siccardi, A.; Littman, D.; et al. In Vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 1997, 3, 1259–1265. [Google Scholar] [CrossRef]
- Connor, R.; Sheridan, K.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1–infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef]
- Fouchier, R.A.M.; Meyaard, L.; Brouwer, M.; Hovenkamp, E.; Schuitemaker, H. Broader tropism and higher cytopathicity for CD4+T cells of a syncytium-inducing compared to a non-syncytium-inducing HIV-1 isolate as a mechanism for accelerated CD4+T cell declinein vivo. Virology 1996, 219, 87–95. [Google Scholar] [CrossRef]
- Koot, M.; Van Leeuwen, R.; De Goede, R.E.Y.; Keet, I.P.M.; Danner, S.; Schattenkerk, J.E.; Schuitemaker, H. Conversion Rate towards a Syncytium-Inducing (SI) phenotype during different stages of human immunodeficiency virus type 1 infection and prognostic value of, S.I. phenotype for survival after AIDS diagnosis. J. Infect. Dis. 1999, 179, 254–258. [Google Scholar] [CrossRef]
- Biti, R.; Ffrench, R.; Young, J.; Bennetts, B.; Stewart, G.; Liang, T. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat. Med. 1997, 3, 252–253. [Google Scholar] [CrossRef]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.; Smith, M.; Allikmets, R.; Goedert, J.; Buchbinder, S.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 1996, 273, 1856–1862. [Google Scholar] [CrossRef]
- Liu, R.; Paxton, W.; Choe, S.; Ceradini, D.; Martin, S.; Horuk, R.; MacDonald, M.; Stuhlmann, H.; Koup, R.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.; Rucker, J.; Liesnard, C.; Farber, C.-M.; Saragosti, S.; Lapouméroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Naif, H.; Cunningham, A.; Alali, M.; Li, S.; Nasr, N.; Buhler, M.; Schols, D.; de Clercq, E.; Stewart, G. A human immunodeficiency virus type 1 isolate from an infected person homozygous for CCR5Delta32 exhibits dual tropism by infecting macrophages and MT2 cells via CXCR4. J. Virol. 2002, 76, 3114–3124. [Google Scholar] [CrossRef] [PubMed]
- Gorry, P.; Zhang, C.; Wu, S.; Kunstman, K.; Trachtenberg, E.; Phair, J.; Wolinsky, S.; Gabuzda, D. Persistence of dual-tropic HIV-1 in an individual homozygous for the CCR5Δ32 allele. Lancet 2002, 359, 1832–1834. [Google Scholar] [CrossRef]
- Carter, C.; McNamara, L.; Onafuwa-Nuga, A.; Shackleton, M.; Riddell, J.; Bixby, D.; Savona, M.; Morrison, S.; Collins, K.L. HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe 2011, 9, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Nixon, C.; Vatakis, D.; Reichelderfer, S.; Dixit, D.; Kim, S.; Uittenbogaart, C.; Zack, J.A. HIV-1 infection of hematopoietic progenitor cells In Vivo in humanized mice. Blood 2013, 122, 2195–2204. [Google Scholar] [CrossRef]
- Bonecchi, R.; Bianchi, G.; Bordignon, P.; D’Ambrosio, D.; Lang, R.; Borsatti, A.; Sozzani, S.; Allavena, P.; Gray, P.; Mantovani, A.; et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 1998, 187, 129–134. [Google Scholar] [CrossRef]
- Han, Y.; Lassen, K.; Monie, D.; Sedaghat, A.; Shimoji, S.; Liu, X.; Pierson, T.; Margolick, J.; Siliciano, R.; Siliciano, J.D. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J. Virol. 2004, 78, 6122–6133. [Google Scholar] [CrossRef]
- Pierson, T.; Hoffman, T.; Blankson, J.; Finzi, D.; Chadwick, K.; Margolick, J.; Buck, C.; Siliciano, J.; Doms, R.; Siliciano, R.F. Characterization of chemokine receptor utilization of viruses in the latent reservoir for human immunodeficiency virus type 1. J. Virol. 2000, 74, 7824–7833. [Google Scholar] [CrossRef]
- Rabin, R.; Park, M.; Fang, L.; Swofford, R.; Stephany, D.; Farber, J.M. The chemokine receptor CCR8 is preferentially expressed in Th2 but not Th1 cells. J. Immunol. 1999, 162, 3840–3850. [Google Scholar]
