Herpesviruses and the Unfolded Protein Response



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
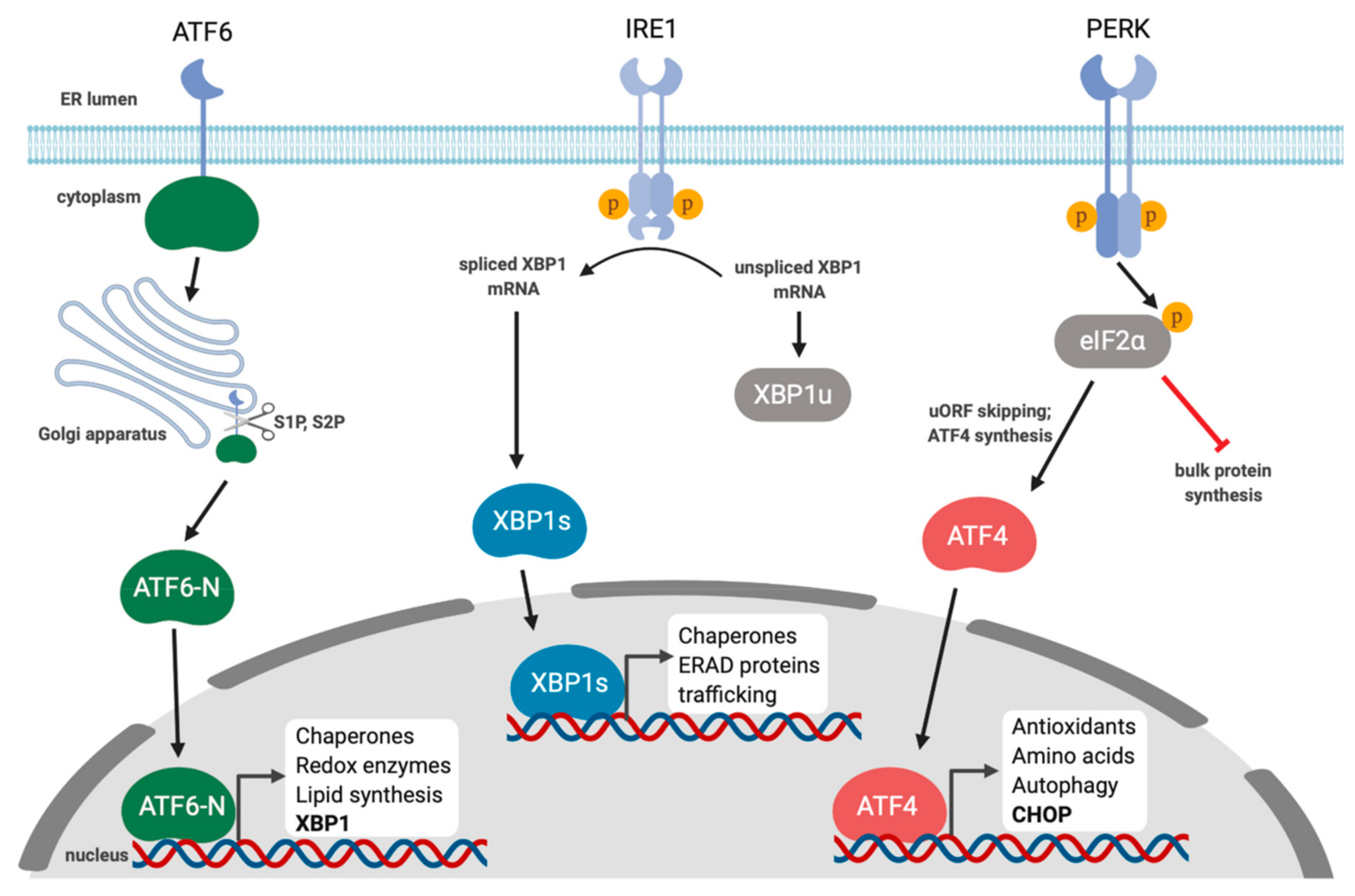
1. Overview of the Unfolded Protein Response
2. IRE1 Is a Kinase and an Endoribonuclease
3. PERK and the Integrated Stress Response
4. ATF6 Is Activated by Regulated Intramembrane Proteolysis
5. Crosstalk among the Branches of the UPR
6. The UPR and Cell Fate
7. The UPR in Health and Disease
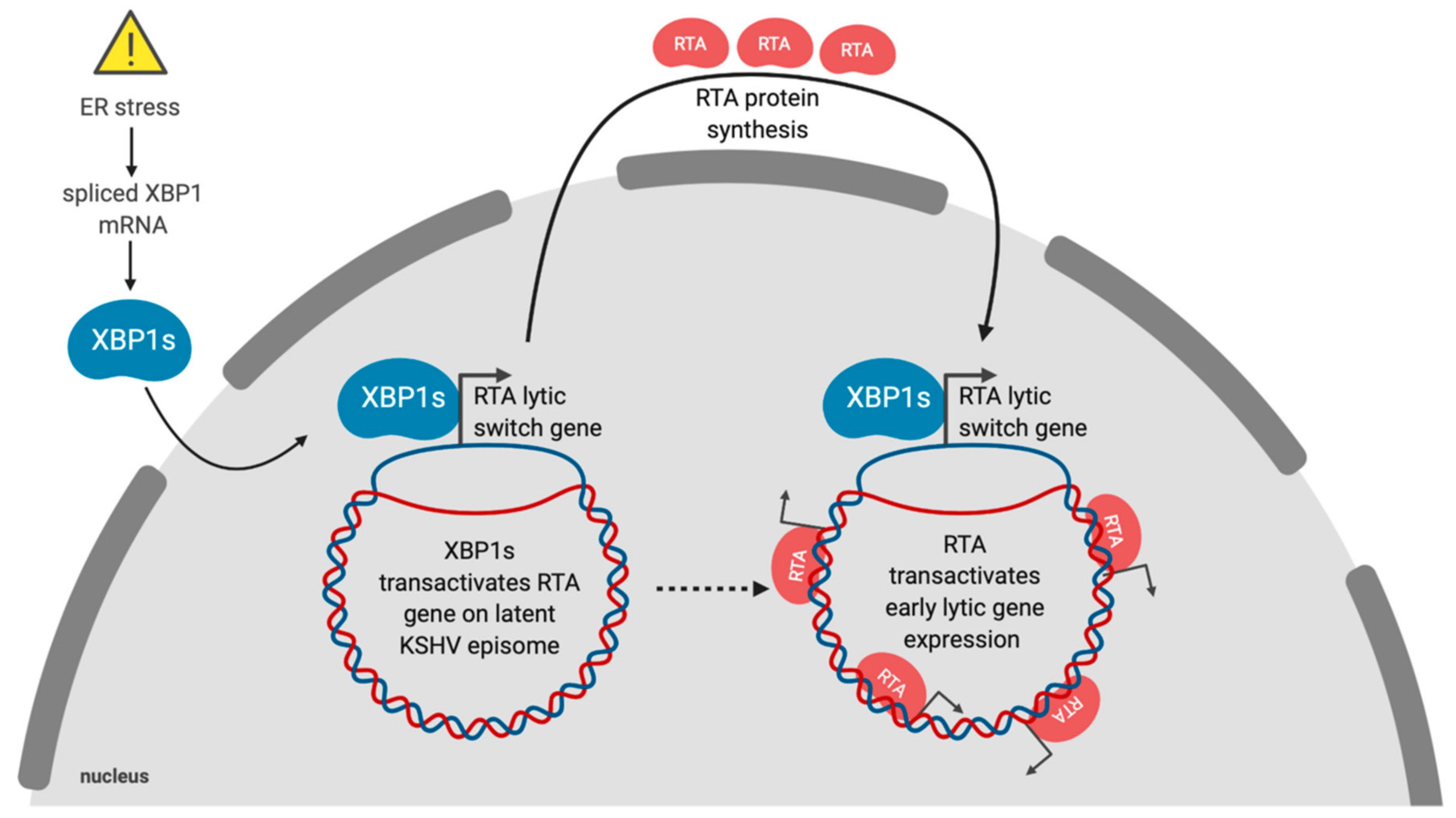
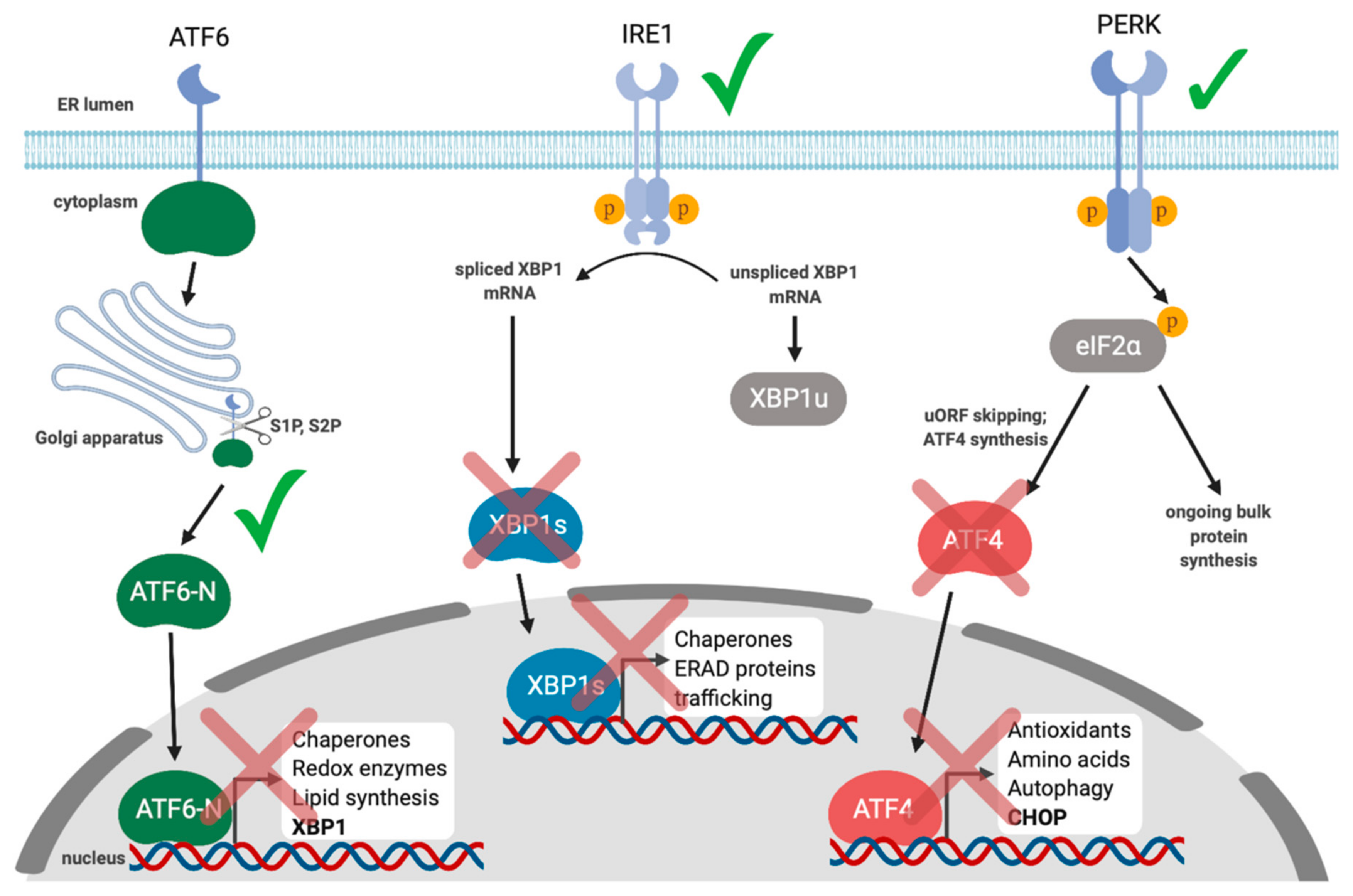
8. Gammaherpesviruses and the UPR
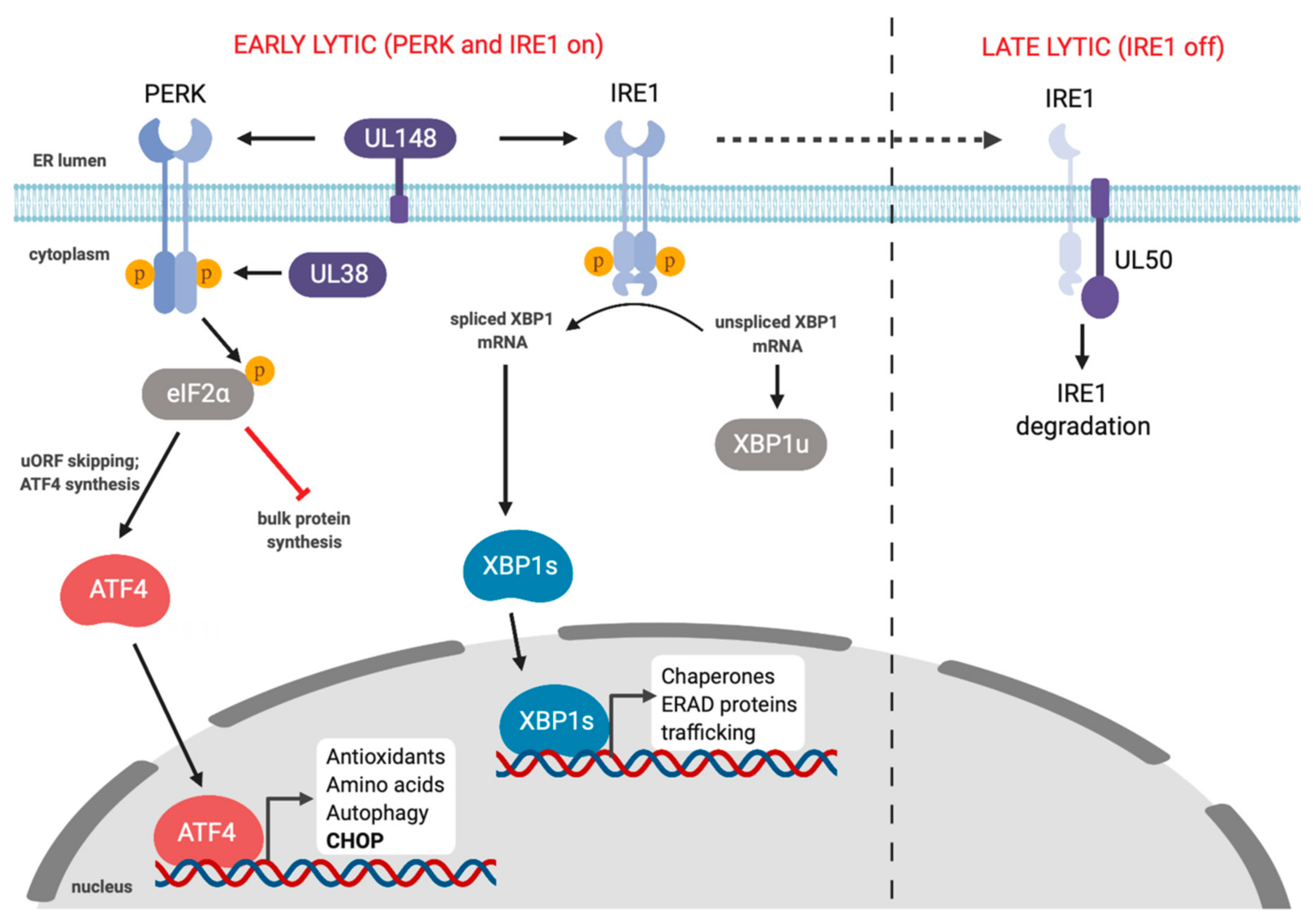
9. Cytomegalovirus (CMV) and the UPR
10. HSV-1 and the UPR
11. Conclusions and Outstanding Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Vattem, K.M.; Wek, R.C. Dimerization and Release of Molecular Chaperone Inhibition Facilitate Activation of Eukaryotic Initiation Factor-2 Kinase in Response to Endoplasmic Reticulum Stress. J. Biol. Chem. 2002, 277, 18728–18735. [Google Scholar] [CrossRef] [PubMed]
- Gething, M.J.; McCammon, K.; Sambrook, J. Expression of wild-type and mutant forms of influenza hemagglutinin: The role of folding in intracellular transport. Cell 1986, 46, 939–950. [Google Scholar] [CrossRef]
- Peluso, R.W.; Lamb, R.A.; Choppin, P.W. Infection with paramyxoviruses stimulates synthesis of cellular polypeptides that are also stimulated in cells transformed by Rous sarcoma virus or deprived of glucose. Proc. Natl. Acad. Sci. USA 1978, 75, 6120–6124. [Google Scholar] [CrossRef]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar] [CrossRef]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in IRE1β-deficient mice. J. Clin. Investig. 2001, 107, 585–593. [Google Scholar] [CrossRef]
- Lee, K.P.K.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the Dual Enzyme Ire1 Reveals the Basis for Catalysis and Regulation in Nonconventional RNA Splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef]
- Welihinda, A.A.; Kaufman, R.J. The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J. Biol. Chem. 1996, 271, 18181–18187. [Google Scholar] [CrossRef]
- Sidrauski, C.; Walter, P. The Transmembrane Kinase Ire1p Is a Site-Specific Endonuclease That Initiates mRNA Splicing in the Unfolded Protein Response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Jurkin, J.; Henkel, T.; Nielsen, A.F.; Minnich, M.; Popow, J.; Kaufmann, T.; Heindl, K.; Hoffmann, T.; Busslinger, M.; Martinez, J. The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 2014, 33, 2922–2936. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liang, F.-X.; Wang, X. A Synthetic Biology Approach Identifies the Mammalian UPR RNA Ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Kosmaczewski, S.G.; Edwards, T.J.; Han, S.M.; Eckwahl, M.J.; Meyer, B.I.; Peach, S.; Hesselberth, J.R.; Wolin, S.L.; Hammarlund, M. The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep. 2014, 15, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential Contributions of ATF6 and XBP1 to the Activation of Endoplasmic Reticulum Stress-Responsive cis-Acting Elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, K.; Takahashi, M.; Nishimoto, M.; Hiyama, T.B.; Higo, T.; Umehara, T.; Sakamoto, K.; Ito, T.; Yokoyama, S. Crystal structure of eukaryotic translation initiation factor 2B. Nature 2016, 531, 122–125. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci. Rep. 2016, 6, 32886. [Google Scholar] [CrossRef]
- Krishnamoorthy, T.; Pavitt, G.D.; Zhang, F.; Dever, T.E.; Hinnebusch, A.G. Tight Binding of the Phosphorylated Subunit of Initiation Factor 2 (eIF2) to the Regulatory Subunits of Guanine Nucleotide Exchange Factor eIF2B Is Required for Inhibition of Translation Initiation. Mol. Cell. Biol. 2001, 21, 5018–5030. [Google Scholar] [CrossRef]
