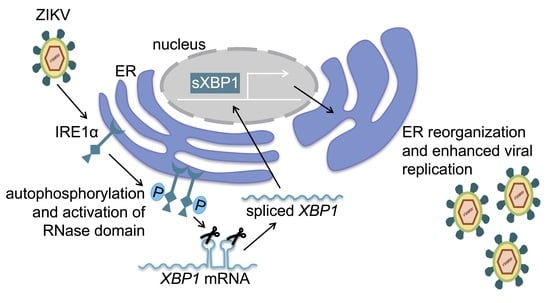
IRE1α Promotes Zika Virus Infection via XBP1

 and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cells and Viruses
2.3. Expression Analysis
2.4. Immunofluorescence Microscopy
2.5. Western Blot
2.6. Plaque Assay
2.7. Mouse Model of ZIKV Infection
2.8. Statistics
3. Results
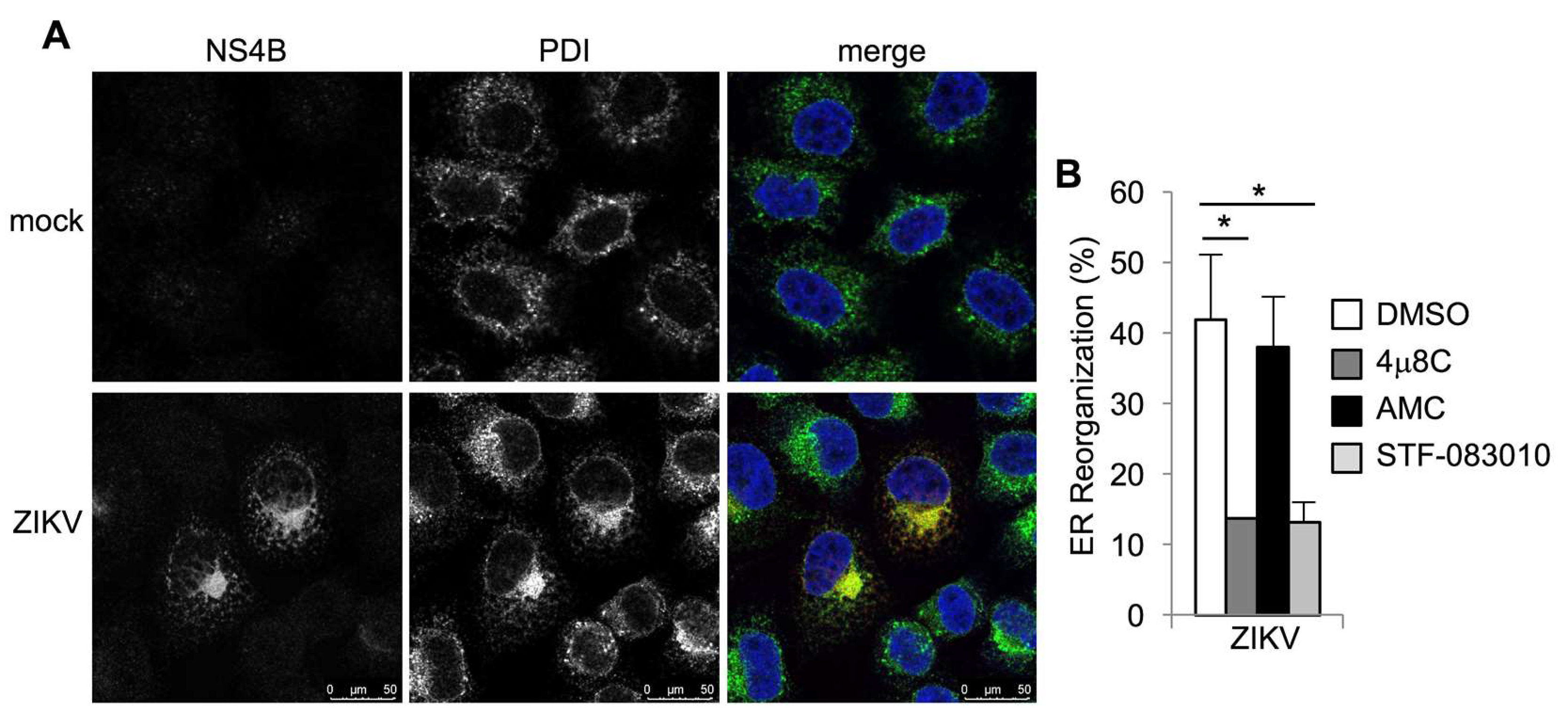
3.1. Zika Virus Infection Stimulates IRE1α Activation and Induction of XBP1 Targets
3.2. IRE1α Inhibitors Prevent Zika-Induced Cell Death
3.3. IRE1α and XBP1 Promote Zika Virus Replication in Cultured Cells
3.4. IRE1α and XBP1 Promote Zika Virus Infection in Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wikan, N.; Smith, D.R. Zika virus: History of a newly emerging arbovirus. Lancet. Infect. Dis. 2016, 16, e119–e126. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, M.K.; Carletti, T.; Marcello, A. Cellular Targets for the Treatment of Flavivirus Infections. Front. Cell. Infect. Microbiol. 2018, 8, 398. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Flaviviridae Replication Organelles: Oh, What a Tangled Web We Weave. Annu. Rev. Virol. 2015, 2, 289–310. [Google Scholar] [CrossRef]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef]
- Blazquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martin-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. [Google Scholar] [CrossRef]
- Li, S.; Kong, L.; Yu, X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit. Rev. Microbiol. 2015, 41, 150–164. [Google Scholar] [CrossRef]
- Karagoz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2019, 11, a033886. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Coelho, D.S.; Domingos, P.M. Physiological roles of regulated Ire1 dependent decay. Front. Genet. 2014, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef]
- Fink, S.L.; Jayewickreme, T.R.; Molony, R.D.; Iwawaki, T.; Landis, C.S.; Lindenbach, B.D.; Iwasaki, A. IRE1alpha promotes viral infection by conferring resistance to apoptosis. Sci. Signal. 2017, 10, 482. [Google Scholar] [CrossRef]
- Pena, J.; Harris, E. Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 2011, 286, 14226–14236. [Google Scholar] [CrossRef]
- Yu, C.Y.; Hsu, Y.W.; Liao, C.L.; Lin, Y.L. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 2006, 80, 11868–11880. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Sen, U.; Vrati, S. Regulated IRE1-dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J. Gen. Virol. 2014, 95, 71–79. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef]
- Ambrose, R.L.; Mackenzie, J.M. ATF6 signaling is required for efficient West Nile virus replication by promoting cell survival and inhibition of innate immune responses. J. Virol. 2013, 87, 2206–2214. [Google Scholar] [CrossRef]
- Carletti, T.; Zakaria, M.K.; Faoro, V.; Reale, L.; Kazungu, Y.; Licastro, D.; Marcello, A. Viral priming of cell intrinsic innate antiviral signaling by the unfolded protein response. Nat. Commun. 2019, 10, 3889. [Google Scholar] [CrossRef]