- Jacobsen, S.E.W.; Nerlov, C. Haematopoiesis in the era of advanced single-cell technologies. Nat. Cell Biol. 2019, 21, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, S.; Xia, J.; Liu, F. Special issue: Stem cell biology hematopoietic hierarchy-an updated roadmap. Trends Cell Biol. 2018, 28, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takizawa, H.; Suda, T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development 2014, 141, 4656–4666. [Google Scholar] [CrossRef] [PubMed]
- Painter, M.; Zaikos, T.; Collins, K.L. Quiescence promotes latent hiv infection and resistance to reactivation from latency with histone deacetylase inhibitors. J. Virol. 2017, 91, e01080-17. [Google Scholar] [CrossRef]
- Liudahl, S.; Coussens, L.M. To help or to harm: Dynamic roles of CD4+ T helper cells in solid tumor microenvironments. In Immunology, Volume 1, Immunotoxicology, Immunopathology, and Immunotherapy; Academic Press: Cambridge, MA, USA, 2018; pp. 97–116. [Google Scholar]
- Pepper, M.; Jenkins, M.K. Origins of CD4+ effector and central memory T cells. Nat. Immunol. 2011, 12, 467–471. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Gao, H.; Xing, S.; Capoferri, A.; Durand, C.; Rabi, S.; Laird, G.; Kim, M.; Hosmane, N.; et al. Transcriptional reprogramming during effector-to-memory transition renders CD4+ T cells permissive for latent HIV-1 infection. Immunity 2017, 47, 766–775. [Google Scholar] [CrossRef]
- Mahnke, Y.; Brodie, T.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef]
- Macallan, D.; Wallace, D.; Zhang, Y.; De Lara, C.; Worth, A.; Ghattas, H.; Griffin, G.; Beverley, P.C.L.; Tough, D.F. Rapid turnover of effector-memory CD4(+) T cells in healthy humans. J. Exp. Med. 2004, 200, 255–260. [Google Scholar] [CrossRef]
- Den Braber, I.; Mugwagwa, T.; Vrisekoop, N.; Westera, L.; Mö, R.; Bregje De Boer, A.; Willems, N.; Schrijver, E.H.R.; Spierenburg, G.; Gaiser, K.; et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 2012, 36, 288–297. [Google Scholar] [CrossRef]
- Riou, C.; Yassine-Diab, B.; Van grevenynghe, J.; Somogyi, R.; Greller, L.; Gagnon, D.; Gimmig, S.; Wilkinson, P.; Shi, Y.; Cameron, M.; et al. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J. Exp. Med. 2007, 204, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Van Grevenynghe, J.; Procopio, F.; He, Z.; Chomont, N.; Riou, C.; Zhang, Y.; Gimmig, S.; Boucher, G.; Wilkinson, P.; Shi, Y.; et al. Transcription factor FOXO3a controls the persistence of memory CD4+ T cells during HIV infection. Nat. Med. 2008, 14, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Pekalski, M.; Ferreira, R.; Coulson, R.M.R.; Cutler, A.; Guo, H.; Smyth, D.; Downes, K.; Dendrou, C.; Castro Dopico, X.; Esposito, L.; et al. postthymic expansion in human CD4 naive T cells defined by expression of functional high-affinity IL-2 receptors. J. Immunol. 2013, 190, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Albuquerque, A.; Matoso, P.; Charmeteau-de-Muylder, B.; Cheynier, R.; Ligeiro, D.; Abecasis, M.; Anjos, R.; Barata, J.; Victorino, R.M.M.; et al. IL-7-induced proliferation of human naive CD4 T-cells relies on continued thymic activity. Front. Immunol. 2017, 8, 20. [Google Scholar] [CrossRef]
- Vandergeeten, C.; Fromentin, R.; DaFonseca, S.; Lawani, M.; Sereti, I.; Lederman, M.; Ramgopal, M.; Routy, J.-P.; Sékaly, R.-P.; Chomont, N. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 2013, 121, 4321–4329. [Google Scholar] [CrossRef]
- Wang, Z.; Gurule, E.; Brennan, T.; Gerold, J.; Kwon, K.; Hosmane, N.; Kumar, M.; Beg, S.; Capoferri, A.; Ray, S.; et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc. Natl. Acad. Sci. USA 2018, 115, E2575–E2584. [Google Scholar] [CrossRef]
- Bosque, A.; Famiglietti, M.; Weyrich, A.; Goulston, C.; Planelles, V. homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog. 2011, 7, e1002288. [Google Scholar] [CrossRef]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef]
- Dobrowolski, C.; Valadkhan, S.; Graham, A.; Shukla, M.; Ciuffi, A.; Telenti, A.; Karn, J. Entry of polarized effector cells into quiescence forces HIV latency. MBio 2019, 10, e00337-19. [Google Scholar] [CrossRef]