- Pavitt, G.D.; Ramaiah, K.V.; Kimball, S.R.; Hinnebusch, A.G. eIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange. Genes Dev. 1998, 12, 514–526. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Cui, W.; Li, J.; Ron, D.; Sha, B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 423–428. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Brewer, J.W.; Alan Diehl, J.; Hendershot, L.M. Two Distinct Stress Signaling Pathways Converge Upon the CHOP Promoter During the Mammalian Unfolded Protein Response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef]
- Hai, T.; Hartman, M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 2001, 273, 1–11. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Wek, S.A.; McGrath, B.C.; Lu, D.; Hai, T.; Harding, H.P.; Wang, X.; Ron, D.; Cavener, D.R.; Wek, R.C. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell. Biol. 2004, 24, 1365–1377. [Google Scholar] [CrossRef]
- Gachon, F.; Gaudray, G.; Thébault, S.; Basbous, J.; Koffi, J.A.; Devaux, C.; Mesnard, J. The cAMP response element binding protein-2 (CREB-2) can interact with the C/EBP-homologous protein (CHOP). FEBS Lett. 2001, 502, 57–62. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Sekine, Y.; Zyryanova, A.; Crespillo-Casado, A.; Fischer, P.M.; Harding, H.P.; Ron, D. Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 2015, 348, 1027–1030. [Google Scholar] [CrossRef]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 2015, 4, 25719440. [Google Scholar] [CrossRef]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.-H.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2013, 2, e00498. [Google Scholar] [CrossRef] [PubMed]
- Zyryanova, A.F.; Weis, F.; Faille, A.; Akeel, A.A.; Crespillo-Casado, A.; Sekine, Y.; Harding, H.P.; Allen, F.; Parts, L.; Fromont, C.; et al. Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 2018, 359, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- Jousse, C.; Bruhat, A.; Carraro, V.; Urano, F.; Ferrara, M.; Ron, D.; Fafournoux, P. Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5’UTR. Nucleic Acids Res. 2001, 29, 4341–4351. [Google Scholar] [CrossRef]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of eIF2 Facilitates Ribosomal Bypass of an Inhibitory Upstream ORF to Enhance CHOP Translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef]
- Lee, Y.-Y.; Cevallos, R.C.; Jan, E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef]
- Haze, K.; Okada, T.; Yoshida, H.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 2001, 355, 19–28. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Zhu, C.; Johansen, F.E.; Prywes, R. Interaction of ATF6 and serum response factor. Mol. Cell. Biol. 1997, 17, 4957–4966. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable Binding of ATF6 to BiP in the Endoplasmic Reticulum Stress Response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of Disulfide Bridges Formed in the Luminal Domain of ATF6 in Sensing Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Kokame, K.; Kato, H.; Miyata, T. Identification of ERSE-II, a New cis—Acting Element Responsible for the ATF6-dependent Mammalian Unfolded Protein Response. J. Biol. Chem. 2001, 276, 9199–9205. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-Independent Activation of XBP1s and/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Endoplasmic Reticulum Stress-Induced Formation of Transcription Factor Complex ERSF Including NF-Y (CBF) and Activating Transcription Factors 6 and 6 That Activates the Mammalian Unfolded Protein Response. Mol. Cell. Biol. 2001, 21, 1239–1248. [Google Scholar] [CrossRef]
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. ATF6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell. Biol. 2000, 20, 5096–5106. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.-Y.; Kaufman, R.J. ATF6α Optimizes Long-Term Endoplasmic Reticulum Function to Protect Cells from Chronic Stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Shiu, R.P.C.; Pouyssegur, J.; Pastan, I. Glucose depletion accounts for the induction of two transformation-sensitive membrane proteins in Rous sarcoma virus-transformed chick embryo fibroblasts. Proc. Natl. Acad. Sci. USA 1977, 74, 3840–3844. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Nadanaka, S.; Okada, T.; Okawa, K.; Mori, K. Luminal domain of ATF6 alone is sufficient for sensing endoplasmic reticulum stress and subsequent transport to the Golgi apparatus. Cell Struct. Funct. 2011, 36, 35–47. [Google Scholar] [CrossRef]
