To Go or Stay: The Development, Benefit, and Detriment of Tissue-Resident Memory CD8 T Cells during Central Nervous System Viral Infections


Abstract
:1. Introduction
2. CD8 T cell Response to Viral Infections of the Brain
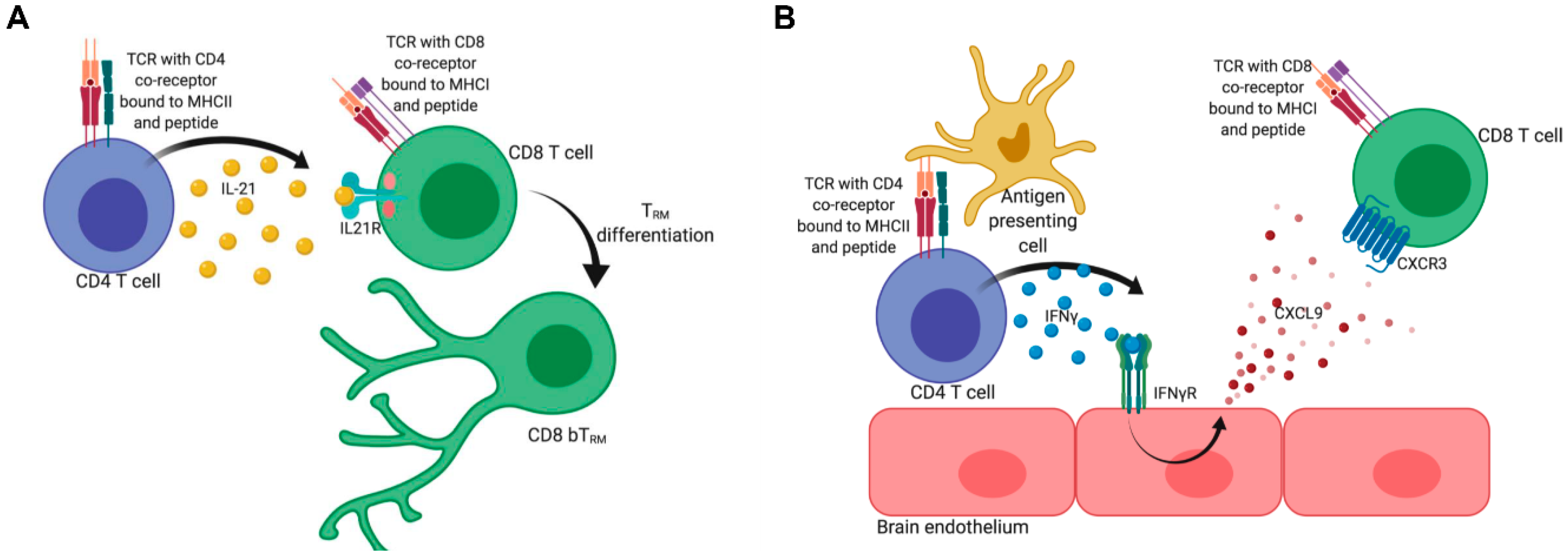
3. Development of CD4 and CD8 TRM
4. Phenotype and Transcriptomes of CD8 TRM
5. PD-1 Expression on CD8 bTRM
6. Factors Influencing the Differentiation of CD8 bTRM
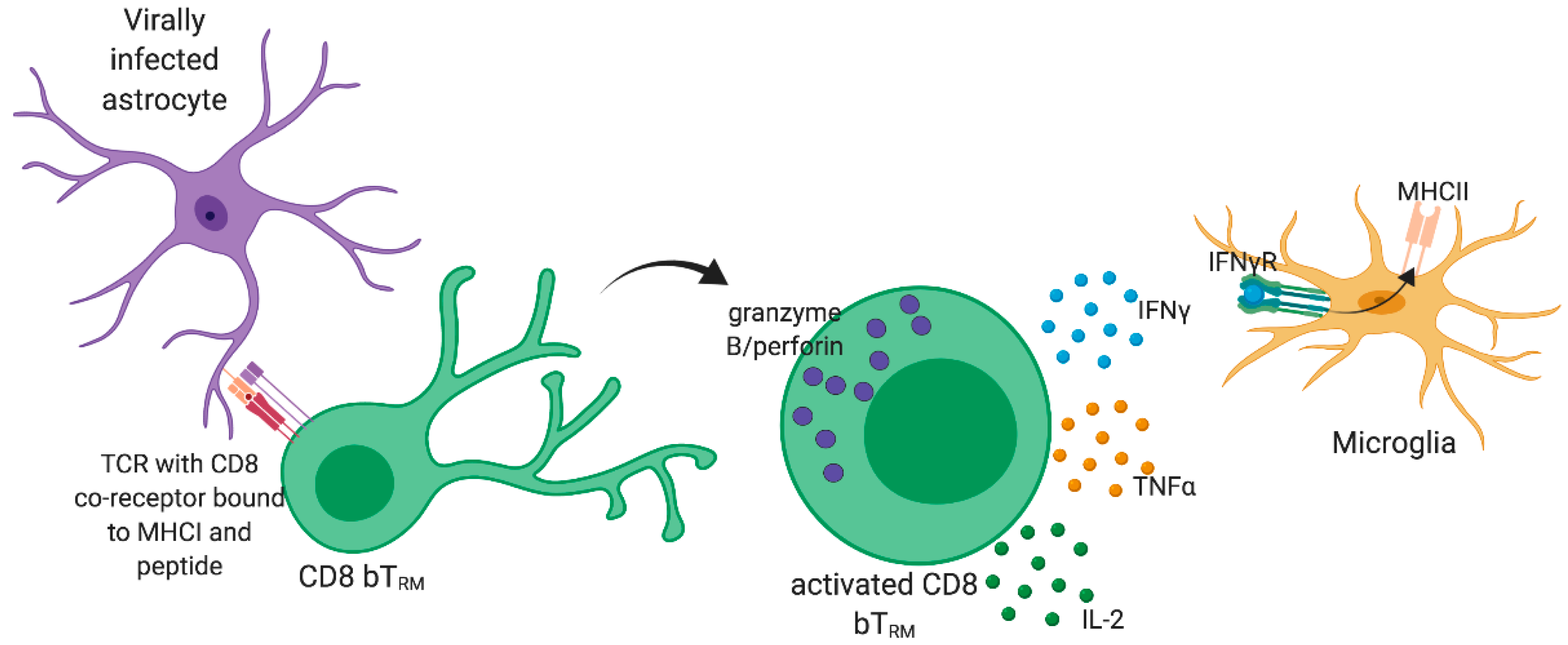
7. Importance of CD8 bTRM in the Control of Virus Reinfection and Viral Latency in the CNS
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Zinselmeyer, B.H.; Corps, K.N.; McGavern, D.B. In vivo dynamics of innate immune sentinels in the CNS. Intravital 2012, 1, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Invest. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Schnell, M.J.; Rall, G.F. Keeping it in check: chronic viral infection and antiviral immunity in the brain. Nat. Rev. Neurosci. 2016, 17, 766–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinchington, P.R.; Leger, A.J.; Guedon, J.M.; Hendricks, R.L. Herpes simplex virus and varicella zoster virus, the house guests who never leave. Herpesviridae 2012, 3. [Google Scholar] [CrossRef]
- Parra, B.; Hinton, D.R.; Marten, N.W.; Bergmann, C.C.; Lin, M.T.; Yang, C.S.; Stohlman, S.A. IFN-γ is required for viral clearance from central nervous system oligodendroglia. J. Immunol. 1999, 162, 1641–1647. [Google Scholar] [PubMed]
- Klein, R.S.; Hunter, C.A. Protective and pathological immunity during central nervous system infections. Immunity 2017, 46, 891–909. [Google Scholar] [CrossRef]
- Byrnes, A.; Durbin, J.; Griffin, D. Control of Sindbis virus infection by antibody in interferon-deficient mice. J. Virol. 2000, 74, 3905–3908. [Google Scholar] [CrossRef]
- Savarin, C.; Bergmann, C.C. Fine tuning the cytokine storm by IFN and IL-10 following neurotropic coronavirus encephalomyelitis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Wakim, L.M.; Woodward-Davis, A.; Bevan, M.J. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc. Natl. Acad. Sci. USA 2010, 107, 17872–17879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soung, A.; Klein, R.S. Viral encephalitis and neurologic diseases: focus on astrocytes. Trends Mol. Med. 2018, 24, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Garber, C.; Funk, K.E.; Salimi, H.; Soung, A.; Kanmogne, M.; Manivasagam, S.; Agner, S.; Cain, M. Neuroinflammation during RNA viral infections. Annu. Rev. Immunol. 2019, 37, 73–95. [Google Scholar] [CrossRef]
- Korn, T.; Kallies, A. T cell responses in the central nervous system. Nat. Rev. Immunol. 2017, 17, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Du Pasquier, R.A.; Clark, K.W.; Smith, P.S.; Joseph, J.T.; Mazullo, J.M.; De Girolami, U.; Letvin, N.L.; Koralnik, I.J. JCV-specific cellular immune response correlates with a favorable clinical outcome in HIV-infected individuals with progressive multifocal leukoencephalopathy. J. Neurovirol. 2001, 7, 318–322. [Google Scholar] [PubMed]