- Archin, N.; Liberty, A.; Kashuba, A.; Choudhary, S.; Kuruc, J.; Crooks, A.; Parker, D.; Anderson, E.; Kearney, M.; Strain, M.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.; Quigley, M.; Almeida, J.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell–like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Anderson, E.; Maldarelli, F. The role of integration and clonal expansion in HIV infection: Live long and prosper. Retrovirology 2018, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-C.; Shan, L.; Hosmane, N.; Wang, J.; Laskey, S.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.; Siliciano, J.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Hosmane, N.; Kwon, K.; Bruner, K.; Capoferri, A.; Beg, S.; Rosenbloom, D.I.S.; Keele, B.; Ho, Y.-C.; Siliciano, J.; Siliciano, R.F. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1, Potential role in latent reservoir dynamics. J. Exp. Med. 2017, 214, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Bui, J.; Sobolewski, M.; Keele, B.; Spindler, J.; Musick, A.; Wiegand, A.; Luke, B.; Shao, W.; Hughes, S.; Coffin, J.; et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog. 2017, 13, e1006283. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.; Mitrophanous, K.; Kingsman, S.; Kingsman, A.J. Minimal requirement for a lentivirus vector based on human immunodeficiency virus type 1. J. Virol. 1998, 72, 811–816. [Google Scholar] [CrossRef]
- Symons, J.; Cameron, P.; Lewin, S.R. HIV integration sites and implications for maintenance of the reservoir. Curr. Opin. HIV AIDS 2018, 13, 152–159. [Google Scholar] [CrossRef]
- Pinzone, M.; Vanbelzen, D.; Weissman, S.; Bertuccio, M.; Cannon, L.; Venanzi-Rullo, E.; Migueles, S.; Jones, R.; Mota, T.; Joseph, S.; et al. Longitudinal HIV sequencing reveals reservoir expression leading to decay which is obscured by clonal expansion. Nat Commun. 2019, 10, 728. [Google Scholar] [CrossRef]
- Purcell, D.; Martin, M.A. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 1993, 67, 6365–6378. [Google Scholar] [CrossRef]
- Schwartz, S.; Felber, B.; Benko, D.; Fenyö, E.; Pavlakis, G.N. Cloning and functional analysis of multiply spliced mRNA species of human immunodeficiency virus type 1. J. Virol. 1990, 64, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Reyon, D.; Khayter, C.; Regan, M.; Keith Joung, J.; Sander, J.D. Engineering designer transcription activator-like effector nucleases (TALENs) by REAL or REAL-Fast assembly. Curr. Protoc. Mol. Biol. 2012, 100, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Ocwieja, K.; Sherrill-Mix, S.; Mukherjee, R.; Custers-Allen, R.; David, P.; Brown, M.; Wang, S.; Link, D.; Olson, J.; Travers, K.; et al. Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 2012, 40, 10345–10355. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Santoni de Sio, F.; Rudilosso, L.; Gallina, P.; Calabria, A.; Beretta, S.; Merelli, I.; Bruzzesi, E.; Passerini, L.; Nozza, S.; et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat. Commun. 2017, 8, 498. [Google Scholar] [CrossRef]
- Wagner, T.; McLaughlin, S.; Garg, K.; Cheung, C.Y.K.; Larsen, B.; Styrchak, S.; Huang, H.; Edlefsen, P.; Mullins, J.; Frenkel, L.M. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef]
- Coffin, J.; Wells, D.; Zerbato, J.; Kuruc, J.; Guo, S.; Luke, B.; Eron, J.; Bale, M.; Spindler, J.; Simonetti, F.; et al. Clones of infected cells arise early in HIV-infected individuals. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Scully, E.; Gandhi, M.; Johnston, R.; Hoh, R.; Lockhart, A.; Dobrowolski, C.; Pagliuzza, A.; Milush, J.; Baker, C.; Girling, V.; et al. Sex-based differences in human immunodeficiency virus type 1 reservoir activity and residual immune activation. J. Infect. Dis. 2019, 219, 1084–1094. [Google Scholar] [CrossRef]
- Das, B.; Dobrowolski, C.; Luttge, B.; Valadkhan, S.; Chomont, N.; Johnston, R.; Bacchetti, P.; Hoh, R.; Gandhi, M.; Deeks, S.; et al. Estrogen receptor-1 is a key regulator of HIV-1 latency that imparts gender-specific restrictions on the latent reservoir. Proc. Natl. Acad. Sci. USA 2018, 115, E7795–E7804. [Google Scholar] [CrossRef]