- Gallagher, C.M.; Walter, P. Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. Elife 2016, 5, e11880. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef]
- Asada, R.; Kanemoto, S.; Kondo, S.; Saito, A.; Imaizumi, K. The signalling from endoplasmic reticulum-resident bZIP transcription factors involved in diverse cellular physiology. J. Biochem. 2011, 149, 507–518. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef]
- Chang, T.-K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between two branches of the unfolded protein response determines apoptotic cell fate. Mol. Cell 2018, 71, 629–636. [Google Scholar] [CrossRef]
- Byrd, A.E.; Aragon, I.V.; Brewer, J.W. MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J. Cell Biol. 2012, 196, 689–698. [Google Scholar] [CrossRef]
- Amin-Wetzel, N.; Saunders, R.A.; Kamphuis, M.J.; Rato, C.; Preissler, S.; Harding, H.P.; Ron, D. A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell 2017, 171, 1625–1637. [Google Scholar] [CrossRef]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK eIF2 kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.J.; Tang, N.; Wambach, M.; Barber, G.N.; Katze, M.G. The P58 cellular inhibitor complexes with the interferon-induced, double-stranded RNA-dependent protein kinase, PKR, to regulate its autophosphorylation and activity. J. Biol. Chem. 1996, 271, 1702–1707. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kang, S.-W.; Goodman, A.G.; Garrison, J.L.; Taunton, J.; Katze, M.G.; Kaufman, R.J.; Hegde, R.S. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 2007, 18, 3681–3691. [Google Scholar] [CrossRef] [PubMed]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1alpha-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef]
- Majumder, M.; Huang, C.; Snider, M.D.; Komar, A.A.; Tanaka, J.; Kaufman, R.J.; Krokowski, D.; Hatzoglou, M. A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Mol. Cell. Biol. 2012, 32, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of Grp78/BiP by Translational Block. J. Biol. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef]
- Starck, S.R.; Tsai, J.C.; Chen, K.; Shodiya, M.; Wang, L.; Yahiro, K.; Martins-Green, M.; Shastri, N.; Walter, P. Translation from the 5’ untranslated region shapes the integrated stress response. Science 2016, 351, aad3867. [Google Scholar] [CrossRef]
- Komar, A.A.; Gross, S.R.; Barth-Baus, D.; Strachan, R.; Hensold, J.O.; Goss Kinzy, T.; Merrick, W.C. Novel Characteristics of the Biological Properties of the Yeast Saccharomyces cerevisiae Eukaryotic Initiation Factor 2A. J. Biol. Chem. 2005, 280, 15601–15611. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.-P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.-P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 Cleavage of BID Is a Critical Apoptotic Signal Downstream of Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2008, 28, 3943–3951. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef]
- Glab, J.A.; Doerflinger, M.; Nedeva, C.; Jose, I.; Mbogo, G.W.; Paton, J.C.; Paton, A.W.; Kueh, A.J.; Herold, M.J.; Huang, D.C.; et al. DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 2017, 24, 944–950. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wang, H.-G. CHOP Is Involved in Endoplasmic Reticulum Stress-induced Apoptosis by Enhancing DR5 Expression in Human Carcinoma Cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Li, G.; Mongillo, M.; Chin, K.-T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α–mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Davies, E.; Madesh, M. Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 445–454. [Google Scholar] [CrossRef]