- Durrant, D.M.; Daniels, B.P.; Klein, R.S. IL-1R1 signaling regulates CXCL12-mediated T cell localization and fate within the central nervous system during west nile virus encephalitis. J. Immunol. 2014, 193, 4095–4106. [Google Scholar] [CrossRef]
- Scarpazza, C.; Prosperini, L.; De Rossi, N.; Moiola, L.; Sormani, M.P.; Gerevini, S.; Capra, R.; Italian PML Group. To do or not to do? Plasma exchange and timing of steroid administration in progressive multifocal leukoencephalopathy. Ann. Neurol. 2017, 82, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Ifergan, I.; Kebir, H.; Alvarez, J.I.; Marceau, G.; Bernard, M.; Bourbonnière, L.; Poirier, J.; Duquette, P.; Talbot, P.J.; Arbour, N.; et al. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on α4 integrin. Brain 2011, 134, 3560–3577. [Google Scholar] [CrossRef]
- Mockus, T.E.; Shwetank; Lauver, M.D.; Ren, H.M.; Netherby, C.S.; Salameh, T.; Kawasawa, Y.I.; Yue, F.; Broach, J.R.; Lukacher, A.E. CD4 T cells control development and maintenance of brain-resident CD8 T cells during polyomavirus infection. PLoS Pathog. 2018, 14, e1007365. [Google Scholar] [CrossRef]
- Griffin, D.E. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003, 3, 493–502. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Immune surveillance of the CNS following infection and injury. Trends Immunol. 2015, 36, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Chesler, D.A.; Reiss, C.S. The role of IFN-© in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002, 13, 441–454. [Google Scholar] [CrossRef]
- Nansen, A.; Christensen, J.P.; Röpke, C.; Marker, O.; Scheynius, A.; Thomsen, A.R. Role of interferon-g in the pathogenesis of LCMV-induced meningitis: unimpaired leukocyte recruitment, but deficient macrophage activation in interferon-© knock-out mice. J. Neuroimmunol. 1998, 86, 202–212. [Google Scholar] [CrossRef]
- Pinschewer, D.D.; Schedensack, M.; Bergthaler, A.; Horvath, E.; Brück, W.; Löhning, M.; Merkler, D. T cells can mediate viral clearance from ependyma but not from brain parenchyma in a major histocompatibility class I- and perforin-independent manner. Brain 2010, 133, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishna, C.; Cantin, E.M. IFNγ inhibits G-CSF induced neutrophil expansion and invasion of the CNS to prevent viral encephalitis. PLoS Pathog. 2018, 14, e1006822. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Baruch, K.; Schwartz, M. Type I/II interferon balance in the regulation of brain physiology and pathology. Trends Immunol. 2016, 37, 181–192. [Google Scholar] [CrossRef]
- Yau, B.; Mitchell, A.J.; Too, L.K.; Ball, H.J.; Hunt, N.H. Interferon-γ-induced nitric oxide synthase-2 contributes to blood/brain barrier dysfunction and acute mortality in experimental Streptococcus pneumoniae meningitis. J. Interferon. Cytokine Res. 2016, 36, 86–99. [Google Scholar] [CrossRef]