- Medigeshi, G.R.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; Defilippis, V.; Fruh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 2007, 81, 10849–10860. [Google Scholar] [CrossRef]
- Yu, C.; Achazi, K.; Niedrig, M. Tick-borne encephalitis virus triggers inositol-requiring enzyme 1 (IRE1) and transcription factor 6 (ATF6) pathways of unfolded protein response. Virus Res. 2013, 178, 471–477. [Google Scholar] [CrossRef]
- Tan, Z.; Zhang, W.; Sun, J.; Fu, Z.; Ke, X.; Zheng, C.; Zhang, Y.; Li, P.; Liu, Y.; Hu, Q.; et al. ZIKV infection activates the IRE1-XBP1 and ATF6 pathways of unfolded protein response in neural cells. J. Neuroinflammation 2018, 15, 275. [Google Scholar] [CrossRef]
- Gladwyn-Ng, I.; Cordon-Barris, L.; Alfano, C.; Creppe, C.; Couderc, T.; Morelli, G.; Thelen, N.; America, M.; Bessieres, B.; Encha-Razavi, F.; et al. Stress-induced unfolded protein response contributes to Zika virus-associated microcephaly. Nat. Neurosci. 2018, 21, 63–71. [Google Scholar] [CrossRef]
- Hou, S.; Kumar, A.; Xu, Z.; Airo, A.M.; Stryapunina, I.; Wong, C.P.; Branton, W.; Tchesnokov, E.; Gotte, M.; Power, C.; et al. Zika Virus Hijacks Stress Granule Proteins and Modulates the Host Stress Response. J. Virol. 2017, 91, 16. [Google Scholar] [CrossRef]
- Govero, J.; Esakky, P.; Scheaffer, S.M.; Fernandez, E.; Drury, A.; Platt, D.J.; Gorman, M.J.; Richner, J.M.; Caine, E.A.; Salazar, V.; et al. Zika virus infection damages the testes in mice. Nature 2016, 540, 438–442. [Google Scholar] [CrossRef]
- Gao, W.; Li, W.; Xiao, T.; Liu, X.S.; Kaelin, W.G., Jr. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL-/- clear cell renal carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1027–1032. [Google Scholar] [CrossRef]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; Van den Pol, A.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef]
- Shaily, S.; Upadhya, A. Zika virus: Molecular responses and tissue tropism in the mammalian host. Rev. Med Virol. 2019, 29, e2050. [Google Scholar] [CrossRef]
- Van Schadewijk, A.; Van’t Wout, E.F.; Stolk, J.; Hiemstra, P.S. A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell Stress Chaperones 2012, 17, 275–279. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, B.; Ye, L.; Kong, L.; Jing, W.; Yang, X.; Wu, Z.; Ye, L. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 2005, 43, 529–536. [Google Scholar]
- Ambrose, R.L.; Mackenzie, J.M. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev. 2017, 31, 1417–1438. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef]
- Lee, A.H.; Chu, G.C.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. Embo J. 2005, 24, 4368–4380. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. Cytoarchitecture of Zika virus infection in human neuroblastoma and Aedes albopictus cell lines. Virology 2017, 501, 54–62. [Google Scholar] [CrossRef]
- Monel, B.; Compton, A.A.; Bruel, T.; Amraoui, S.; Burlaud-Gaillard, J.; Roy, N.; Guivel-Benhassine, F.; Porrot, F.; Genin, P.; Meertens, L.; et al. Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. Embo J. 2017, 36, 1653–1668. [Google Scholar] [CrossRef]