- Michalak, M.; Robert Parker, J.M.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Akiyama, M.; Liew, C.W.; Lu, S.; Hu, J.; Martinez, R.; Hambro, B.; Kennedy, R.T.; Kulkarni, R.N. X-Box Binding Protein 1 Is Essential for Insulin Regulation of Pancreatic—Cell Function. Diabetes 2013, 62, 2439–2449. [Google Scholar] [CrossRef]
- Bettigole, S.E.; Lis, R.; Adoro, S.; Lee, A.-H.; Spencer, L.A.; Weller, P.F.; Glimcher, L.H. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat. Immunol. 2015, 16, 829–837. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.-J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef]
- Elphick, D.A.; Mahida, Y.R. Paneth cells: Their role in innate immunity and inflammatory disease. Gut 2005, 54, 1802–1809. [Google Scholar] [CrossRef]
- Iwakoshi, N.N.; Lee, A.-H.; Vallabhajosyula, P.; Otipoby, K.L.; Rajewsky, K.; Glimcher, L.H. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 2003, 4, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.-H.; Qian, S.-B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.R.; Sukhdeo, K.; Protopopova, M.; Sinha, R.; Enos, M.; Carrasco, D.E.; Zheng, M.; Mani, M.; Henderson, J.; Pinkus, G.S.; et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007, 11, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Sandoval, T.A.; Chae, C.-S.; Chopra, S.; Tan, C.; Rutkowski, M.R.; Raundhal, M.; Chaurio, R.A.; Payne, K.K.; Konrad, C.; et al. IRE1α–XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 2018, 562, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef]
- Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef]
- Tavernier, S.J.; Osorio, F.; Vandersarren, L.; Vetters, J.; Vanlangenakker, N.; Van Isterdael, G.; Vergote, K.; De Rycke, R.; Parthoens, E.; van de Laar, L.; et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat. Cell Biol. 2017, 19, 698–710. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.-I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef]
- Kamimura, D.; Bevan, M.J. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J. Immunol. 2008, 181, 5433–5441. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Zheng, Z.; Chang, L.; Zhao, Y.-S.; Tan, C.; Dandekar, A.; Zhang, Z.; Lin, Z.; Gui, M.; Li, X.; et al. Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. EMBO J. 2013, 32, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.W.; Cui, D.; Arellano, J.; Dorweiler, B.; Harding, H.; Fitzgerald, K.A.; Ron, D.; Tabas, I. Adaptive suppression of the ATF4–CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat. Cell Biol. 2009, 11, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.W.; Kutzler, L.; Kimball, S.R.; Tabas, I. Toll-like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat. Cell Biol. 2012, 14, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Roulston, A.; Marcellus, R.C.; Branton, P.E. Viruses and apoptosis. Annu. Rev. Microbiol. 1999, 53, 577–628. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Speck, S.H.; Ganem, D. Viral Latency and Its Regulation: Lessons from the γ-Herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef]
- Bhende, P.M.; Dickerson, S.J.; Sun, X.; Feng, W.-H.; Kenney, S.C. X-Box-Binding Protein 1 Activates Lytic Epstein-Barr Virus Gene Expression in Combination with Protein Kinase, D.J. Virology 2007, 81, 7363–7370. [Google Scholar] [CrossRef]
- Matar, C.G.; Rangaswamy, U.S.; Wakeman, B.S.; Iwakoshi, N.; Speck, S.H. Murine gammaherpesvirus 68 reactivation from B cells requires IRF4 but not XBP-1. J. Virol. 2014, 88, 11600–11610. [Google Scholar] [CrossRef]