- Drevets, D.A.; Dillon, M.J.; Schawang, J.E.; Stoner, J.A.; Leenen, P.J. IFN-© triggers CCR2-independent monocyte entry into the brain during systemic infection by virulent Listeria monocytogenes. Brain Behav. Immun. 2010, 24, 919–929. [Google Scholar] [CrossRef]
- Shwetank; Frost, E.; Mockus, T.; Ren, H.; Toprak, M.; Lauver, M.; Netherby-Winslow, C.; Jin, G.; Cosby, J.; Evavold, B.; et al. PD-1 dynamically regulates inflammation and development of brain-resident memory CD8 T cells during persistent viral encephalitis. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Kreutzfeldt, M.; Bergthaler, A.; Fernandez, M.; Brück, W.; Steinbach, K.; Vorm, M.; Coras, R.; Blümcke, I.; Bonilla, W.V.; Fleige, A.; et al. Neuroprotective intervention by interferon-γ blockade prevents CD8+ T cell-mediated dendrite and synapse loss. J. Exp. Med. 2013, 210, 2087–2103. [Google Scholar] [CrossRef]
- Wu, G.F.; Dandekar, A.A.; Pewe, L.; Perlman, S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J. Immunol. 2000, 165, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.F.; Dandekar, A.A.; Pewe, L.; Perlman, S. The role of CD4 and CD8 T cells in MHV-JHM-induced demyelination. Adv. Exp. Med. Biol. 2001, 494, 341–347. [Google Scholar] [PubMed]
- Savarin, C.; Bergmann, C.C. Viral-induced suppression of self-reactive T cells: Lessons from neurotropic coronavirus-induced demyelination. J. Neuroimmunol. 2017, 308, 12–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Gold, R.; Adams, O.; Lassmann, H. Progressive multifocal leukoencephalopathy and immune reconstitution inflammatory syndrome (IRIS). Acta Neuropathol. 2015, 130, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Baerwald, K.D.; Popko, B. Developing and mature oligodendrocytes respond differently to the immune cytokine interferon-g. J. Neurosci. Res. 1998, 52, 230–239. [Google Scholar] [CrossRef]
- Lim, J.L.; van der Pol, S.M.; Baron, W.; McCord, J.M.; de Vries, H.E.; van Horssen, J. Protandim protects oligodendrocytes against an oxidative insult. Antioxidants 2016, 55, 30. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.M.; Watson, N.B.; Minchenberg, S.B.; Massa, P.T. The influence of macrophage growth factors on Theiler’s murine encephalomyelitis virus (TMEV) infection and activation of macrophages. Cytokine 2018, 102, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.C.; Beura, L.K.; Masopust, D. Tissue resident memory T cells and viral immunity. Curr. Opin. Virol. 2017, 22, 44–50. [Google Scholar] [CrossRef]
- Williams, M.; Bevan, M. Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007, 25, 171–192. [Google Scholar] [CrossRef]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef]
- Caldeira-Dantas, S.; Furmanak, T.; Smith, C.; Quinn, M.; Teos, L.; Ertel, A.; Kurup, D.; Tandon, M.; Alevizos, I.; Snyder, C. The chemokine receptor CXCR3 promotes CD8+ T cell accumulation in uninfected salivary glands but is not necessary after murine cytomegalovirus infection. J. Immunol. 2018, 200, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Vezys, V.; Marzo, A.; Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef] [PubMed]
