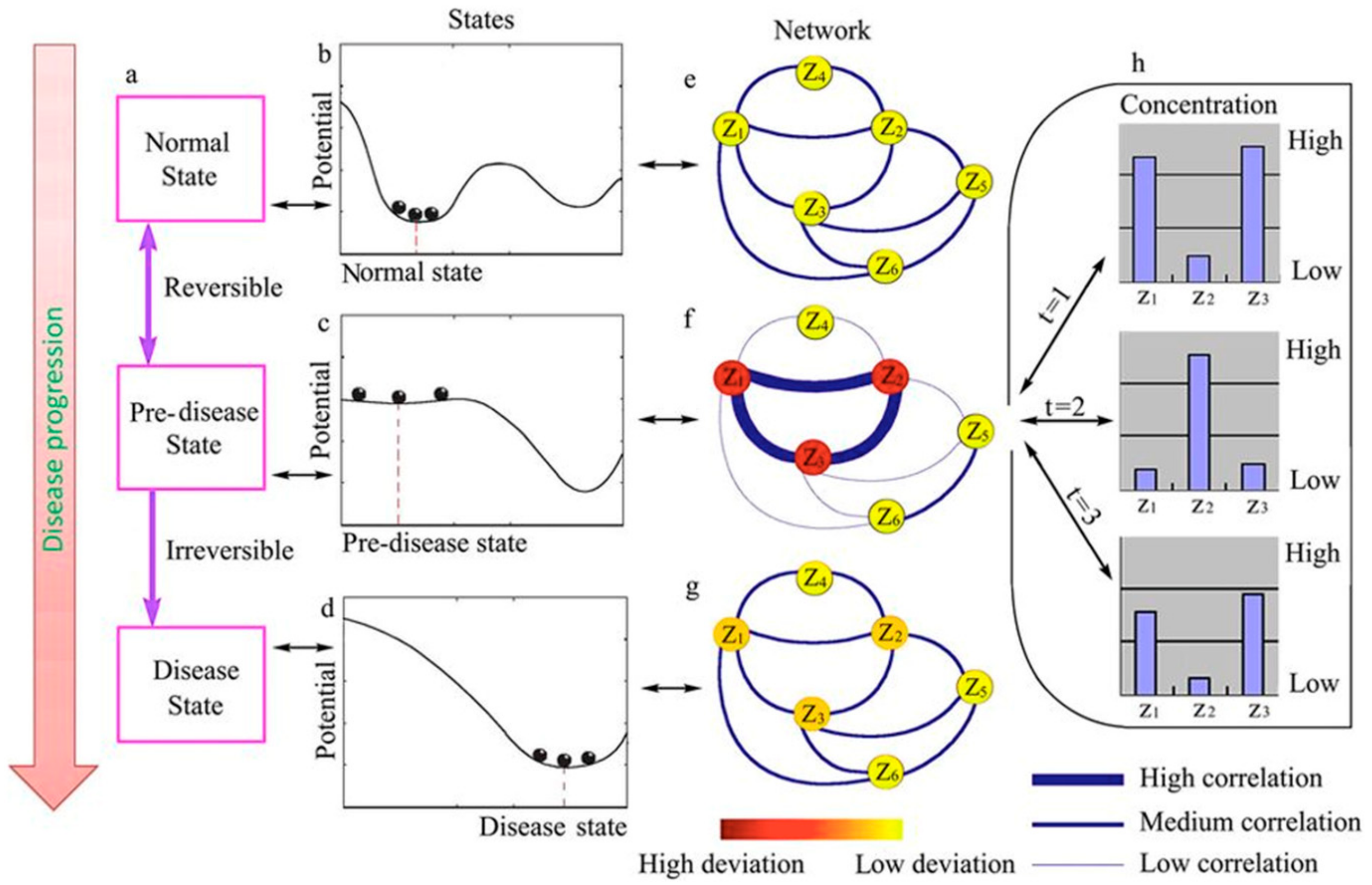
Identifying Early Warning Signals for the Sudden Transition from Mild to Severe Tobacco Etch Disease by Dynamical Network Biomarkers



Abstract
1. Introduction
2. Materials and Methods
2.1. Description of the Nicotiana tabacum—TEV Pathosystem and Transcriptomic Data
2.2. Identification of Genes with Biphasic Gene Expression Pattern along Disease Categories
2.3. Criteria for Identification and Statistical Evaluation of DNB
2.4. Functional Enrichment Analysis of DNBs
2.5. Network Analyses
3. Results and Discussion
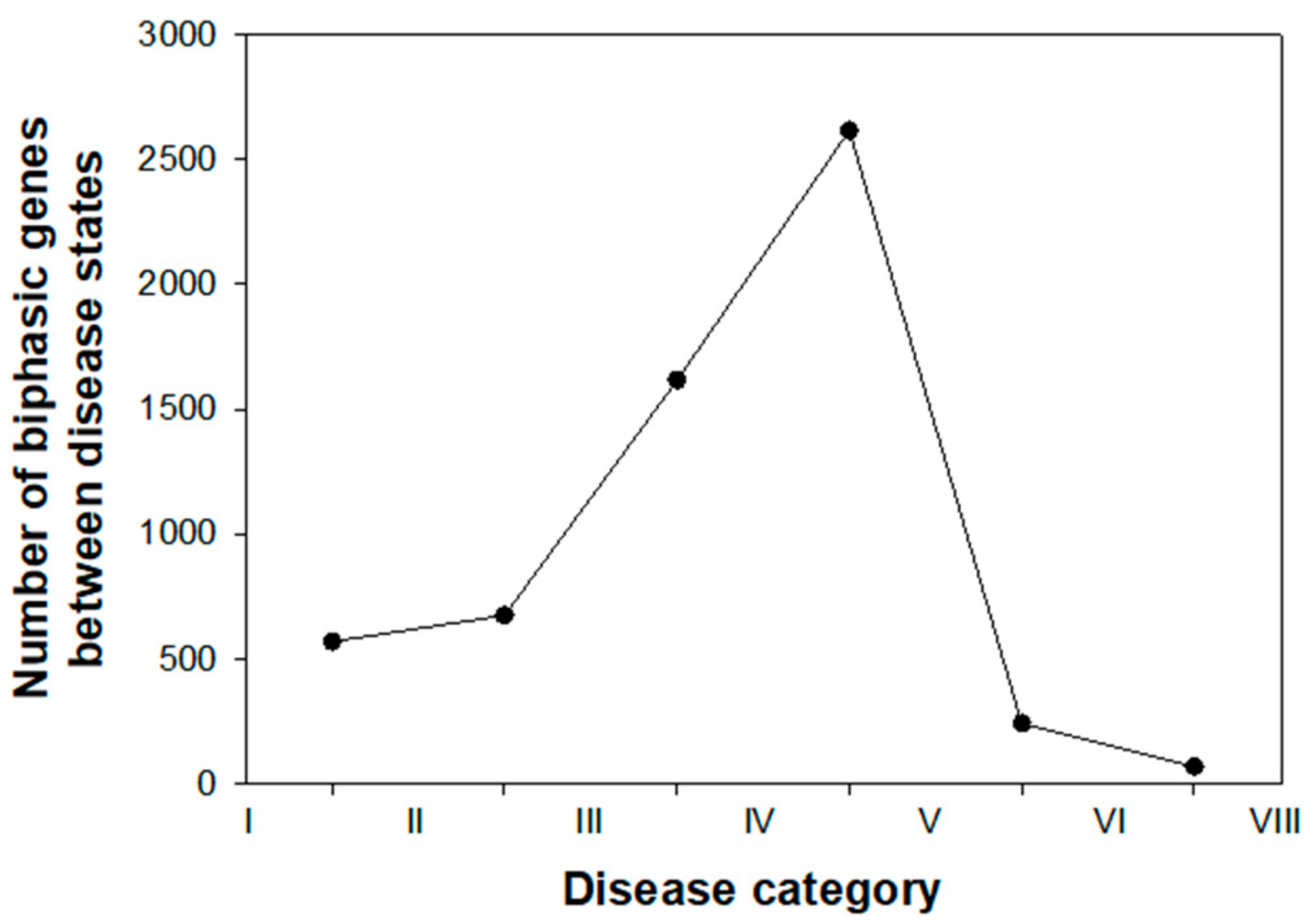
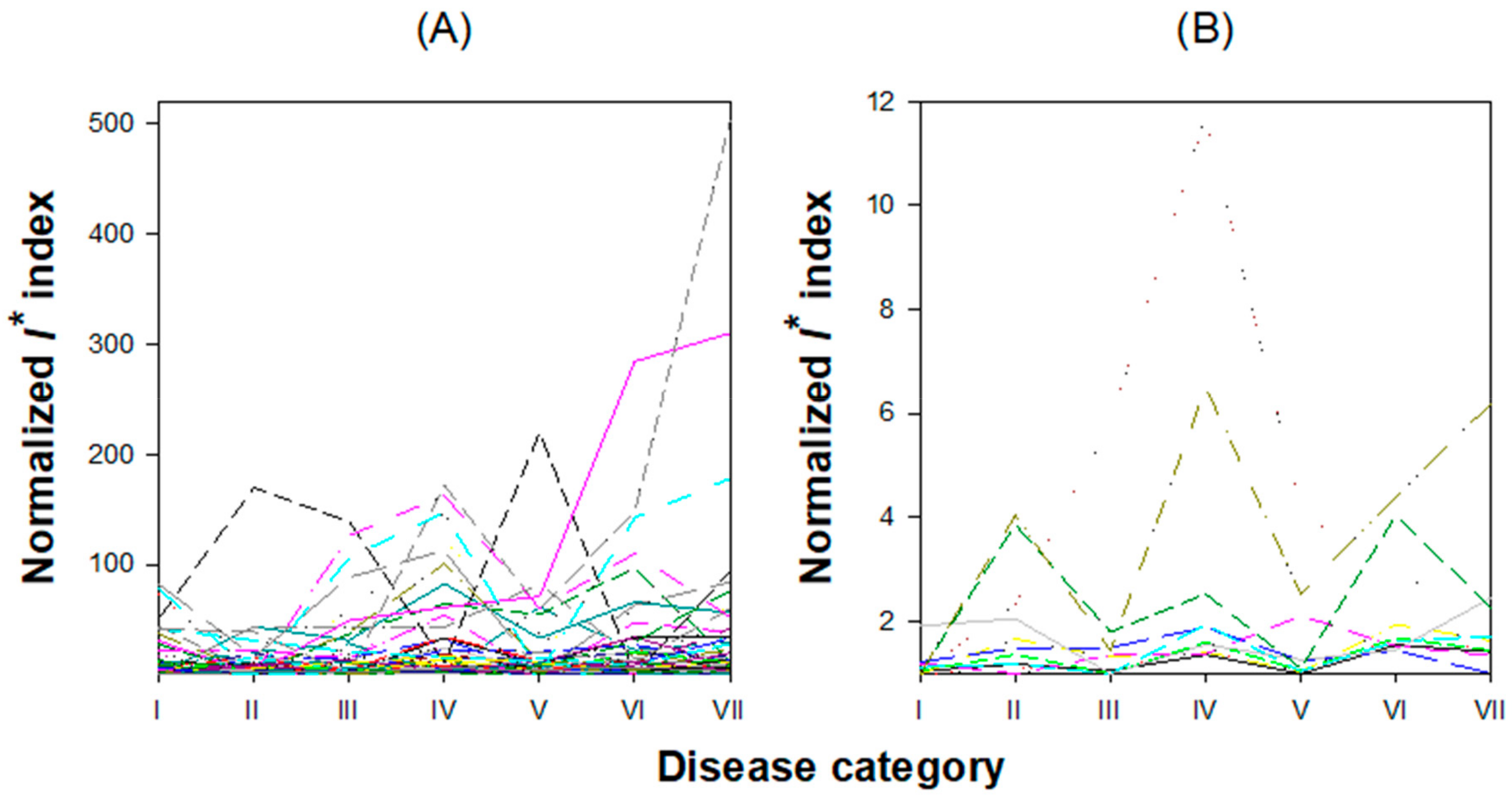
3.1. Distribution and Characterization of Genes Showing Biphasic Expression Profiles across Disease Categories
3.1.1. Characterization of Early Biphasic Genes
3.1.2. Characterization of Intermediate Biphasic Genes
3.1.3. Characterization of Late Biphasic Genes
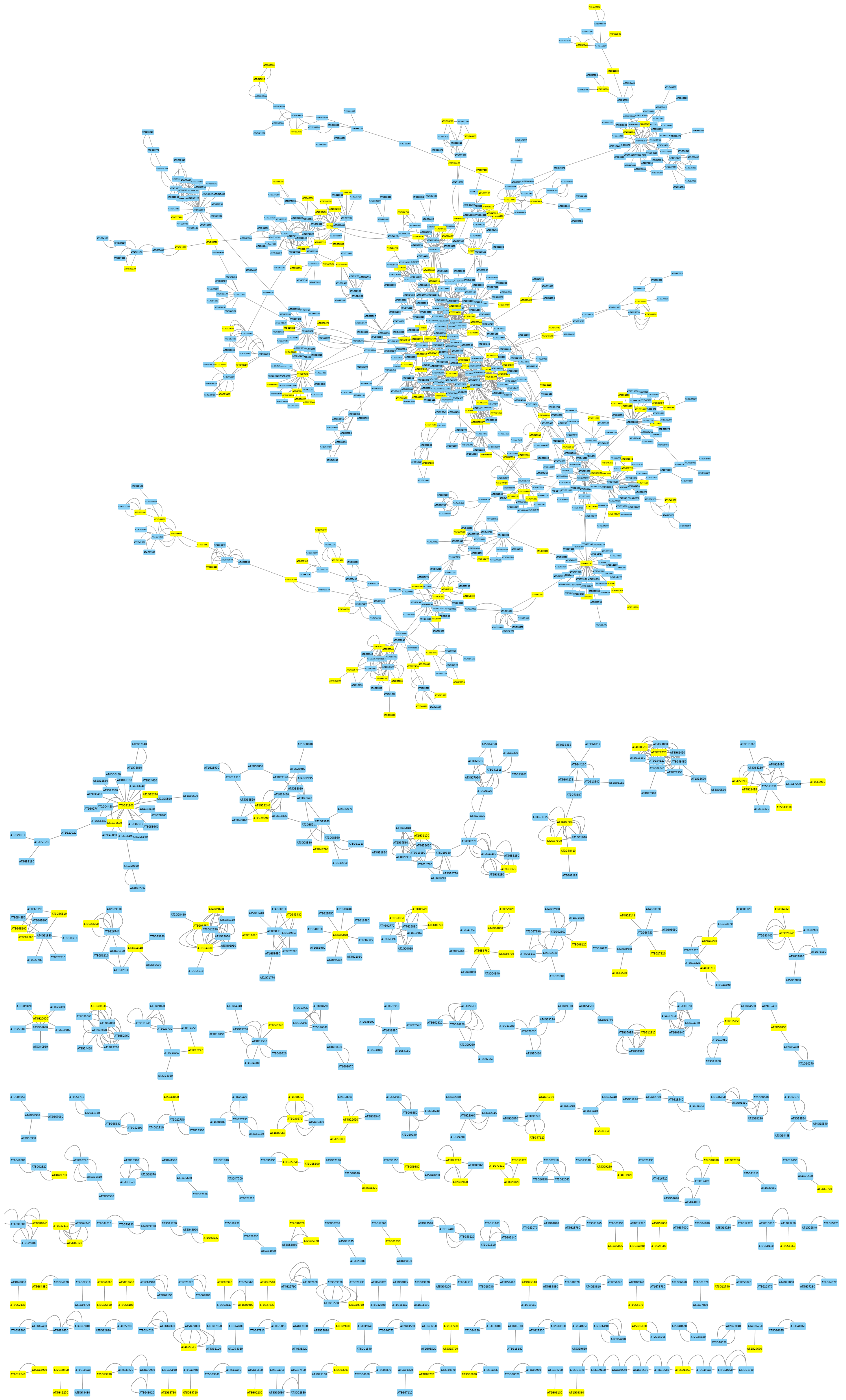
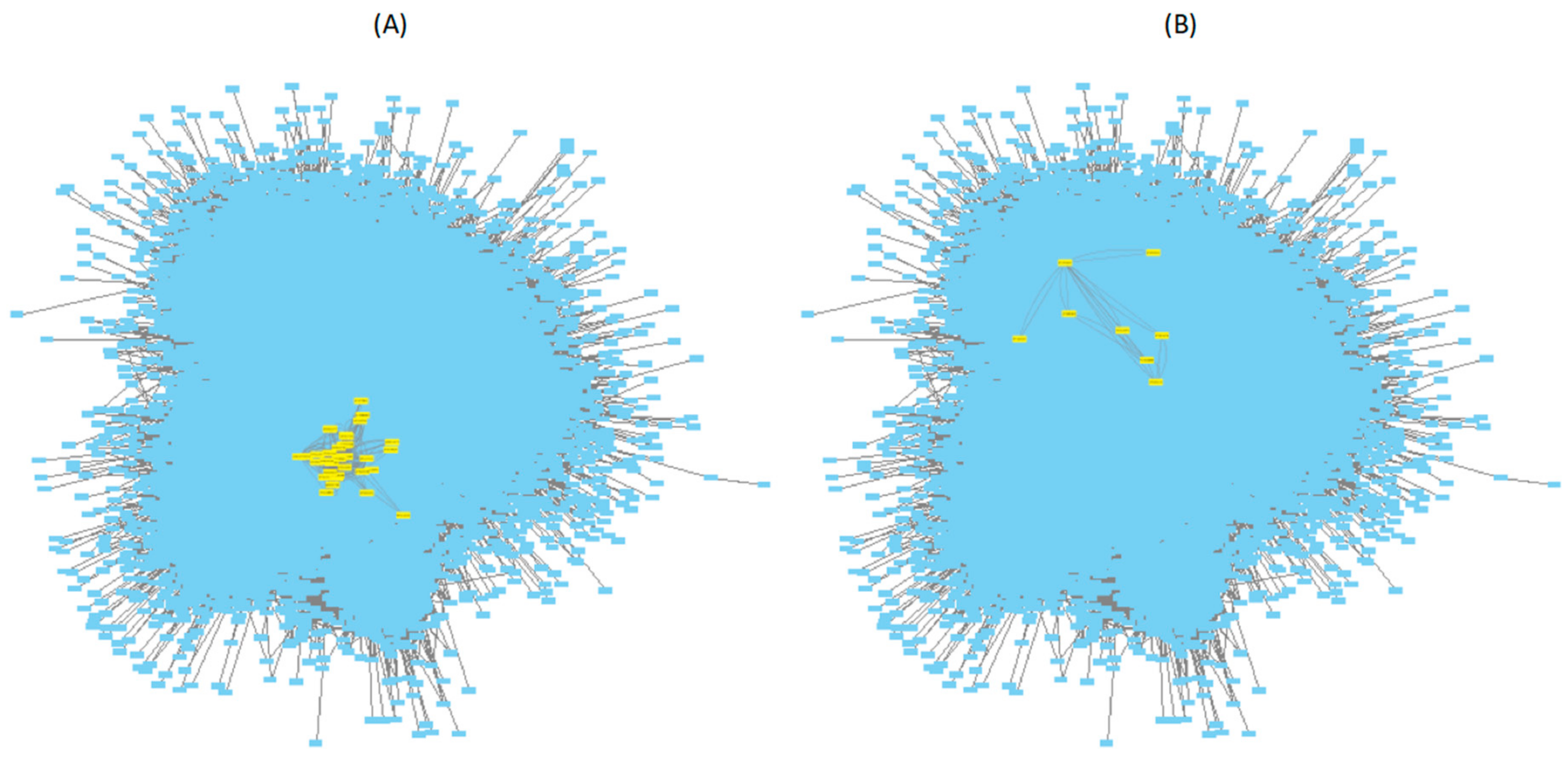
3.2. Mapping Biphasic Genes into A. thaliana AI-1 PPIN
Inference of PPIN-Based DNBs
3.3. Mapping Biphasic Genes into A. thaliana TRN
Inference of TRN-Based DNBs
3.4. Topological Properties of the DNBs Subnetworks
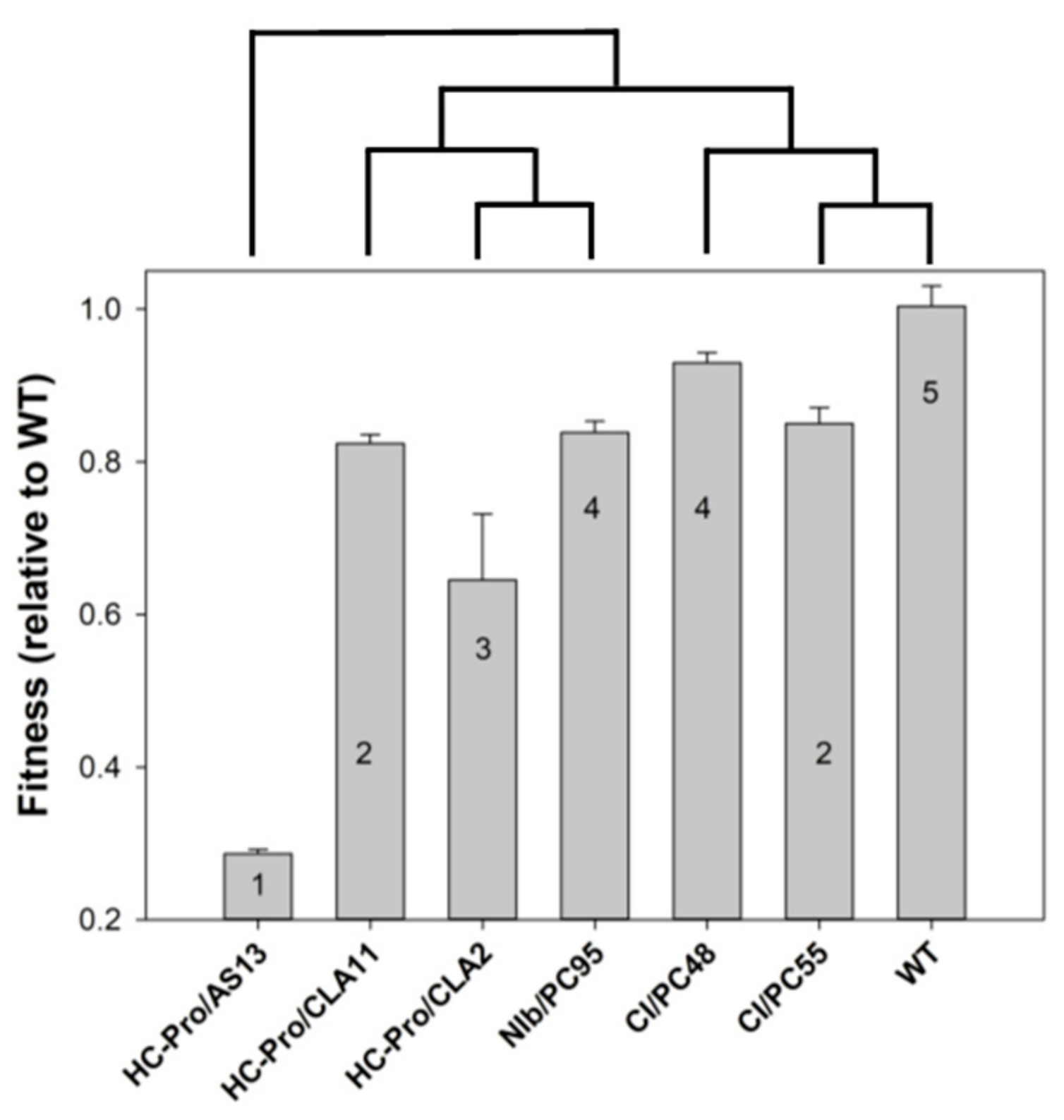
3.5. Mutations in Different Viral Proteins and Their Effect on the Likelihood of Disease Progression
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scheffer, M.; Bascompte, J.; Brock, W.A.; Brovkin, V.; Carpenter, S.R.; Dakos, V.; Held, H.; van Nes, E.H.; Rietkerk, M.; Sugihara, G. Early-warning signals for critical transitions. Nature 2009, 46, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, M.; Carpenter, S.R.; Lenton, T.M.; Bascompte, J.; Brock, W.; Dakos, V.; van de Koppel, J.; van de Leemput, I.A.; Levin, S.A.; van Nes, E.H.; et al. Anticipating critical transitions. Science 2012, 338, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.; Manrubia, S. Tipping points and early warning signals in the genomic composition of populations induced by environmental changes. Sci. Rep. 2015, 5, 9664. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, R. Catastrophe Theory for Scientists and Engineers; Dover: New York, NY, USA, 1981. [Google Scholar]
- Weissmann, H.; Shnerb, N.M. Predicting catastrophic shifts. J. Theor. Biol 2016, 397, 128–134. [Google Scholar] [CrossRef]