- Wilson, S.J.; Tsao, E.H.; Webb, B.L.J.; Ye, H.; Dalton-Griffin, L.; Tsantoulas, C.; Gale, C.V.; Du, M.-Q.; Whitehouse, A.; Kellam, P. X Box Binding Protein XBP-1s Transactivates the Kaposi’s Sarcoma—Associated Herpesvirus (KSHV) ORF50 Promoter, Linking Plasma Cell Differentiation to KSHV Reactivation from Latency. J. Virol. 2007, 81, 13578–13586. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Feng, J.; Harada, J.N.; Chanda, S.K.; Kenney, S.C.; Sun, R. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi’s sarcoma-associated herpesvirus. FEBS Lett. 2007, 581, 3485–3488. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Dalton-Griffin, L.; Wilson, S.J.; Kellam, P. X-box binding protein 1 contributes to induction of the Kaposi’s sarcoma-associated herpesvirus lytic cycle under hypoxic conditions. J. Virol. 2009, 83, 7202–7209. [Google Scholar] [CrossRef]
- Cai, Q.; Lan, K.; Verma, S.C.; Si, H.; Lin, D.; Robertson, E.S. Kaposi’s Sarcoma-Associated Herpesvirus Latent Protein LANA Interacts with HIF-1 To Upregulate RTA Expression during Hypoxia: Latency Control under Low Oxygen Conditions. J. Virol. 2006, 80, 7965–7975. [Google Scholar] [CrossRef]
- Hsiao, J.-R.; Chang, K.-C.; Chen, C.-W.; Wu, S.-Y.; Su, I.-J.; Hsu, M.-C.; Jin, Y.-T.; Tsai, S.-T.; Takada, K.; Chang, Y. Endoplasmic Reticulum Stress Triggers XBP-1-Mediated Up-regulation of an EBV Oncoprotein in Nasopharyngeal Carcinoma. Cancer Res. 2009, 69, 4461–4467. [Google Scholar] [CrossRef]
- Lee, D.Y.; Sugden, B. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 2008, 111, 2280–2289. [Google Scholar] [CrossRef]
- Chadburn, A.; Hyjek, E.M.; Tam, W.; Liu, Y.; Rengifo, T.; Cesarman, E.; Knowles, D.M. Immunophenotypic analysis of the Kaposi sarcoma herpesvirus (KSHV; HHV-8)-infected B cells in HIV+ multicentric Castleman disease (MCD). Histopathology 2008, 53, 513–524. [Google Scholar] [CrossRef]
- Hassman, L.M.; Ellison, T.J.; Kedes, D.H. KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. J. Clin. Investig. 2011, 121, 752–768. [Google Scholar] [CrossRef]
- Jenner, R.G.; Maillard, K.; Cattini, N.; Weiss, R.A.; Boshoff, C.; Wooster, R.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. USA 2003, 100, 10399–10404. [Google Scholar] [CrossRef] [PubMed]
- Kati, S.; Tsao, E.H.; Günther, T.; Weidner-Glunde, M.; Rothämel, T.; Grundhoff, A.; Kellam, P.; Schulz, T.F. Activation of the B-cell antigen receptor triggers reactivation of latent Kaposi’s Sarcoma-associated Herpesvirus. J. Virol. 2013, 87, 8004–8016. [Google Scholar] [CrossRef] [PubMed]
- Chadburn, A.; Said, J.; Gratzinger, D.; Chan, J.K.C.; de Jong, D.; Jaffe, E.S.; Natkunam, Y.; Goodlad, J.R. HHV8/KSHV-Positive Lymphoproliferative Disorders and the Spectrum of Plasmablastic and Plasma Cell Neoplasms: 2015 SH/EAHP Workshop Report-Part 3. Am. J. Clin. Pathol. 2017, 147, 171–187. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.; Chen, H.-S.; Hu, C.-C.A.; Lieberman, P.M. Deregulation of KSHV latency conformation by ER-stress and caspase-dependent RAD21-cleavage. PLoS Pathog. 2017, 13, e1006596. [Google Scholar] [CrossRef]
- Chen, H.-S.; De Leo, A.; Wang, Z.; Kerekovic, A.; Hills, R.; Lieberman, P.M. BET-Inhibitors Disrupt Rad21-Dependent Conformational Control of KSHV Latency. PLoS Pathog. 2017, 13, e1006100. [Google Scholar] [CrossRef]
- O’Flaherty, B.M.; Soni, T.; Wakeman, B.S.; Speck, S.H. The murine gammaherpesvirus immediate-early Rta synergizes with IRF4, targeting expression of the viral M1 superantigen to plasma cells. PLoS Pathog. 2014, 10, e1004302. [Google Scholar] [CrossRef]
- Clambey, E.T.; Virgin, H.W., 4th; Speck, S.H. Disruption of the murine gammaherpesvirus 68 M1 open reading frame leads to enhanced reactivation from latency. J. Virol. 2000, 74, 1973–1984. [Google Scholar] [CrossRef]
- Feng, J.; Gong, D.; Fu, X.; Wu, T.; Wang, J.; Chang, J.; Zhou, J.; Lu, G.; Wang, Y.; Sun, R. M1 of Murine Gamma-Herpesvirus 68 Induces Endoplasmic Reticulum Chaperone Production. Sci. Rep. 2015, 5, 17228. [Google Scholar] [CrossRef]
- Johnston, B.P.; Pringle, E.S.; McCormick, C. KSHV activates unfolded protein response sensors but suppresses downstream transcriptional responses to support lytic replication. PLoS Pathog. 2019, 15, e1008185. [Google Scholar] [CrossRef]