- Scully, E.P. Sex differences in HIV infection. Curr. HIV AIDS Rep. 2018, 15, 136–146. [Google Scholar] [CrossRef]
- Joseph, S.; Swanstrom, R.; Kashuba, A.D.M.; Cohen, M.S. Bottlenecks in HIV-1 transmission: Insights from the study of founder viruses. Nat. Rev. Microbiol. 2015, 13, 414–425. [Google Scholar] [CrossRef]
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.S.; Willey, R.L.; Sato, H.; Chang, L.J.; Blumenthal, R.; Martin, M.A. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 1993, 67, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Orensteint, J.; Dimitrov, D.; Martin, M. Cell-to-cell spread of HIV-1 occurs within minutes and may not involve the participation of virus particles. Virology 1992, 186, 712–724. [Google Scholar] [CrossRef]
- Collins, D.; Lubow, J.; Lukic, Z.; Mashiba, M.; Collins, K.L. Vpr promotes macrophage-dependent HIV-1 infection of CD4+ T lymphocytes. PLoS Pathog. 2015, 11, e1005054. [Google Scholar] [CrossRef] [PubMed]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.A.; Williams, J.; Schiffner, T.; Gärtner, K.; Ochsenbauer, C.; Kappes, J.; Russell, R.; Frater, J.; Sattentau, Q.J. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J. Virol. 2014, 88, 2025–2034. [Google Scholar] [CrossRef]
- Park, R.; Wang, T.; Koundakjian, D.; Hultquist, J.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef]
- Schiffner, T.; Sattentau, Q.; Duncan, C.J.A. Cell-to-cell spread of HIV-1 and evasion of neutralizing antibodies. Vaccine 2013, 31, 5789–5797. [Google Scholar] [CrossRef]
- Lorenzo-Redondo, R.; Fryer, H.; Bedford, T.; Kim, E.-Y.; Archer, J.; Kosakovsky Pond, S.; Chung, Y.-S.; Penugonda, S.; Chipman, J.; Fletcher, C.V.; et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef]
- Hatano, H.; Hayes, T.; Dahl, V.; Sinclair, E.; Lee, T.-H.; Hoh, R.; Lampiris, H.; Hunt, P.; Palmer, S.; Mccune, J.; et al. A randomized, controlled trial of raltegravir intensification in antiretroviral-treated, HIV-infected patients with a suboptimal CD4 1 T cell response. J. Infect. Dis. 2011, 203, 960–968. [Google Scholar] [CrossRef]
- Dinoso, J.; Kim, S.; Wiegand, A.; Palmer, S.; Gange, S.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.; Finucane, M.; Nettles, R.; Quinn, T.; Broman, K.; Ray, S.; Persaud, D.; Siliciano, R.F. Genotypic analysis of HIV-1 drug resistance at the limit of detection: Virus production without evolution in treated adults with undetectable HIV loads. J. Infect. Dis. 2004, 189, 1452–1465. [Google Scholar] [CrossRef]
- Hermankova, M.; Ray, S.; Ruff, C.; Powell-Davis, M.; Ingersoll, R.; D’Aquila, R.; Quinn, T.; Siliciano, J.; Siliciano, R.; Persaud, D. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. J. Am. Med. Assoc. 2001, 286, 196–207. [Google Scholar] [CrossRef]
- Anderson, J.; Archin, N.; Ince, W.; Parker, D.; Wiegand, A.; Coffin, J.; Kuruc, J.; Eron, J.; Swanstrom, R.; Margolis, D.M. Clonal sequences recovered from plasma from patients with residual HIV-1 viremia and on intensified antiretroviral therapy are identical to replicating viral RNAs recovered from circulating resting CD4+ T cells. J. Virol. 2011, 85, 5220–5223. [Google Scholar] [CrossRef]
- Evering, T.; Mehandru, S.; Racz, P.; Tenner-Racz, K.; Poles, M.; Figueroa, A.; Mohri, H.; Markowitz, M. Absence of HIV-1 evolution in the gut-associated lymphoid tissue from patients on combination Antiviral therapy initiated during primary infection. PLoS Pathog. 2012, 8, e1002506. [Google Scholar] [CrossRef] [PubMed]
- Joos, B.; Fischer, M.; Kuster, H.; Pillai, S.; Wong, J.; Böni, J.; Hirschel, B.; Weber, R.; Trkola, A.; Günthard, H. Swiss HIV cohort study TSHC HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16725–16730. [Google Scholar] [CrossRef]
- Conway, J.; Perelson, A.S. Residual viremia in treated HIV+ individuals. PLoS Comput. Biol. 2016, 12, e1004677. [Google Scholar] [CrossRef]