- Strogatz, S.H. Nonlinear Dynamics and Chaos: With Applications to Physics, Biology, Chemistry and Engineering; Perseus Books Publishing: Reading, MA, USA, 1994. [Google Scholar]
- Van Nes, E.H.; Scheffer, M. Slow recovery from perturbations as a generic indicator of a nearby catastrophic shift. Am. Nat. 2007, 169, 738–747. [Google Scholar] [CrossRef]
- Donangelo, R.; Fort, H.; Dakos, V.; Scheffer, M.; van Nes, E.H. Early warning signals for catastrophic shifts in ecosystems: Comparison between spatial and temporal indicators. Int. J. Bifurc. Chaos 2010, 20, 315–321. [Google Scholar] [CrossRef]
- Dutta, P.S.; Sharma, Y.; Abbott, K.C. Robustness of early warning signals for catastrophic and non-catastrophic transitions. Oikos 2018, 127, 1251–1263. [Google Scholar] [CrossRef]
- Chen, L.; Liu, R.; Liu, Z.P.; Li, M.; Aihara, K. Detecting early-warning signals for sudden deterioration of complex diseases by dynamical network biomarkers. Sci. Rep. 2012, 2, 342. [Google Scholar] [CrossRef]
- Huang, Y.; Zaas, A.K.; Rao, A.; Dobigeon, N.; Woolf, P.J.; Veldman, T.; Øien, N.C.; McClain, M.T.; Varkey, J.B.; Nicholson, B.; et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza A infection. PLoS Genet. 2011, 7, e1002234. [Google Scholar] [CrossRef]
- Liu, R.; Yu, X.; Liu, X.; Xu, D.; Aihara, K.; Chen, L. Identifying critical transitions of complex diseases based on a single simple. Bioinformatics 2014, 30, 1579–1586. [Google Scholar] [CrossRef]
- Liu, X.; Chang, X.; Liu, R.; Yu, X.; Chen, L.; Aihara, K. Quantifying critical states of complex diseases using single-sample dynamic network biomarkers. PLoS Comput. Biol. 2017, 13, e1005633. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, Z.; Zhu, Y.; Wang, J.; Mei, Y.; Cheng, Y. Pathway mapping and development of disease-specific biomarkers: Protein-based network biomarkers. J. Cell Mol. Med. 2015, 19, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Liu, R.; Li, Y.; Chen, L. Detecting critical state before phase transition of complex biological systems by hidden Markov model. Bioinformatics 2016, 32, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, Y.; Liu, X.; Liu, R.; Chen, L. Detecting the tipping points in a three-state model of complex diseases by temporal differential networks. J. Trans. Med. 2017, 15, 217. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Gelenbe, E. Anomaly detection in gene expression via stochastic models of gene regulatory networks. BMC Genom. 2009, 10, S26. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zeng, J.; Zhou, L.; Hu, C.; Yin, P.; Lin, X. A new strategy for analyzing time-series data using dynamic networks: Identifying prospective biomarkers of hepatocellular carcinoma. Sci. Rep. 2016, 6, 32448. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, M.; Liu, Z.P.; Wu, J.; Chen, L.; Aihara, K. Identifying critical transitions and their leading biomolecular networks in complex diseases. Sci. Rep. 2012, 2, 813. [Google Scholar] [CrossRef]
- Liu, R.; Aihara, K.; Chen, L. Dynamical network biomarkers for identifying critical transitions and their driving networks of biological processes. Quant. Biol. 2013, 1, 105–114. [Google Scholar] [CrossRef]
- Liu, R.; Wang, X.; Aihara, K.; Chen, L. Early diagnosis of complex diseases by molecular biomarkers, network biomarkers, and dynamical network biomarkers. Med. Res. Rev. 2014, 34, 455–478. [Google Scholar] [CrossRef]
- Liu, R.; Chen, P.; Aihara, K.; Chen, L. Identifying early-warning signals of critical transitions with strong noise by dynamical network markers. Sci. Rep. 2015, 5, 17501. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Ji, H.; Aihara, K.; Chen, L. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 2016, 44, e164. [Google Scholar] [CrossRef] [PubMed]
- Torshizi, A.D.; Petzold, L. Sparse pathway-induced dynamic network biomarker discovery for early warning signal detection in complex diseases. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 14, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Vafaee, F. Using multi-objective optimization to identify dynamical network biomarkers as early-warning signals of complex diseases. Sci. Rep. 2016, 6, 22023. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, G.; Chen, L. Prediction and early diagnosis of complex diseases by edge-network. Bioinformatics 2014, 30, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zeng, T.; Li, G. Integrative enrichment analysis: A new computational method to detect dysregulated pathways in heterogeneous samples. BMC Genom. 2015, 16, 918. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, J.; Sun, S.; Zhou, X.; Zeng, T.; Chen, L. Individual-specific edge-network analysis for disease prediction. Nucleic Acids Res. 2017, 45, e170. [Google Scholar] [CrossRef]
- Zeng, T.; Zhang, C.C.; Zhang, W.; Liu, R.; Liu, J.; Chen, L. Deciphering early development of complex disease by progressive module network. Methods 2014, 67, 334–343. [Google Scholar] [CrossRef]