- Granato, M.; Romeo, M.A.; Tiano, M.S.; Santarelli, R.; Gonnella, R.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Bortezomib promotes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci. Rep. 2017, 7, 13052. [Google Scholar] [CrossRef]
- Saji, C.; Higashi, C.; Niinaka, Y.; Yamada, K.; Noguchi, K.; Fujimuro, M. Proteasome inhibitors induce apoptosis and reduce viral replication in primary effusion lymphoma cells. Biochem. Biophys. Res. Commun. 2011, 415, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.J.; Duran, E.M.; Kurtoglu, M.; Andreansky, S.; Lampidis, T.J.; Mesri, E.A. Activation of the unfolded protein response by 2-deoxy-D-glucose inhibits Kaposi’s sarcoma-associated herpesvirus replication and gene expression. Antimicrob. Agents Chemother. 2012, 56, 5794–5803. [Google Scholar] [CrossRef] [PubMed]
- Shigemi, Z.; Baba, Y.; Hara, N.; Matsuhiro, J.; Kagawa, H.; Watanabe, T.; Fujimuro, M. Effects of ER stress on unfolded protein responses, cell survival, and viral replication in primary effusion lymphoma. Biochem. Biophys. Res. Commun. 2016, 469, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef]
- Chandriani, S.; Ganem, D. Host Transcript Accumulation during Lytic KSHV Infection Reveals Several Classes of Host Responses. PLoS ONE 2007, 2, e811. [Google Scholar] [CrossRef]
- Abernathy, E.; Gilbertson, S.; Alla, R.; Glaunsinger, B. Viral Nucleases Induce an mRNA Degradation-Transcription Feedback Loop in Mammalian Cells. Cell Host Microbe 2015, 18, 243–253. [Google Scholar] [CrossRef]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A Comprehensive Annotation of the Kaposi’s Sarcoma-Associated Herpesvirus Genome Using Next-Generation Sequencing Reveals Novel Genomic and Functional Features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef]
- Kronstad, L.M.; Brulois, K.F.; Jung, J.U.; Glaunsinger, B.A. Dual Short Upstream Open Reading Frames Control Translation of a Herpesviral Polycistronic mRNA. PLoS Pathog. 2013, 9, e1003156. [Google Scholar] [CrossRef]
- Reinke, A.W.; Baek, J.; Ashenberg, O.; Keating, A.E. Networks of bZIP protein-protein interactions diversified over a billion years of evolution. Science 2013, 340, 730–734. [Google Scholar] [CrossRef]
- Lin, S.F.; Robinson, D.R.; Kung, H.J.; Miller, G. Kaposi’s sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J. Virol. 1999, 73, 1909–1917. [Google Scholar]
- Yang, W.-S.; Hsu, H.-W.; Campbell, M.; Cheng, C.-Y.; Chang, P.-C. K-bZIP Mediated SUMO-2/3 Specific Modification on the KSHV Genome Negatively Regulates Lytic Gene Expression and Viral Reactivation. PLoS Pathog. 2015, 11, e1005051. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Izumiya, Y.; Wu, C.Y.; Fitzgerald, L.D.; Campbell, M.; Ellison, T.J.; Lam, K.S.; Luciw, P.A.; Kung, H.J. Kaposi’s Sarcoma-associated Herpesvirus (KSHV) encodes a SUMO E3 ligase that is SIM-dependent and SUMO-2/3-specific. J. Biol. Chem. 2010, 285, 5266–5273. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Kobayashi, K.; Kim, K.Y.; Pochampalli, M.; Izumiya, C.; Shevchenko, B.; Wang, D.-H.; Huerta, S.B.; Martinez, A.; Campbell, M.; et al. Kaposi’s Sarcoma-Associated Herpesvirus K-Rta Exhibits SUMO-Targeting Ubiquitin Ligase (STUbL) Like Activity and Is Essential for Viral Reactivation. PLoS Pathog. 2013, 9, e1003506. [Google Scholar] [CrossRef] [PubMed]
- Giffin, L.; Yan, F.; Ben Major, M.; Damania, B. Modulation of Kaposi’s sarcoma-associated herpesvirus interleukin-6 function by hypoxia-upregulated protein 1. J. Virol. 2014, 88, 9429–9441. [Google Scholar] [CrossRef] [PubMed]
- Behnke, J.; Feige, M.J.; Hendershot, L.M. BiP and its nucleotide exchange factors Grp170 and Sil1: Mechanisms of action and biological functions. J. Mol. Biol. 2015, 427, 1589–1608. [Google Scholar] [CrossRef]
- Chen, D.; Xiang, Q.; Nicholas, J. Human herpesvirus 8 interleukin-6 interacts with calnexin cycle components and promotes protein folding. J. Virol. 2017, 91, e00965-17. [Google Scholar] [CrossRef]