- Kearney, M.; Wiegand, A.; Shao, W.; McManus, W.; Bale, M.; Luke, B.; Maldarelli, F.; Mellors, J.; Coffin, J.M. Ongoing HIV replication during ART reconsidered. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2017; p. 4. [Google Scholar] [CrossRef]
- Rosenbloom, D.I.S.; Hill, A.; Laskey, S.; Siliciano, R.F. Re-evaluating evolution in the HIV reservoir. Nature 2017, 551, E6–E9. [Google Scholar] [CrossRef]
- Yukl, S.; Shergill, A.; McQuaid, K.; Gianella, S.; Lampiris, H.; Hare, C.; Pandori, M.; Sinclair, E.; Günthard, H.; Fischer, M.; et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS 2010, 24, 2451–2460. [Google Scholar] [CrossRef]
- Buzón, M.; Massanella, M.; Llibre, J.; Esteve, A.; Dahl, V.; Puertas, M.; Gatell, J.; Domingo, P.; Paredes, R.; Sharkey, M.; et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat. Med. 2010, 16, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.; Zhou, Y.; Kieffer, T.; Ruff, C.; Buck, C.; Siliciano, R.F. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J. Virol. 2002, 76, 8518–8531. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.; Stanwick, T.; Dempsey, M.; Stevenson, M. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 1991, 254, 423–427. [Google Scholar] [CrossRef]
- Hu, W.-S.; Hughes, S.H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2, a006882. [Google Scholar] [CrossRef]
- Lesbats, P.; Engelman, A.; Cherepanov, P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef]
- Josefsson, L.; King, M.; Makitalo, B.; Brännström, J.; Shao, W.; Maldarelli, F.; Kearney, M.; Hu, W.-S.; Chen, J.; Gaines, H.; et al. Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc. Natl. Acad. Sci. USA 2011, 108, 11199–11204. [Google Scholar] [CrossRef]
- Josefsson, L.; Palmer, S.; Faria, N.; Lemey, P.; Casazza, J.; Ambrozak, D.; Kearney, M.; Shao, W.; Kottilil, S.; Sneller, M.; et al. single cell analysis of lymph node tissue from HIV-1 infected patients reveals that the majority of CD4+ T-cells contain one HIV-1 DNA molecule. PLoS Pathog. 2013, 9, e1003432. [Google Scholar] [CrossRef]
- Serrao, E.; Ballandras-Colas, A.; Cherepanov, P.; Maertens, G.; Engelman, A.N. Key determinants of target DNA recognition by retroviral intasomes. Retrovirology 2015, 12, 39. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.; Munroe, D.J. Weak palindromic consensus sequences are a common feature found at the integration target sites of many retroviruses. J. Virol. 2005, 79, 5211–5214. [Google Scholar] [CrossRef]
- Schröder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Lewinski, M.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; et al. Retroviral DNA integration: Viral and cellular determinants of target-site selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef]
- Ikeda, T.; Shibata, J.; Yoshimura, K.; Koito, A.; Matsushita, S. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. J. Infect. Dis. 2007, 195, 716–725. [Google Scholar] [CrossRef]
- Einkauf, K.; Lee, G.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.; Jiang, C.; Lian, X.; Chowdhury, F.; et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Invest. 2019, 129, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Casartelli, N.; Pellin, D.; Rizzi, E.; Souque, P.; Severgnini, M.; Di Serio, C.; Fricke, T.; Diaz-Griffero, F.; Zimmer, C.; et al. Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 2015, 6, 6483. [Google Scholar] [CrossRef] [PubMed]
- Di Nunzio, F.; Danckaert, A.; Fricke, T.; Perez, P.; Fernandez, J.; Perret, E.; Roux, P.; Shorte, S.; Charneau, P.; Diaz-Griffero, F.; et al. Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PLoS ONE 2012, 7, e46037. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.; Machado, A.K. Transportin-1 binds to the HIV-1 capsid via a nuclear localization signal and triggers uncoating. Nat. Microbiol. 2019, 4, 1840–1850. [Google Scholar] [CrossRef]