- Cervera, H.; Ambrós, S.; Bernet, G.P.; Rodrigo, G.; Elena, S.F. Viral fitness correlates with the magnitude and direction of the perturbation induced in the host’s transcriptome: The tobacco etch potyvirus-tobacco case study. Mol. Biol. Evol. 2018, 35, 1599–1615. [Google Scholar] [CrossRef]
- Carrasco, P.; de la Iglesia, F.; Elena, S.F. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in tobacco etch virus. J. Virol. 2007, 81, 12979–12984. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Martín, S.; Daròs, J.A.; Elena, S.F. From hypo- to hypersuppression: Effect of amino acid substitutions on the RNA-silencing activity of the tobacco etch potyvirus HC-Pro. Genetics 2008, 180, 1039–1049. [Google Scholar] [CrossRef]
- Fernández-Pozo, N.; Menda, N.; Edwards, J.D.; Saha, S.; Tecle, I.Y.; Strickler, S.R.; Bombarely, A.; Fisher-York, T.; Pujar, A.; Foerster, H.; et al. The Sol Genomics network (SGN)—From genotype to phenotype to breeding. Nucleic Acids Res. 2015, 43, D1036–D1041. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; García-Hernández, M.; et al. The Arabidopsis information resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from gene ontology and reactome pathways, and data analysis tools enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Statist. 2001, 29, 1165–1188. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 11. [Google Scholar] [CrossRef]
- Arabidopsis Interactome Mapping Consortium. Evidence for network evolution in an Arabidopsis interactome map. Science 2011, 333, 601–607. [Google Scholar] [CrossRef]
- Carrera, J.; Rodrigo, G.; Jaramillo, A.; Elena, S.F. Reverse-engineering the Arabidopsis thaliana transcriptional network under hanging environmental conditions. Genome Biol. 2009, 10, R96. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Munné-Bosch, S. Ethylene response factors: A key regulatory hub in hormone and stress signaling. Plant Physiol. 2015, 169, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.A.; Kumari, P.H.; Kumar, G.S.; Mohanalatha, C.; Kishor, P.B.K. Osmotin: A plant sentinel and a possible agonist of mammalian adiponectin. Front. Plant Sci. 2015, 6, 163. [Google Scholar]