- Hu, D.; Wang, V.; Yang, M.; Abdullah, S.; Davis, D.A.; Uldrick, T.S.; Polizzotto, M.N.; Veeranna, R.P.; Pittaluga, S.; Tosato, G.; et al. Induction of Kaposi’s Sarcoma-Associated Herpesvirus-Encoded Viral Interleukin-6 by X-Box Binding Protein 1. J. Virol. 2016, 90, 368–378. [Google Scholar] [CrossRef]
- Chang, P.-J.; Hung, C.-H.; Wang, S.-S.; Tsai, P.-H.; Shih, Y.-J.; Chen, L.-Y.; Huang, H.-Y.; Wei, L.-H.; Yen, J.-B.; Lin, C.-L.; et al. Identification and characterization of two novel spliced genes located in the orf47-orf46-orf45 gene locus of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2014, 88, 10092–10109. [Google Scholar] [CrossRef]
- Sanchez, V.; Sztul, E.; Britt, W.J. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-golgi-intermediate compartment. J. Virol. 2000, 74, 3842–3851. [Google Scholar] [CrossRef]
- Das, S.; Vasanji, A.; Pellett, P.E. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 2007, 81, 11861–11869. [Google Scholar] [CrossRef]
- Das, S.; Pellett, P.E. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 2011, 85, 5864–5879. [Google Scholar] [CrossRef] [PubMed]
- Karleusa, L.; Mahmutefendic, H.; Tomas, M.I.; Zagorac, G.B.; Lucin, P. Landmarks of endosomal remodeling in the early phase of cytomegalovirus infection. Virology 2018, 515, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Pierciey, F.J.; Maguire, T.G.; Alwine, J.C. PKR-Like Endoplasmic Reticulum Kinase Is Necessary for Lipogenic Activation during HCMV Infection. PLoS Pathog. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Terhune, S.; Torigoi, E.; Moorman, N.; Silva, M.; Qian, Z.; Shenk, T.; Yu, D. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 2007, 81, 3109–3123. [Google Scholar] [CrossRef]
- Qian, Z.; Xuan, B.; Gualberto, N.; Yu, D. The human cytomegalovirus protein pUL38 suppresses endoplasmic reticulum stress-mediated cell death independently of its ability to induce mTORC1 activation. J. Virol. 2011, 85, 9103–9113. [Google Scholar] [CrossRef]
- Xuan, B.; Qian, Z.; Torigoi, E.; Yu, D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 2009, 83, 3463–3474. [Google Scholar] [CrossRef]
- Siddiquey, M.N.A.; Zhang, H.; Nguyen, C.C.; Domma, A.J.; Kamil, J.P. The Human Cytomegalovirus Endoplasmic Reticulum-Resident Glycoprotein UL148 Activates the Unfolded Protein Response. J. Virol. 2018, 92, e00896-18. [Google Scholar] [CrossRef]
- Zhang, H.; Read, C.; Nguyen, C.C.; Siddiquey, M.N.A.; Shang, C.; Hall, C.M.; von Einem, J.; Kamil, J.P. A cytomegalovirus immunevasin triggers integrated stress response-dependent reorganization of the endoplasmic reticulum. bioRxiv 2019, 641068. [Google Scholar] [CrossRef]
- Nguyen, C.C.; Siddiquey, M.N.A.; Zhang, H.; Li, G.; Kamil, J.P. Human Cytomegalovirus Tropism Modulator UL148 Interacts with SEL1L, a Cellular Factor That Governs Endoplasmic Reticulum-Associated Degradation of the Viral Envelope Glycoprotein gO. J. Virol. 2018, 92, e00688-18. [Google Scholar] [CrossRef]
- Nguyen, C.C.; Domma, A.J.; Zhang, H.; Kamil, J.P. ER reorganization and intracellular retention of CD58 are functionally independent properties of the human cytomegalovirus ER resident glycoprotein UL148. J. Virol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wiertz, E.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef]
- Van den Boomen, D.J.H.; Timms, R.T.; Grice, G.L.; Stagg, H.R.; Skodt, K.; Dougan, G.; Nathan, J.A.; Lehner, P.J. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc. Natl. Acad. Sci. USA 2014, 111, 11425–11430. [Google Scholar] [CrossRef]
- Morito, D.; Nagata, K. Pathogenic Hijacking of ER-Associated Degradation: Is ERAD Flexible? Mol. Cell 2015, 59, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, B.; Iwakoshi, N.N.; Lilley, B.N.; Lee, A.-H.; Glimcher, L.H.; Ploegh, H.L. Human cytomegalovirus protein US11 provokes an unfolded protein response that may facilitate the degradation of class I major histocompatibility complex products. J. Virol. 2005, 79, 2768–2779. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Xuan, B.; Chapa, T.J.; Gualberto, N.; Yu, D. Murine Cytomegalovirus Targets Transcription Factor ATF4 To Exploit the Unfolded-Protein Response. J. Virol. 2012, 86, 6712–6723. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.; Burkhart, J.M.; Hinte, F.; Tirosh, B.; Mohr, H.; Zahedi, R.P.; Sickmann, A.; Ruzsics, Z.; Budt, M.; Brune, W. Cytomegalovirus Downregulates IRE1 to Repress the Unfolded Protein Response. PLoS Pathog. 2013, 9, e1003544. [Google Scholar] [CrossRef]