- Burdick, R.; Delviks-Frankenberry, K.; Chen, J.; Janaka, S.; Sastri, J.; Hu, W.-S.; Pathak, V.K. Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 2017, 13, e1006570. [Google Scholar] [CrossRef]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.; Mellors, J.; Kearney, M.; et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef]
- Mitchell, R.; Beitzel, B.; Schroder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Biggar, R.; Chaturvedi, A.; Goedert, J.; Engels, E.A. AIDS-related cancer and severity of immunosuppression in persons with AIDS. JNCI J. Natl. Cancer Inst. 2007, 99, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ramírez, R.; Shiels, M.; Dubrow, R.; Engels, E.A. Cancer risk in HIV-infected people in the USA from 1996 to 2012: A population-based, registry-linkage study. Lancet HIV 2017, 4, 495–504. [Google Scholar] [CrossRef]
- Katano, H.; Sato, Y.; Hoshino, S.; Tachikawa, N.; Oka, S.; Morishita, Y.; Ishida, T.; Watanabe, T.; Rom, W.; Mori, S.; et al. Integration of HIV-1 caused STAT3-associated B cell lymphoma in an AIDS patient. Microbes Infect. 2007, 9, 1581–1589. [Google Scholar] [CrossRef]
- Kuo, H.-H.; Ahmad, R.; Lee, G.; Gao, C.; Chen, H.-R.; Ouyang, Z.; Szucs, M.; Kim, D.; Tsibris, A.; Chun, T.-W.; et al. Anti-apoptotic protein BIRC5 maintains survival of HIV-1-infected CD4+ T Cells. Immunity 2018, 48, 1183–1194.e5. [Google Scholar] [CrossRef]
- Archin, N.; Bateson, R.; Tripathy, M.; Crooks, A.; Yang, K.-H.; Dahl, N.; Kearney, M.; Anderson, E.; Coffin, J.; Strain, M.; et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J. Infect. Dis. 2014, 210, 728–735. [Google Scholar] [CrossRef]
- Zaikos, T.; Painter, M.; Kettinger, N.T.S.; Terry, V.; Collins, K.L. Class 1-Selective Histone Deacetylase (HDAC) inhibitors enhance HIV latency reversal while preserving the activity of HDAC isoforms necessary for maximal HIV gene expression. J. Virol. 2018, 92, 2110–2127. [Google Scholar] [CrossRef]
- Shao, W.; Shan, J.; Kearney, M.; Wu, X.; Maldarelli, F.; Mellors, J.; Luke, B.; Coffin, J.; Hughes, S.H. Retrovirus Integration Database (RID): A public database for retroviral insertion sites into host genomes. Retrovirology 2016, 13, 47. [Google Scholar] [CrossRef]
- Patro, S.; Coffin, J.M. Combined HIV-1 sequence and integration site analysis informs viral dynamics and allows reconstruction of replicating viral ancestors. Proc. Natl. Acad. Sci. USA 2019, 116, 25891–25899. [Google Scholar] [CrossRef]
- Liu, H.; Dow, E.; Arora, R.; Kimata, J.; Bull, L.; Arduino, R.; Rice, A.P. Integration of human immunodeficiency virus type 1 in untreated infection occurs preferentially within genes. J. Virol. 2006, 80, 7765–7768. [Google Scholar] [CrossRef][Green Version]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.; Bixby, C.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.; Dewar, R.; et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.; Salazar, M.; Keele, B.; Learn, G.; Giorgi, E.; Li, H.; Decker, J.; Wang, S.; Baalwa, J.; Kraus, M.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Rutsaert, S.; Bosman, K.; Trypsteen, W.; Nijhuis, M.; Vandekerckhove, L. Digital PCR as a tool to measure HIV persistence. Retrovirology 2018, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.; Wang, Z.; Simonetti, F.; Bender, A.; Kwon, K.; Sengupta, S.; Fray, E.; Beg, S.; Antar, A.A.R.; Jenike, K.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120. [Google Scholar] [CrossRef]
- Procopio, F.; Fromentin, R.; Kulpa, D.; Brehm, J.; Bebin, A.-G.; Strain, M.; Richman, D.; O’Doherty, U.; Palmer, S.; Hecht, F.; et al. A novel assay to measure the magnitude of the inducible viral reservoir in hiv-infected individuals. EBioMedicine 2015, 2, 874–883. [Google Scholar] [CrossRef]
- Siliciano, J.; Siliciano, R.F. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol. Biol. 2005, 304, 3–15. [Google Scholar] [CrossRef]
- Wang, Z.; Simonetti, F.; Siliciano, R.; Laird, G.M. Measuring replication competent HIV-1, advances and challenges in defining the latent reservoir. Retrovirology 2018, 15, 21. [Google Scholar] [CrossRef]
