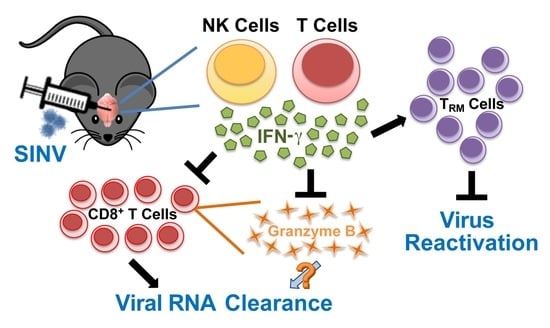
Interferon-Gamma Modulation of the Local T Cell Response to Alphavirus Encephalomyelitis


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Virus and Infection of Cells
2.3. Virus Infection of Mice
2.4. Immunoblot Analysis
2.5. Gene Expression Measurement by Real-Time PCR
2.6. Mononuclear Cell Isolation
2.7. Flow Cytometry
2.8. Brain and Spinal Cord Histology
2.9. Statistics
3. Results
3.1. IFN-γ Facilitates Virus Clearance from Neurons In Vitro
3.2. IFN-γ Induces Neuronal Expression of Antiviral ISGs
3.3. Source of IFN-γ during In Vivo SINV CNS Infection
3.4. Effect of IFN-γ Signaling on ISG Expression in the CNS of SINV-Infected Mice
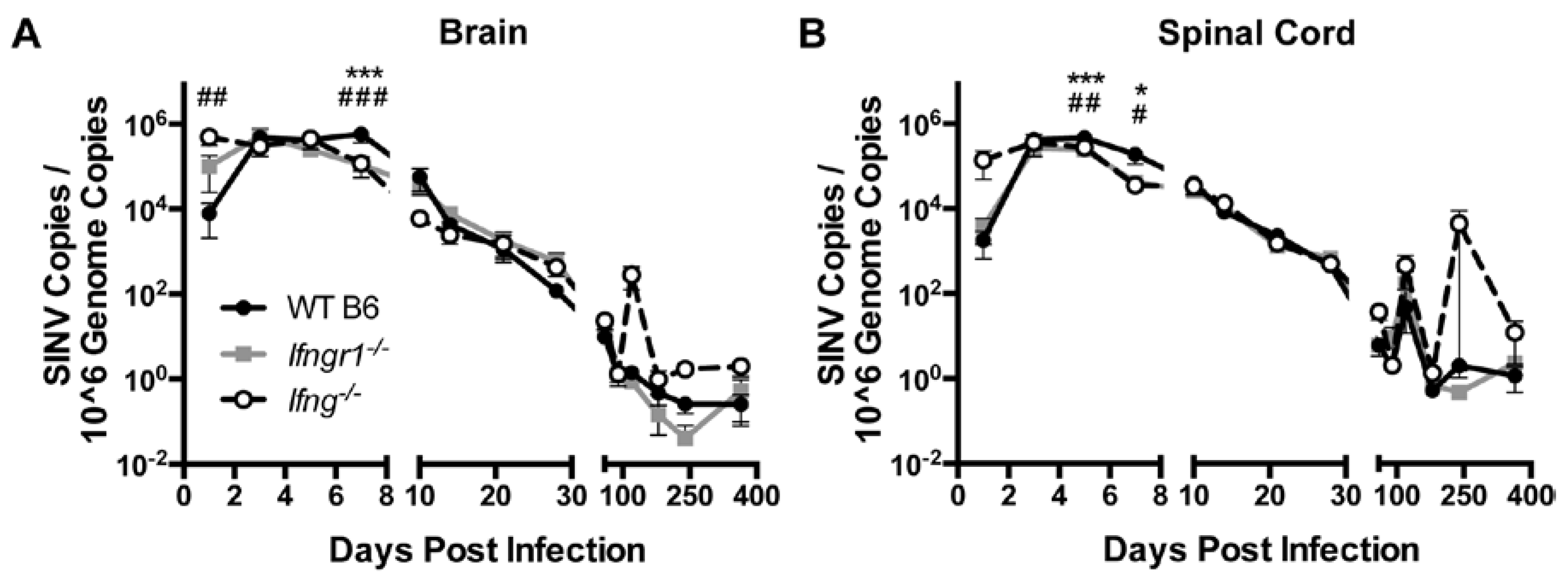
3.5. Effect of IFN-γ Signaling on Viral RNA Clearance from the CNS of SINV-Infected Mice
3.6. Effect of IFN-γ Signaling on CNS Pathology in Response to SINV Infection
3.7. The Effect of IFN-γ Signaling on Proliferation and Infiltration of Immune Cells into the CNS
3.8. The Effect of IFN-γ Signaling on the Function of Brain CD4+ T Cells during SINV Infection
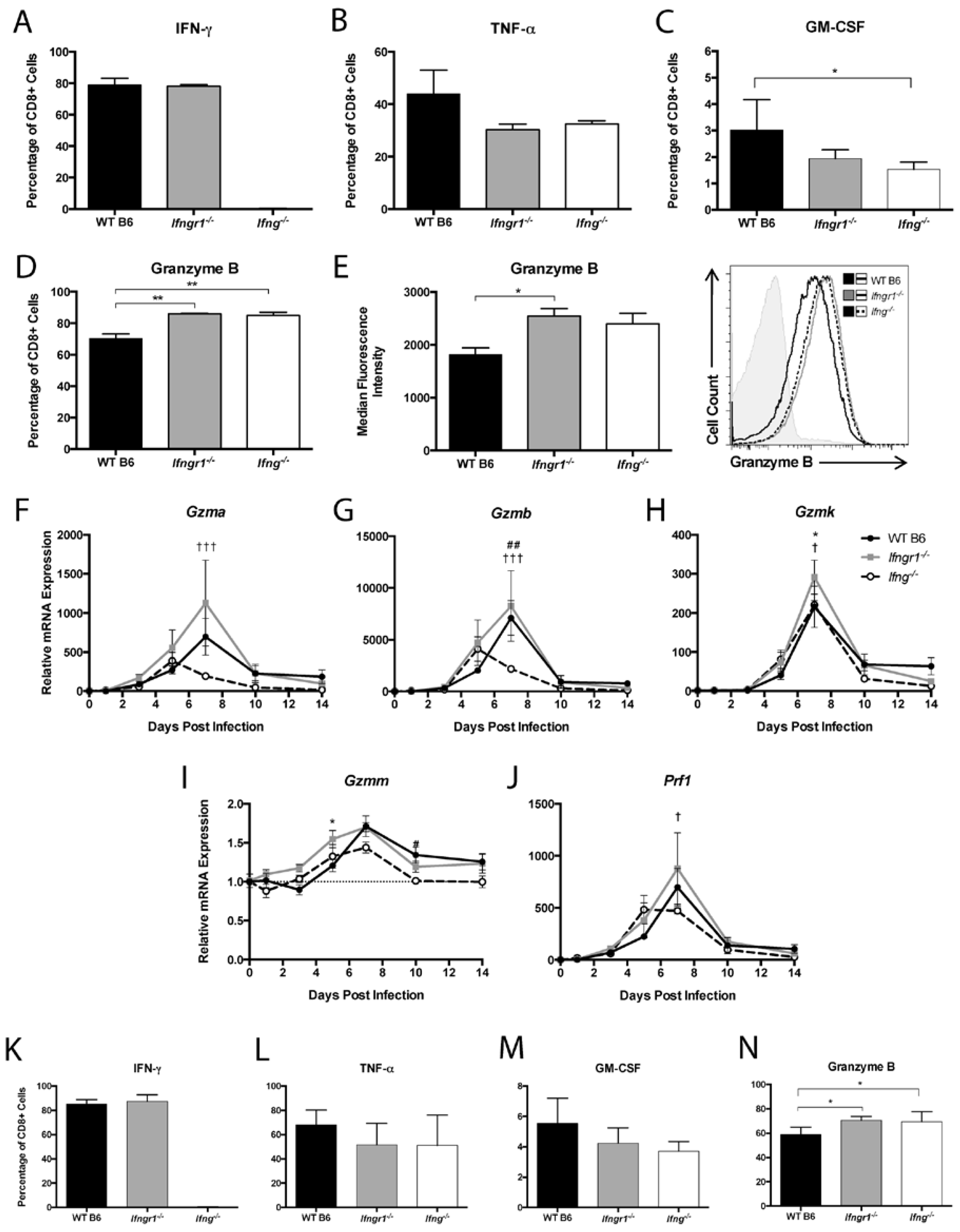
3.9. The Effect of IFN-γ Signaling on the Function of CD8+ T Cells in the CNS during SINV Infection
3.10. Effect of IFN-γ Signaling on CD8+ T Cell and NK Cell Degranulation and Cytotoxic Function during SINV Infection
3.11. The Effect of IFN-γ Signaling on Establishment of CD8+ Tissue-Resident Memory (TRM) Cells in the CNS after SINV Infection
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griffin, D.E.; Levine, B.; Tyor, W.R.; Irani, D.N. The immune response in viral encephalitis. Semin. Immunol. 1992, 4, 111–119. [Google Scholar] [PubMed]
- Neumann, H.; Cavalié, A.; Jenne, D.E.; Wekerle, H. Induction of MHC class I genes in neurons. Science 1995, 269, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Griffin, D.E. The role of CD8(+) T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J. Virol. 2000, 74, 6117–6125. [Google Scholar] [CrossRef]
- Griffin, D.E. Alphaviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 652–686. [Google Scholar]
- Metcalf, T.U.; Griffin, D.E. Alphavirus-Induced Encephalomyelitis: Antibody-Secreting Cells and Viral Clearance from the Nervous System. J. Virol. 2011, 85, 11490–11501. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Hardwick, J.M.; Trapp, B.D.; Crawford, T.O.; Bollinger, R.C.; Griffin, D.E. Antibody-mediated clearance of alphavirus infection from neurons. Science 1991, 254, 856–860. [Google Scholar] [CrossRef]