- Beura, L.K.; Fares-Frederickson, N.J.; Steinert, E.M.; Scott, M.C.; Thompson, E.A.; Fraser, K.A.; Schenkel, J.M.; Vezys, V.; Masopust, D. CD4+ resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J. Exp. Med. 2019, 216, 1214–1229. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Becker, E.; Spocinska, M.; Slawik, M.; Parga-Vidal, L.; Stark, R.; Wiendl, M.; Atreya, R.; Rath, T.; Leppkes, M.; et al. Hobit- and Blimp-1-driven CD4+ tissue-resident memory T cells control chronic intestinal inflammation. Nat. Immunol. 2019, 20, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.I.; Westerhof, L.M.; MacLeod, M.K.L. The roles of resident, central and effector memory CD4 T-cells in protective immunity following infection or vaccination. Immunology 2018, 154, 574–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iijima, N.; Iwasaki, A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 2014, 346, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.; Jiang, X.; Zaid, A.; Macleod, B.L.; Li, J.; Park, C.O.; Haque, A.; Bedoui, S.; Heath, W.R.; Mueller, S.N.; et al. Skin CD4+ memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Kallies, A. Transcriptional regulation of tissue-resident lymphocytes. Trends Immunol. 2017, 38, 94–103. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Steinbach, K.; Vincenti, I.; Kreutzfeldt, M.; Page, N.; Muschaweckh, A.; Wagner, I.; Drexler, I.; Pinschewer, D.; Korn, T.; Merkler, D. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J. Exp. Med. 2016, 213, 1571–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Hu, S.; Sheng, W.S.; Chauhan, P.; Lokensgard, J.R. Reactive glia promote development of CD103. Immun. Inflamm. Dis. 2018, 6, 332–344. [Google Scholar] [CrossRef]
- Landrith, T.A.; Sureshchandra, S.; Rivera, A.; Jang, J.C.; Rais, M.; Nair, M.G.; Messaoudi, I.; Wilson, E.H. CD103+ CD8 T cells in the Toxoplasma-infected brain exhibit a tissue-resident memory transcriptional profile. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.B.; Da Costa, A.; Lund, J.M. Regulatory T cells shape the resident memory T cell response to virus infection in the tissues. J. Immunol. 2014, 192, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Woodward-Davis, A.; Liu, R.; Hu, Y.; Villadangos, J.; Smyth, G.; Bevan, M.J. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 2012, 189, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Hunzeker, J.; Bonneau, R.H. Modulation of microglia and CD8+ T cell activation during the development of stress-induced herpes simplex virus type-1 encephalitis. Brain Behav. Immun. 2007, 21, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Brizić, I.; Šušak, B.; Arapović, M.; Huszthy, P.C.; Hiršl, L.; Kveštak, D.; Juranić Lisnić, V.; Golemac, M.; Pernjak Pugel, E.; Tomac, J.; et al. Brain-resident memory CD8+ T cells induced by congenital CMV infection prevent brain pathology and virus reactivation. Eur. J. Immunol. 