- Thalmann, M.; Santelia, D. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 2017, 214, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Doyle, E.A.; Lane, A.M.; Sides, J.M.; Mudgett, M.B.; Monroe, J.D. An α-amylase (At4g25000) in Arabidopsis leaves is secreted and induced by biotic and abiotic stress. Plant Cell Environ. 2007, 30, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Mudgil, Y.; Shiu, S.H.; Stone, S.L.; Salt, J.N.; Goring, D.R. A large complement of the predicted Arabidopsis ARM repeat proteins are members of the U-box E3 ubiquitin ligase family. Plant Physiol. 2004, 134, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Dreher, K.; Callis, J. Ubiquitin, hormones and biotic stress in plants. Ann. Bot. 2007, 99, 787–822. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, Y. Posttranslationally modified small-peptide signaling in plants. Annu. Rev. Plant Biol. 2014, 65, 385–413. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M. The rhomboid-like superfamily: Molecular mechanisms and biological roles. Annu Rev. Cell Dev. Biol. 2014, 30, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, P.; Shanklin, J.; Shah, J.; Whittle, J.; Klessing, D.F. A fatty acid desaturase modulates the activation of defense signaling pathways in plants. Proc. Natl. Acad. Sci. USA 2001, 98, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, X.; Sun, C.; Rong, J. Identification and expression profiling of the Regulator of Chromosome Condensation 1 (RCC1) gene family in Gossypium hirsutum L. under abiotic stress and hormone treatment. Int. J. Mol. Sci. 2019, 20, 1727. [Google Scholar] [CrossRef]
- Ascencio-Ibáñez, J.T.; Sozzani, R.; Lee, T.J.; Chu, T.M.; Wolfinger, R.D.; Cella, R.; Hanley-Bowdoin, L. Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol. 2008, 148, 436–454. [Google Scholar] [CrossRef]
- Champion, A.; Kreis, M.; Mockaitis, K.; Picaud, A.; Henry, Y. Arabidopsis kinome: After the casting. Funct. Integr. Genom. 2004, 4, 163–187. [Google Scholar] [CrossRef]
- Takahashi, N.; Kuroda, H.; Kuromori, T.; Hirayama, T.; Seki, M.; Shinozaki, K.; Shimada, H.; Matsui, M. Expression and interaction analysis of Arabidopsis Skp1-related genes. Plant Cell Physiol. 2004, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Michard, E.; Lima, P.T.; Borges, F.; Silva, A.C.; Portes, M.T.; Carvalho, J.E.; Gilliham, M.; Liu, L.H.; Obermeyer, G.; Feijó, J.A. Glutamate receptor-like genes form Ca2+ channels in pollen tubes and are regulated by pistil D-serine. Science 2011, 6028, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Yang, T.; Poovaiah, B.W. Calcium signaling-mediated plant response to cold stress. Int. J. Mol. Sci. 2018, 19, 3896. [Google Scholar] [CrossRef]