- Binder, G.K.; Griffin, D.E. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science 2001, 293, 303–306. [Google Scholar] [CrossRef]
- Burdeinick-Kerr, R.; Wind, J.; Griffin, D.E. Synergistic Roles of Antibody and Interferon in Noncytolytic Clearance of Sindbis Virus from Different Regions of the Central Nervous System. J. Virol. 2007, 81, 5628–5636. [Google Scholar] [CrossRef]
- Baxter, V.K.; Griffin, D.E. Interferon gamma modulation of disease manifestation and the local antibody response to alphavirus encephalomyelitis. J. Gen. Virol. 2016, 97, 2908–2925. [Google Scholar] [CrossRef]
- Levine, B.; Griffin, D.E. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J. Virol. 1992, 66, 6429–6435. [Google Scholar] [CrossRef]
- Tyor, W.R.; Griffin, D.E. Virus specificity and isotype expression of intraparenchymal antibody-secreting cells during Sindbis virus encephalitis in mice. J. Neuroimmunol. 1993, 48, 37–44. [Google Scholar] [CrossRef]
- Kulcsar, K.A.; Baxter, V.K.; Greene, I.P.; Griffin, D.E. Interleukin 10 modulation of pathogenic Th17 cells during fatal alphavirus encephalomyelitis. Proc. Natl. Acad. Sci. USA 2014, 111, 16053–16058. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.F.; Griffin, D.E. Contribution of T cells to mortality in neurovirulent Sindbis virus encephalomyelitis. J. Neuroimmunol. 2002, 127, 106–114. [Google Scholar] [CrossRef]
- Martin, N.M.; Griffin, D.E. Interleukin-10 modulation of virus clearance and disease in mice with alphaviral encephalomyelitis. J. Virol. 2018, 92, e01517-17. [Google Scholar] [CrossRef]
- Tau, G.; Rothman, P. Biologic functions of the IFN-gamma receptors. Allergy 1999, 54, 1233–1251. [Google Scholar] [CrossRef]
- Burdeinick-Kerr, R.; Govindarajan, D.; Griffin, D.E. Noncytolytic clearance of Sindbis virus infection from neurons by gamma interferon is dependent on Jak/Stat signaling. J. Virol. 2009, 83, 3429–3435. [Google Scholar] [CrossRef] [PubMed]
- Farrar, M.A.; Schreiber, R.D. The molecular cell biology of interferon-gamma and its receptor. Annu. Rev. Immunol. 1993, 11, 571–611. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef]