- Massanella, M.; Yek, C.; Lada, S.; Nakazawa, M.; Shefa, N.; Huang, K.; Richman, D.D. Improved assays to measure and characterize the inducible HIV reservoir. EBioMedicine 2018, 36, 113–121. [Google Scholar] [CrossRef]
- Wonderlich, E.; Subramanian, K.; Cox, B.; Wiegand, A.; Lackman-Smith, C.; Bale, M.; Stone, M.; Hoh, R.; Kearney, M.; Maldarelli, F.; et al. Effector memory differentiation increases detection of replication-competent HIV-l in resting CD4+ T cells from virally suppressed individuals. PLoS Pathog. 2019, 15, e1008074. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Description | Advantages | Limitations |
|---|---|---|---|
| Modified TC-Seq [7,122] | Sonicate genomic DNA into small fragments. End-repair DNA fragments. Add dA-linker to 3’ ends. Attach linkers to 3’ ends using dT. Perform Nested PCR on fragments. Attach Illumina sequencing adaptors. Paired-end Illumina sequencing across integration sites. |
|
|
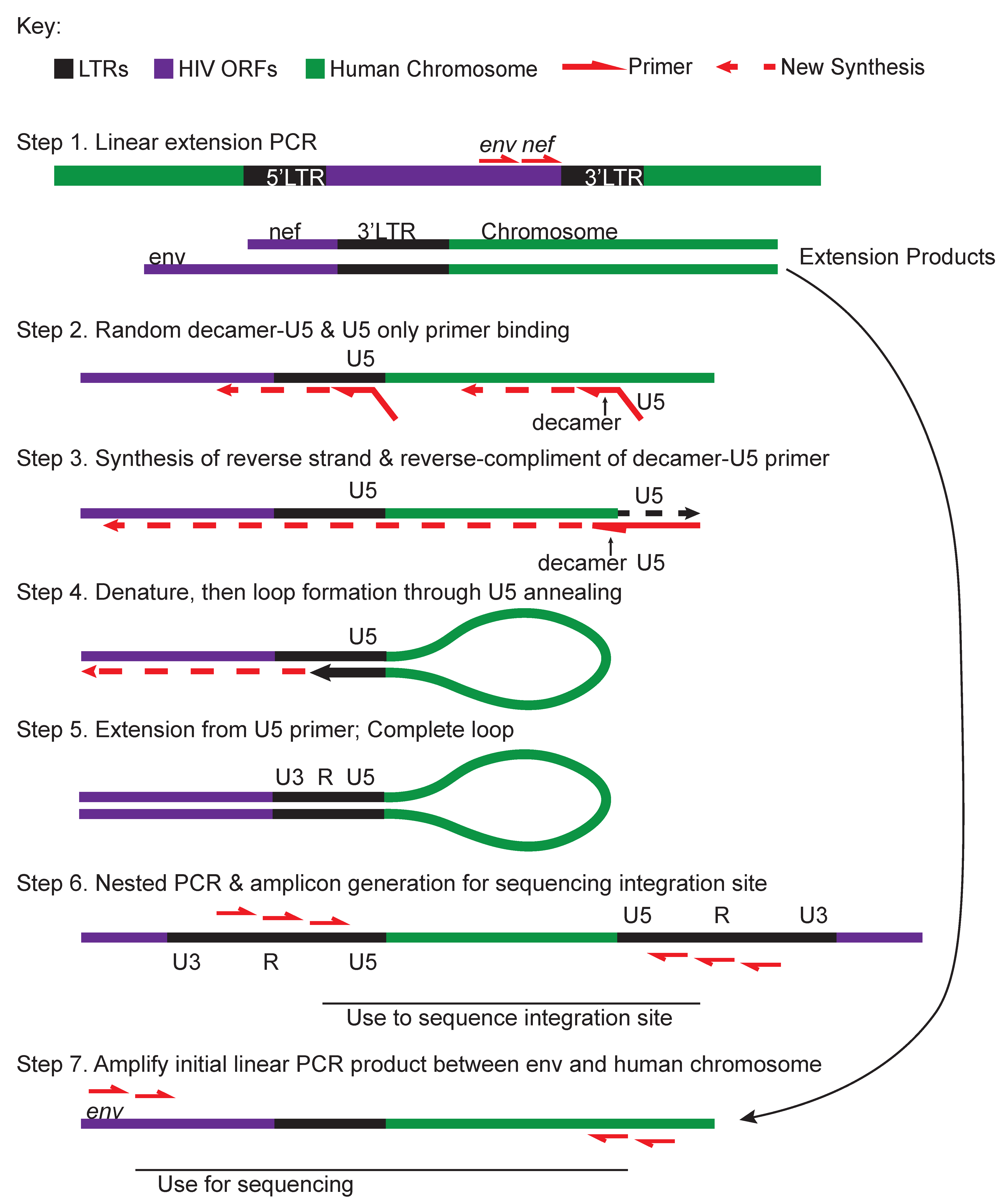
| ISLA (Integration site loop amplification) (Figure 5) [76] | Linear extension PCR products from the 3’end of HIV into chromosome are amplified with a random decamer complementary to the host genome with a U5-priming sequence-tail. The U5 primer is used as a reverse primer to convert the ssPCR into a dsPCR of 3’HIV-chromosome product. Linearized products produce a genetic barbell with LTR sequence on either side of human sequence which can be PCR amplified and prepared for NextGen sequencing. |
|
|
| Circular Template [11,132] | PCR amplification and sequencing across the insertion site starting from genomic DNA from HIV infected cells. A restriction enzyme (PstI) creates small provirus-host DNA fragments that can be ligated together into circular units and amplified using HIV-specific primers oriented in opposing directions across the integration site, then sequenced by Sanger sequencing. |
|
|
| Technique | Description | Advantages | Limitations |
|---|---|---|---|
| FLIPS (Full-length individual proviral sequencing) [15] | An assay used to capture nearly full-length genomes by two non-overlapping, nested-PCR reactions amplifying from the U5 region in the 5’ and 3’ LTRs. PCR products are sequenced by paired-end Illumina sequencing using the Nextera library preparation kit (Illumina). Reads are mapped by de novo genome assembly. |
|
|
| SCS Assay (single-cell sequencing) [108] | CD4+ T cells are FACS sorted from HIV-infected patients into a 96-well plate such that there is no more than one cell in any one well. Once the cell in each well is lysed, the DNA is distributed from one well into 10. PCR is used to amplify HIV DNA between gag-pol, identified through gel-electrophoresis, and sequenced. Sequenced products can then be aligned to a reference HIV genome. |
|
|
| SPS/SGS (Single-genome (provirus) sequencing and single genome amplification) [67,133,134]. | Virions collected from the peripheral blood or cultured media containing HIV virion-producing cells are collected, lysed, and converted to cDNA. The cDNA is serially diluted and used for qPCR with primer probe directed at pol (SGS) or env (SGA). Once HIV-containing samples are identified, the cDNA is then subjected to PCR amplification between gag-pol or env and sequenced. |
|
|
| Technique | Description | Advantages | Limitations |
|---|---|---|---|
| Matched integration site and proviral sequencing (MIP-Seq) [115] | Genomic DNA is isolated from CD4+ T cells, quantified using ddPCR for viral gag, and diluted to single proviral genomes based on ddPCR and Poisson distribution. This is followed by multiple displacement amplification (MDA) and whole genome amplification (WGA), generating 1,000-10,000 copies of gDNA. Amplified gDNA is divided such that some is used for NFL sequencing [14] and some for integration site analysis (ISLA; Table 1; Figure 5) or other methods. |
|
|