- Zhou, H.; Duan, H.; Liu, Y.; Sun, X.; Zhao, J.; Lin, H. Patellin protein family functions in plant development and stress response. J. Plant Physiol. 2019, 234–235, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Peiro, A.; Izquierdo-García, A.; Sánchez-Navarro, J.A.; Pallás, V.; Mulet, J.M.; Aparicio, F. Patellins 3 and 6, two members of the plant patellin family, interact with the movement protein of alfalfa mosaic virus and interfere with viral movement. Mol. Plant Pathol. 2014, 15, 881–891. [Google Scholar] [CrossRef]
- Chen, K.; Fan, B.; Du, L.; Chen, Z. Activation of hypersensitive cell death by pathogen-induced receptor-like protein kinases from Arabidopsis. Plant Mol. Biol. 2004, 56, 271–283. [Google Scholar] [CrossRef]
- Suzuki, N.; Bajad, S.; Shuman, J.; Shulaev, V.; Mittler, R. The transcriptional co-activator MBF1c is a key regulator of thermotolerance in Arabidopsis thaliana. J. Biol. Chem. 2008, 283, 9269–9275. [Google Scholar] [CrossRef]
- Wang, X.; Du, Y.; Yu, D. Trehalose phosphate synthase 5-dependent trehalose metabolism modulates basal defense responses in Arabiodopsis thaliana. J. Integr. Plant Biol. 2019, 61, 509–527. [Google Scholar] [CrossRef]
- Sánchez-Fernández, R.; Davies, T.G.; Coleman, J.O.; Rea, P.A. The Arabidopsis thaliana ABC protein superfamily, a complete inventory. J. Biol. Chem. 2001, 276, 30231–30244. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.; Xie, B.; Hong, Z.; Yang, Q. Arabidopsis NUCLEOSTEMIN-LIKE 1 (NSN1) regulates cell cycling potentially by cooperating with nucleosome assembly protein AtNAP1;1. BMC Plant Biol. 2018, 18, 99. [Google Scholar] [CrossRef]
- Reumann, S.; Babujee, L.; Ma, C.; Wienkoop, S.; Siemsen, T.; Antonicelli, G.E.; Rasche, N.; Lüder, F.; Weckwerth, W.; Jahn, O. Proteome analysis of Arabidopsis leaf peroxisomes reveals novel targeting peptides, metabolic pathways, and defense mechanisms. Plant Cell 2007, 19, 3170–3193. [Google Scholar] [CrossRef] [PubMed]
- Crocco, C.D.; Ocampo, G.G.; Ploschuk, E.L.; Mantese, A.; Botto, J.F. Heterologous expression of AtBBX21 enhances the rate of photosynthesis and alleviates photoinhibition in Solanum tuberosum. Plant Physiol. 2018, 177, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Palik, Y.; Yang, S.; Choi, G. Phytochrome regulates translation of mRNA in the cytosol. Proc. Natl. Acad. Sci. USA 2012, 109, 1335–13340. [Google Scholar]