2018, 48, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Hu, S.; Sheng, W.S.; Chauhan, P.; Singh, A.; Lokensgard, J.R. The PD-1: PD-L1 pathway promotes development of brain-resident memory T cells following acute viral encephalitis. J. Neuroinflamm. 2017, 14, 82. [Google Scholar] [CrossRef] [PubMed]
- Phares, T.W.; Ramakrishna, C.; Parra, G.I.; Epstein, A.; Chen, L.; Atkinson, R.; Stohlman, S.A.; Bergmann, C.C. Target-dependent B7-H1 regulation contributes to clearance of central nervous system infection and dampens morbidity. J. Immunol. 2009, 182, 5430–5438. [Google Scholar] [CrossRef] [PubMed]
- Shwetank; Abdelsamed, H.A.; Frost, E.L.; Schmitz, H.M.; Mockus, T.E.; Youngblood, B.A.; Lukacher, A.E. Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunol. Cell. Biol. 2017, 95, 953–959. [Google Scholar] [CrossRef]
- Bhadra, R.; Gigley, J.P.; Khan, I.A. PD-1-mediated attrition of polyfunctional memory CD8+ T cells in chronic toxoplasma infection. J. Infect. Dis. 2012, 206, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Frost, E.L.; Kersh, A.E.; Evavold, B.D.; Lukacher, A.E. Cutting edge: Resident memory CD8 T cells express high-affinity TCRs. J. Immunol. 2015, 195, 3520–3524. [Google Scholar] [CrossRef] [PubMed]
- Phares, T.W.; Stohlman, S.A.; Hwang, M.; Min, B.; Hinton, D.R.; Bergmann, C.C. CD4 T cells promote CD8 T cell immunity at the priming and effector site during viral encephalitis. J. Virol. 2012, 86, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Kapil, P.; Atkinson, R.; Ramakrishna, C.; Cua, D.J.; Bergmann, C.C.; Stohlman, S.A. Interleukin-12 (IL-12), but not IL-23, deficiency ameliorates viral encephalitis without affecting viral control. J. Virol. 2009, 83, 5978–5986. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Valenzuela, R.; Netland, J.; Seo, Y.J.; Bevan, M.J.; Grakoui, A.; Suthar, M.S. Dynamics of tissue-specific CD8+ T cells responses during West Nile virus infection. J. Virol. 2018, 92, e00014-18. [Google Scholar] [CrossRef] [PubMed]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef]
- Corgnac, S.; Boutet, M.; Kfoury, M.; Naltet, C.; Mami-Chouaib, F. The emerging role of CD8+ tissue resident memory (TRM) cells in antitumor immune: a unique functional contribution of the CD103 integrin. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Bevan, M.J. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8⁺ T cells responding to infection. Nat. Immunol. 2015, 16, 406–414. [Google Scholar] [CrossRef]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Bevan, M.J.; Fink, P.J. Local inflammatory cues regulate differentiation and persistence of CD8+ tissue-resident memory T cells. Cell Rep. 2017, 19, 114–124. [Google Scholar] [CrossRef]