- Murrell, J.R.; Hunter, D.D. An olfactory sensory neuron line, odora, properly targets olfactory proteins and responds to odorants. J. Neurosci. 1999, 19, 8260–8270. [Google Scholar] [CrossRef]
- Lustig, S.; Jackson, A.C.; Hahn, C.S.; Griffin, D.E.; Strauss, E.G.; Strauss, J.H. Molecular basis of Sindbis virus neurovirulence in mice. J. Virol. 1988, 62, 2329–2336. [Google Scholar] [CrossRef]
- Park, E.; Griffin, D.E. The nsP3 macrodomain is important for Sindbis virus replication in neurons and neurovirulence in mice. Virology 2009, 388, 305–314. [Google Scholar] [CrossRef]
- Jackson, A.C.; Moench, T.R.; Trapp, B.D.; Griffin, D.E. Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab. Investig. 1988, 58, 503–509. [Google Scholar] [PubMed]
- Rowell, J.F.J.; Griffin, D.E.D. The inflammatory response to nonfatal Sindbis virus infection of the nervous system is more severe in SJL than in BALB/c mice and is associated with low levels of IL-4 mRNA and high levels of IL-10-producing CD4+ T cells. J. Immunol. 1999, 162, 1624–1632. [Google Scholar] [PubMed]
- Baxter, V.K.; Glowinski, R.; Braxton, A.M.; Potter, M.C.; Slusher, B.S.; Griffin, D.E. Glutamine antagonist-mediated immune suppression decreases pathology but delays virus clearance in mice during nonfatal alphavirus encephalomyelitis. Virology 2017, 508, 134–149. [Google Scholar] [CrossRef]
- Traver, M.K.; Henry, S.C.; Cantillana, V.; Oliver, T.; Hunn, J.P.; Howard, J.C.; Beer, S.; Pfeffer, K.; Coers, J.; Taylor, G.A. Immunity-related GTPase M (IRGM) proteins influence the localization of guanylate-binding protein 2 (GBP2) by modulating macroautophagy. J. Biol. Chem. 2011, 286, 30471–30480. [Google Scholar] [CrossRef] [PubMed]
- Eskildsen, S. Characterization of the 2“-5-”oligoadenylate synthetase ubiquitin-like family. Nucleic Acids Res. 2003, 31, 3166–3173. [Google Scholar] [CrossRef]
- Mattijssen, S.; Pruijn, G.J.M. Viperin, a key player in the antiviral response. Microbes Infect. 2012, 14, 419–426. [Google Scholar] [CrossRef]
- Karki, S.; Li, M.M.H.; Schoggins, J.W.; Tian, S.; Rice, C.M.; MacDonald, M.R. Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PLoS ONE 2012, 7, e37398. [Google Scholar] [CrossRef]