| Multiple-displacement amplification with single-genome sequencing (MDA-SGS) [131] | Genomic DNA is extracted from PBMCs or other primary cells and diluted across a 96-well plate. Whole-gDNA is amplified in-well using MDA. MDA wells are screened for proviruses of interest using SGS (subgenomic fragments) from P6 through part of RT. Then integration sites are determined using modified TC-Seq (Table 1) followed by NFL amplification using Sanger sequencing or PacBio sequencing. |
|
|
| Technique | Description | Advantages | Limitations |
|---|---|---|---|
| ddPCR (droplet digital PCR) (reviewed [135]) | Target molecules are emulsified into thousands of nanoliter droplets and amplified by PCR using a primer-probe set. Droplets containing HIV genomic material that fluoresces above a certain threshold will be considered positive. The ratio between the positive and negative droplets is used to calculate the absolute number of starting molecules using a Poisson distribution. |
|
|
| IPDA (intact proviral DNA assay) [136] | Uses two amplicons covering the packaging signal (ψ) and env region and ddPCR to designate deleted proviruses as defective. In parallel, multiplex PCR is performed with two unique primer-probe sets targeting ψ and env with unique labeling probes and measured by ddPCR. Primer-probe sets amplify validated, highly conserved regions of the genome. Droplets are scored based on expression of combinations of probe fluorescent patterns. |
|
|
| TILDA(Tat/Rev induced limiting dilution assay) [137] | CD4+ T cells are stimulated in vitro (PMA and ionomycin) to maximally produce tat/rev transcripts. Cells are serially diluted as replicates. Real-time PCR with primer-probe pairs are used to quantify inducible viral RNA. The frequency of cells with inducible HIV RNA can then be calculated from the number of positive wells at each dilution by the maximum likelihood method. |
|
|
| QVOA (quantitative viral outgrowth assay) [138,139] | Culture method to quantify the replication-competent viral reservoir. HIV(+) donor rCD4+ T cells are cultured with irradiated PBMCs and CD4+ T cells from an HIV(-) donor and stimulated (PHA; IL-2). Replication -competent virus can spread to HIV(-) CD4+ T cells, amplifying the infection, allowing detection and quantification of viral outgrowth. |
|
|
| mQVOA (Modified quantitative viral outgrowth assay) [140] | A more sensitive adaptation of the gold-standard assay; CD4+ T cells from patients are serially diluted and stimulated (αCD3/CD28 antibodies). MOLT-4/CCR5 cells are co-cultured with the primary cells. HIV RNA is extracted, and RT-qPCR is performed to amplify pol. The number of wells positive for HIV RNA at each dilution level are used to determine the infection frequency by maximum likelihood estimate. |
|
|
| dQVOA (differentiation culture quantitative viral Outgrowth assay) [141] | Measures the impact of TEM differentiation on induction and outgrowth of replication-competent HIV. rCD4+ T cells from patients are activated through culture with a differentiation cytokine cocktails to drive cells towards the TEM terminally differentiated subset. Cells are distributed at limiting dilutions and cultured in differentiation cytokines, then activated. Titer measured by p24 ELISA. |
|
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Virgilio, M.C.; Collins, K.L. The Impact of Cellular Proliferation on the HIV-1 Reservoir. Viruses 2020, 12, 127. https://doi.org/10.3390/v12020127
Virgilio MC, Collins KL. The Impact of Cellular Proliferation on the HIV-1 Reservoir. Viruses. 2020; 12(2):127. https://doi.org/10.3390/v12020127
Chicago/Turabian StyleVirgilio, Maria C., and Kathleen L. Collins. 2020. "The Impact of Cellular Proliferation on the HIV-1 Reservoir" Viruses 12, no. 2: 127. https://doi.org/10.3390/v12020127
APA StyleVirgilio, M. C., & Collins, K. L. (2020). The Impact of Cellular Proliferation on the HIV-1 Reservoir. Viruses, 12(2), 127. https://doi.org/10.3390/v12020127