- Attanasio, J.; Wherry, E. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity 2016, 44, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Crawford, A.; Shin, H.; Polley, A.; Freeman, G.J.; Wherry, E.J. Tissue-specific differences in PD-1 and PD-L1 expression during chronic viral infection: Implications for CD8 T-cell exhaustion. J. Virol. 2010, 84, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Phares, T.W.; Stohlman, S.A.; Hinton, D.R.; Bergmann, C.C. Enhanced CD8 Tcell anti-viral function and clinical disease in B7-H1-deficient mice requires CD4 T cells during encephalomyelitis. J. Neuroinflamm. 2012, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Shen, Y.; Liang, J.; Huang, Y.; Liu, X.; Jiang, D.; Yang, S.; Zhao, Y.; Yang, K. PDL1 fusion protein protects against experimental cerebral malaria via repressing over-reactive CD8+ T cell responses. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Bord, E.; Broge, T.; Glotzbecker, B.; Mills, H.; Gheuens, S.; Rosenblatt, J.; Avigan, D.; Koralnik, I. Increased program cell death-1 expression on T lymphocytes of patients with progressive multifocal leukoencephalopathy. J. Acquir. Immune Defic. Syndr. 2012, 60, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Hombrink, P.; Helbig, C.; Backer, R.A.; Piet, B.; Oja, A.E.; Stark, R.; Brasser, G.; Jongejan, A.; Jonkers, R.E.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef]
- Schachtele, S.J.; Hu, S.; Sheng, W.S.; Mutnal, M.B.; Lokensgard, J.R. Glial cells suppress postencephalitic CD8+ T lymphocytes through PD-L1. Glia 2014, 62, 1582–1594. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef]
- Berger, J.R.; Pall, L.; Lanska, D.; Whiteman, M. Progressive multifocal leukoencephalopathy in patients with HIV infection. J. Neurovirol. 1998, 4, 59–68. [Google Scholar] [CrossRef]
- Yi, J.S.; Du, M.; Zajac, A.J. A vital role for interleukin-21 in the control of a chronic viral infection. Science 2009, 324, 1572–1576. [Google Scholar] [CrossRef]
- Fröhlich, A.; Marsland, B.J.; Sonderegger, I.; Kurrer, M.; Hodge, M.R.; Harris, N.L.; Kopf, M. IL-21 receptor signaling is integral to the development of Th2 effector responses in vivo. Blood 2007, 109, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Novy, P.; Huang, X.; Leonard, W.; Yang, Y. Intrinsic IL-21 signaling is critical for CD8 T cell survival and memory formation in response to vaccinia viral infection. J. Immunol. 2011, 186, 2729–2738. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Liu, Y.; Weinstein, J.S.; Craft, J.; Kaech, S.M. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity 2011, 35, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Phares, T.W.; DiSano, K.D.; Hinton, D.R.; Hwang, M.; Zajac, A.J.; Stohlman, S.A.; Bergmann, C.C. IL-21 optimizes T cell and humoral responses in the central nervous system during viral encephalitis. J. Neuroimmunol. 2013, 263, 43–54. [Google Scholar] [CrossRef]