- Diamond, M.S. Mechanisms of evasion of the type I interferon antiviral response by flaviviruses. J. Interf. Cytok. Res. 2009, 29, 521–530. [Google Scholar] [CrossRef]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef]
- Uraki, R.; Hwang, J.; Jurado, K.A.; Householder, S.; Yockey, L.J.; Hastings, A.K.; Homer, R.J.; Iwasaki, A.; Fikrig, E. Zika virus causes testicular atrophy. Sci. Adv. 2017, 3, e1602899. [Google Scholar] [CrossRef]
- Ma, W.; Li, S.; Ma, S.; Jia, L.; Zhang, F.; Zhang, Y.; Zhang, J.; Wong, G.; Zhang, S.; Lu, X.; et al. Zika Virus Causes Testis Damage and Leads to Male Infertility in Mice. Cell 2016, 167, 1511–1524. [Google Scholar] [CrossRef]
- Miner, J.J.; Sene, A.; Richner, J.M.; Smith, A.M.; Santeford, A.; Ban, N.; Weger-Lucarelli, J.; Manzella, F.; Ruckert, C.; Govero, J.; et al. Zika Virus Infection in Mice Causes Panuveitis with Shedding of Virus in Tears. Cell Rep. 2016, 16, 3208–3218. [Google Scholar] [CrossRef]
- Li, S.; Ye, L.; Yu, X.; Xu, B.; Li, K.; Zhu, X.; Liu, H.; Wu, X.; Kong, L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology 2009, 391, 257–264. [Google Scholar] [CrossRef]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F.; et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [Google Scholar] [CrossRef]
- Laguesse, S.; Creppe, C.; Nedialkova, D.D.; Prevot, P.P.; Borgs, L.; Huysseune, S.; Franco, B.; Duysens, G.; Krusy, N.; Lee, G.; et al. A Dynamic Unfolded Protein Response Contributes to the Control of Cortical Neurogenesis. Dev. Cell 2015, 35, 553–567. [Google Scholar] [CrossRef]
- Yockey, L.J.; Jurado, K.A.; Arora, N.; Millet, A.; Rakib, T.; Milano, K.M.; Hastings, A.K.; Fikrig, E.; Kong, Y.; Horvath, T.L.; et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 2018, 3, 19. [Google Scholar] [CrossRef]
- Sutarjono, B. Can We Better Understand How Zika Leads to Microcephaly? A Systematic Review of the Effects of the Zika Virus on Human Brain Organoids. J. Infect. Dis. 2019, 219, 734–745. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef] [PubMed]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef]
- Tang, C.H.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolpikova, E.P.; Tronco, A.R.; Den Hartigh, A.B.; Jackson, K.J.; Iwawaki, T.; Fink, S.L. IRE1α Promotes Zika Virus Infection via XBP1. Viruses 2020, 12, 278. https://doi.org/10.3390/v12030278
Kolpikova EP, Tronco AR, Den Hartigh AB, Jackson KJ, Iwawaki T, Fink SL. IRE1α Promotes Zika Virus Infection via XBP1. Viruses. 2020; 12(3):278. https://doi.org/10.3390/v12030278
Chicago/Turabian StyleKolpikova, Elena P., Ana R. Tronco, Andreas B. Den Hartigh, Konner J. Jackson, Takao Iwawaki, and Susan L. Fink. 2020. "IRE1α Promotes Zika Virus Infection via XBP1" Viruses 12, no. 3: 278. https://doi.org/10.3390/v12030278
APA StyleKolpikova, E. P., Tronco, A. R., Den Hartigh, A. B., Jackson, K. J., Iwawaki, T., & Fink, S. L. (2020). IRE1α Promotes Zika Virus Infection via XBP1. Viruses, 12(3), 278. https://doi.org/10.3390/v12030278