- Kakizaki, T.; Kitashiba, H.; Zou, Z.; Li, F.; Fukino, N.; Ohara, T.; Nishio, T.; Ishida, M. A 2-oxoglutarate-dependent dioxygenase mediates the bioshynthesis of glucoraphasatin in radish. Plant Physiol. 2017, 173, 1583–1593. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Wang, M.; Zheng, Q.; Shen, Q.; Guo, S. The critical role of potassium in plant stress response. Int. J. Mol. Sci. 2013, 14, 7370–7390. [Google Scholar] [CrossRef]
- Ballut, L.; Drucker, M.; Pugnière, M.; Cambon, F.; Blanc, S.; Roquet, F.; Candresse, T.; Schmid, H.P.; Nicolas, P.; Gall, O.L.; et al. HcPro, a multifunctional protein encoded by a plant RNA virus, targets the 20S proteasome and affects its enzymatic activities. J. Gen. Virol. 2005, 86, 2595–2603. [Google Scholar] [CrossRef]
- Pogorelko, G.V.; Mokryakova, M.; Fursova, O.V.; Abdeeva, I.; Piruzian, E.S.; Bruskin, S.A. Characterization of three Arabidopsis thaliana immunophilin genes involved in the plant defense response against Pseudomonas syringae. Gene 2014, 538, 12–22. [Google Scholar] [CrossRef]
- Scranton, M.A.; Yee, A.; Park, S.Y.; Walling, L.L. Plant leucine aminopeptidases moonlight as molecular chaperones to alleviate stress-induced damage. J. Biol. Chem. 2012, 287, 18408–18417. [Google Scholar] [CrossRef]
- Chen, R.; Sun, S.; Wang, C.; Li, Y.; Liang, Y.; An, F.; Li, C.; Dong, H.; Yang, X.; Zhang, J.; et al. The Arabidopsis PARAQUAT RESISTANT 2 gene encodes an S-nitrosogluthatione reductase that is a key regulator of cell death. Cell Res. 2009, 19, 1377–1387. [Google Scholar] [CrossRef]
- De Chassey, B.; Navratil, V.; Tafforeau, L.; Hiet, M.S.; Aublin-Gex, A.; Agaugué, S.; Meiffren, G.; Pradezynski, F.; Faria, B.F.; Chantier, T.; et al. Hepatitis C virus infection protein network. Mol. Syst. Biol. 2008, 4, 230. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, G.; Carrera, J.; Ruiz-Ferrer, V.; del Toro, F.J.; Llave, C.; Voinnet, O.; Elena, S.F. A meta-analysis reveals the commonalities and differences in Arabidopsis thaliana response to different viral pathogens. PLoS ONE 2012, 7, e40526. [Google Scholar] [CrossRef] [PubMed]