- Tian, Y.; Zajac, A.J. IL-21 and T cell differentiation: Consider the context. Trends Immunol. 2016, 37, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Phares, T.W.; Marques, C.P.; Stohlman, S.A.; Hinton, D.R.; Bergmann, C.C. Factors supporting intrathecal humoral responses following viral encephalomyelitis. J. Virol. 2011, 85, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Stumhofer, J.S.; Silver, J.S.; Hunter, C.A. IL-21 is required for optimal antibody production and T cell responses during chronic Toxoplasma gondii infection. PLoS ONE 2013, 8, e62889. [Google Scholar] [CrossRef]
- Tian, Y.; Cox, M.A.; Kahan, S.M.; Ingram, J.T.; Bakshi, R.K.; Zajac, A.J. A context-dependent role for IL-21 in modulating the differentiation, distribution, and abundance of effector and memory CD8 T cell subsets. J. Immunol. 2016, 196, 2153–2166. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Lu, B.; Gerard, C.; Iwasaki, A. CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature 2009, 462, 510–513. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Zhang, N.; Marshall, H.D.; Staron, M.M.; Guan, T.; Hu, Y.; Cauley, L.S.; Craft, J.; Kaech, S.M. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity 2014, 41, 633–645. [Google Scholar] [CrossRef]
- Villegas-Mendez, A.; Greig, R.; Shaw, T.N.; de Souza, J.B.; Gwyer Findlay, E.; Stumhofer, J.S.; Hafalla, J.C.; Blount, D.G.; Hunter, C.A.; Riley, E.M.; et al. IFN-γ-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J. Immunol. 2012, 189, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Jameson, S.; Masopust, D. Understanding subset diversity in T cell memory. Immunity 2018, 48, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Wynne-Jones, E.; Freestone, D.; Pellicci, D.G.; Mielke, L.A.; Newman, D.M.; Braun, A.; Masson, F.; Kallies, A.; Belz, G.T.; et al. T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity 2015, 43, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Casey, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. IL-15-independent maintenance of tissue-resident and boosted effector memory CD8 T cells. J. Immunol. 2016, 196, 3920–3926. [Google Scholar] [CrossRef] [PubMed]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1PR1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.; Fraser, K.; Vezys, V.; Masopust, D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Zaid, A.; Hor, J.L.; Christo, S.N.; Prier, J.E.; Davies, B.; Alexandre, Y.O.; Gregory, J.L.; Russell, T.A.; Gebhardt, T.; et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018, 19, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Beura, L.; Mitchell, J.; Thompson, E.; Schenkel, J.; Mohammed, J.; Wijeyesinghe, S.; Fonseca, R.; Burbach, B.; Hickman, H.; Vezys, V.; et al. Intravital mucosal imaging of CD8+ resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol. 2018, 19, 173–182. [Google Scholar] [CrossRef]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, S.; Beltman, J.B.; Chodaczek, G.; Hoekstra, M.E.; van Beek, A.E.; Gomez-Eerland, R.; Ritsma, L.; van Rheenen, J.; Marée, A.F.; Zal, T.; et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc. Natl. Acad. Sci. USA 2012, 109, 19739–19744. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.M.; Bonneau, R.H.; Kinchington, P.R.; Hendricks, R.L. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 2003, 18, 593–603. [Google Scholar] [CrossRef]