- Sam, M.D.; Evans, B.T.; Coen, D.M.; Hogle, J.M. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J. Virol. 2009, 83, 2996–3006. [Google Scholar] [CrossRef]
- Drori, A.; Messerle, M.; Brune, W.; Tirosh, B. Lack of XBP-1 impedes murine cytomegalovirus gene expression. PLoS ONE 2014, 9, e110942. [Google Scholar] [CrossRef]
- Hinte, F.; van Anken, E.; Tirosh, B.; Brune, W. Repression of Viral Gene Expression and Replication by the Unfolded Protein Response Effector XBP1u. bioRxiv 2019, 2, 763227. [Google Scholar]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Uemura, A.; Mori, K. pXBP1(U), a negative regulator of the unfolded protein response activator pXBP1(S), targets ATF6 but not ATF4 in proteasome-mediated degradation. Cell Struct. Funct. 2009, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Pierciey, F.J.; Alwine, J.C. Human Cytomegalovirus Induces the Endoplasmic Reticulum Chaperone BiP through Increased Transcription and Activation of Translation by Using the BiP Internal Ribosome Entry Site. J. Virol. 2010, 84, 11479–11486. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Maguire, T.G.; Paton, A.W.; Paton, J.C.; Alwine, J.C. The Endoplasmic Reticulum Chaperone BiP/GRP78 Is Important in the Structure and Function of the Human Cytomegalovirus Assembly Compartment. J. Virol. 2009, 83, 11421–11428. [Google Scholar] [CrossRef]
- Burnett, H.F.; Audas, T.E.; Liang, G.; Lu, R.R. Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones 2012, 17, 473–483. [Google Scholar] [CrossRef]
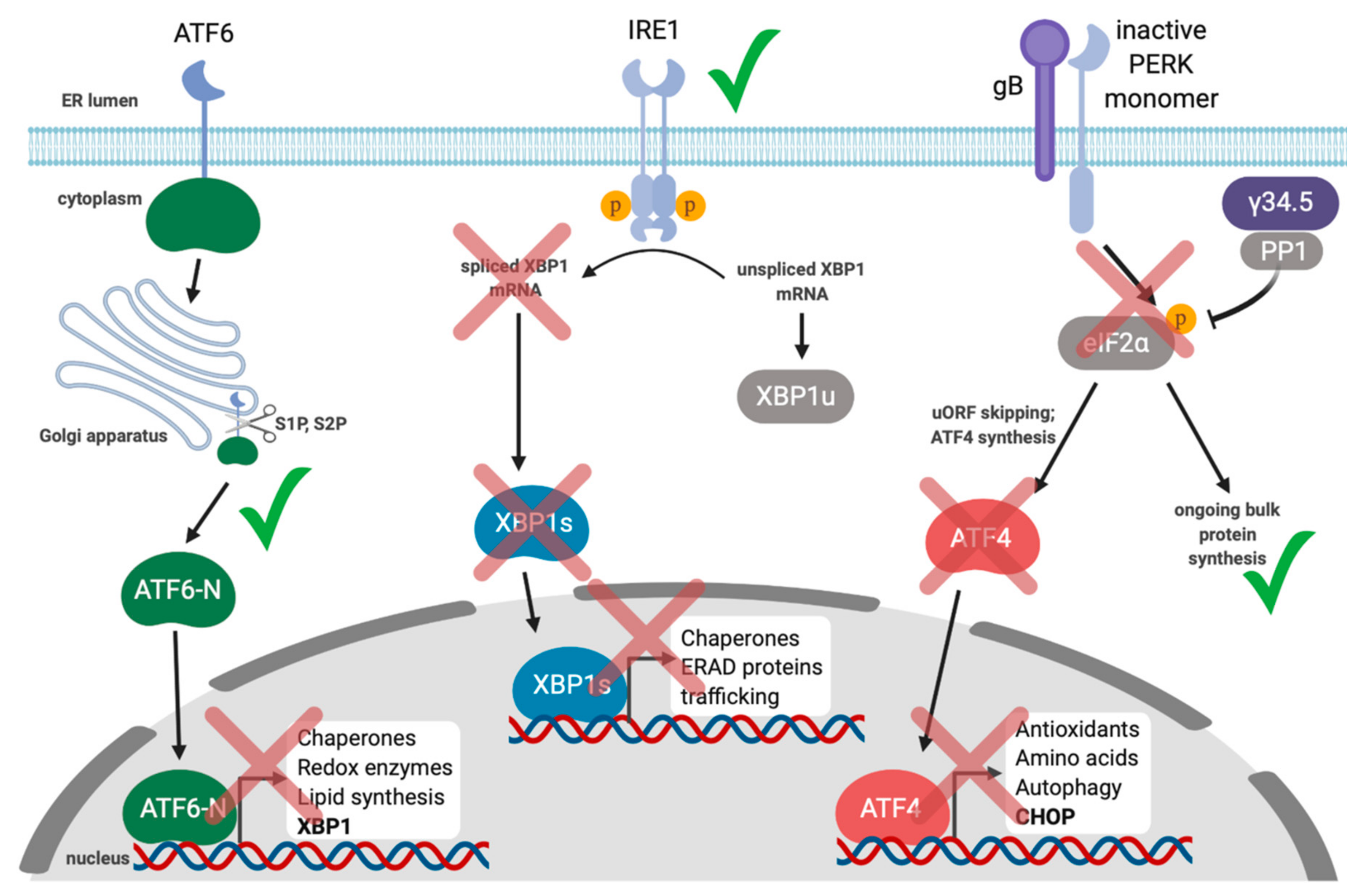
- Cheng, G.; Feng, Z.; He, B. Herpes Simplex Virus 1 Infection Activates the Endoplasmic Reticulum Resident Kinase PERK and Mediates eIF-2° Dephosphorylation by the 134.5 Protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef]
- Mulvey, M.; Arias, C.; Mohr, I. Resistance of mRNA Translation to Acute Endoplasmic Reticulum Stress-Inducing Agents in Herpes Simplex Virus Type 1-Infected Cells Requires Multiple Virus-Encoded Functions. J. Virol. 2006, 80, 7354–7363. [Google Scholar] [CrossRef]
- Mulvey, M.; Arias, C.; Mohr, I. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 2007, 81, 3377–3390. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Jiang, W.; Virgin, H.W., 4th; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.-L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.E.; Jackson, W.; Benetti, L.; Grose, C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J. Virol. 2011, 85, 9414–9424. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Grose, C. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J. Virol. 2014, 88, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Su, A.; Wang, H.; Li, Y.; Wang, X.; Chen, D.; Wu, Z. Opposite Roles of RNase and Kinase Activities of Inositol-Requiring Enzyme 1 (IRE1) on HSV-1 Replication. Viruses 2017, 9, 235. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnston, B.P.; McCormick, C. Herpesviruses and the Unfolded Protein Response. Viruses 2020, 12, 17. https://doi.org/10.3390/v12010017
Johnston BP, McCormick C. Herpesviruses and the Unfolded Protein Response. Viruses. 2020; 12(1):17. https://doi.org/10.3390/v12010017
Chicago/Turabian StyleJohnston, Benjamin P., and Craig McCormick. 2020. "Herpesviruses and the Unfolded Protein Response" Viruses 12, no. 1: 17. https://doi.org/10.3390/v12010017
APA StyleJohnston, B. P., & McCormick, C. (2020). Herpesviruses and the Unfolded Protein Response. Viruses, 12(1), 17. https://doi.org/10.3390/v12010017