- Bosque, G.; Folch-Fortuny, A.; Picó, J.; Ferrer, A.; Elena, S.F. Topology analysis and visualization of Potyvirus protein-protein interaction network. BMC Syst. Biol. 2014, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barceló, C.; Daròs, J.A.; Elena, S.F. HC-Pro hypo- and hypersuppressor mutants: Differences in viral siRNA accumulation in vivo and siRNA binding activity in vitro. Arch. Virol. 2010, 155, 251–254. [Google Scholar] [CrossRef]
- Liu, X.; Liu, R.; Zhao, X.M.; Chen, L. Detecting early-warning signals of type 1 diabetes and its leading biomolecular networks by dynamical network biomarkers. BMC Med. Genom. 2013, 6, S8. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Property | PPIN | PPINDNB | TRN | TRNDNB 1 |
| Shortest path | 3.529 ± 0.739 | 3.650 ± 0.990 * | 3.476 ± 0.274 | 3.355 ± 0.216 * |
| Betweenness centrality | 0.023 ± 0.388 | 0.015 ± 0.104 | 1.144 ± 3.739 × 10−4 | 2.612 ± 7.300 × 10−4 |
| Closeness centrality | 0.292 ± 0.094 | 0.308 ± 0.158 | 0.289 ± 0.022 | 0.299 ± 0.019 * |
| Clustering coefficient | 0.138 ± 0.198 | 0.152 ± 0.242 | 0.213 ± 0.137 | 0.184 ± 0.107 * |
| Degree | 20.138 ± 31.148 | 20.523 ± 41.789 | 58.905 ± 60.034 | 90.513 ± 85.910 * |
| Eccentricity | 7.544 ± 1.300 | 7.530 ± 1.859 | 120.526 ± 82.141 | 5.930 ± 0.443 * |
| Neighborhood connectivity | 65.840 ± 73.970 | 38.605 ± 45.927 * | 58.898 ± 60.027 | 98.598 ± 53.418 * |
| Topological coefficient | 0.161 ± 0.178 | 0.195 ± 0.222 | 0.096 ± 0.083 | 0.065 ± 0.050 * |
| Critical exponent degree distribution 2 | −2.749 ± 0.399 | −2.043 ± 0.366 * | −3.225 ± 0.174 | −2.512 ± 0.210 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarazona, A.; Forment, J.; Elena, S.F. Identifying Early Warning Signals for the Sudden Transition from Mild to Severe Tobacco Etch Disease by Dynamical Network Biomarkers. Viruses 2020, 12, 16. https://doi.org/10.3390/v12010016
Tarazona A, Forment J, Elena SF. Identifying Early Warning Signals for the Sudden Transition from Mild to Severe Tobacco Etch Disease by Dynamical Network Biomarkers. Viruses. 2020; 12(1):16. https://doi.org/10.3390/v12010016
Chicago/Turabian StyleTarazona, Adrián, Javier Forment, and Santiago F. Elena. 2020. "Identifying Early Warning Signals for the Sudden Transition from Mild to Severe Tobacco Etch Disease by Dynamical Network Biomarkers" Viruses 12, no. 1: 16. https://doi.org/10.3390/v12010016
APA StyleTarazona, A., Forment, J., & Elena, S. F. (2020). Identifying Early Warning Signals for the Sudden Transition from Mild to Severe Tobacco Etch Disease by Dynamical Network Biomarkers. Viruses, 12(1), 16. https://doi.org/10.3390/v12010016