- Liu, T.; Khanna, K.M.; Carriere, B.N.; Hendricks, R.L. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J. Virol. 2001, 75, 11178–11184. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, K.; Vincenti, I.; Egervari, K.; Kreutzfeldt, M.; van der Meer, F.; Page, N.; Klimek, B.; Rossitto-Borlat, I.; Di Liberto, G.; Muschaweckh, A.; et al. Brain-resident memory T cells generated early in life predispose to autoimmune disease in mice. Sci. Transl. Med. 2019, 11, eaav5519. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| TRM Marker | Function | Frequency of Marker Expression on CD8 bTRM during Brain Infection | ||
|---|---|---|---|---|
| Acute Infections: | Persistent Infections: | |||
| CD103 | Binds to E-cadherin | VSV: ≤90% LCMV: ≤60% MCMV: <40% | MuPyV, T. Gondii: 40–60% WNV: <20% | [11,19,51,52,53,54] |
| CD69 | Antagonizes S1PR1 expression | VSV, LCMV: >80% MCMV: <60% | MuPyV, T. Gondii: >80% HSV: <40% | [19,51,52,53,55,56,57] |
| PD-1 | Inhibitory receptor, antagonizes TCR engagement | VSV: <1% MCMV: <25% JHMV: 20–45% | MuPyV: >90% T. Gondii: 30–50% WNV: 20–30% | [11,58,59,60,61] |
| CD62L | Lymphocyte-endothelial cell interactions | MCMV: <5% | MuPyV: <5% | [57,62] |
| Ki67 | General marker of cellular proliferation | JHMV: <5% | MuPyV: <20% | [19,63] |
| Granzyme-B | Mediates apoptosis in target cells | LCMV: <60% VSV: >30% JHMV: >20% | WNV: <20% | [11,16,51,63] |
| IFNγ (after stimulation) | Pleiotropic cytokine | JHMV: >30% MCMV: >60% LCMV: >50% | MuPyV: <60% WNV: <10% | [16,51,57,60,64,65] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mockus, T.E.; Ren, H.M.; Shwetank; Lukacher, A.E. To Go or Stay: The Development, Benefit, and Detriment of Tissue-Resident Memory CD8 T Cells during Central Nervous System Viral Infections. Viruses 2019, 11, 842. https://doi.org/10.3390/v11090842
Mockus TE, Ren HM, Shwetank, Lukacher AE. To Go or Stay: The Development, Benefit, and Detriment of Tissue-Resident Memory CD8 T Cells during Central Nervous System Viral Infections. Viruses. 2019; 11(9):842. https://doi.org/10.3390/v11090842
Chicago/Turabian StyleMockus, Taryn E., Heather M. Ren, Shwetank, and Aron E. Lukacher. 2019. "To Go or Stay: The Development, Benefit, and Detriment of Tissue-Resident Memory CD8 T Cells during Central Nervous System Viral Infections" Viruses 11, no. 9: 842. https://doi.org/10.3390/v11090842
APA StyleMockus, T. E., Ren, H. M., Shwetank, & Lukacher, A. E. (2019). To Go or Stay: The Development, Benefit, and Detriment of Tissue-Resident Memory CD8 T Cells during Central Nervous System Viral Infections. Viruses, 11(9), 842. https://doi.org/10.3390/v11090842