- Chiu, H.P.; Chiu, H.; Yang, C.F.; Lee, Y.L.; Chiu, F.L.; Kuo, H.C.; Lin, R.J.; Lin, Y.L. Inhibition of Japanese encephalitis virus infection by the host zinc-finger antiviral protein. PLoS Pathog. 2018, 14, e1007166. [Google Scholar] [CrossRef]
- Lee, E.Y.; Schultz, K.L.W.; Griffin, D.E. Mice deficient in interferon-gamma or interferon-gamma receptor 1 have distinct inflammatory responses to acute viral encephalomyelitis. PLoS ONE 2013, 8, e76412. [Google Scholar] [CrossRef]
- Kulcsar, K.A.; Baxter, V.K.; Abraham, R.; Nelson, A.; Griffin, D.E. Distinct immune responses in resistant and susceptible strains of mice during neurovirulent alphavirus encephalomyelitis. J. Virol. 2015, 89, 8280–8291. [Google Scholar] [CrossRef]
- Charles, P.C.; Trgovcich, J.; Davis, N.L.; Johnston, R.E. Immunopathogenesis and immune modulation of Venezuelan equine encephalitis virus-induced disease in the mouse. Virology 2001, 284, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, S.; Baxter, V.K.; Schultz, K.L.W.; Slusher, B.S.; Griffin, D.E. Protective effects of glutamine antagonist 6-diazo-5-oxo-l-norleucine in mice with alphavirus encephalomyelitis. J. Virol. 2016, 90, 9251–9262. [Google Scholar] [CrossRef]
- Afonina, I.S.; Cullen, S.P.; Martin, S.J. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol. Rev. 2010, 235, 105–116. [Google Scholar] [CrossRef]
- Betts, M.R.; Brenchley, J.M.; Price, D.A.; De Rosa, S.C.; Douek, D.C.; Roederer, M.; Koup, R.A. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 2003, 281, 65–78. [Google Scholar] [CrossRef]
- Lieberman, J. Granzyme A activates another way to die. Immunol. Rev. 2010, 235, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef]
- Burdeinick-Kerr, R.; Griffin, D.E. Gamma interferon-dependent, noncytolytic clearance of Sindbis virus infection from neurons in vitro. J. Virol. 2005, 79, 5374–5385. [Google Scholar] [CrossRef]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Tallóczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Levine, B. Eating oneself and uninvited guests: Autophagy-related pathways in cellular defense. Cell 2005, 120, 159–162. [Google Scholar]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef]
- Grégoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; et al. IRGM Is a Common Target of RNA Viruses that Subvert the Autophagy Network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar] [CrossRef]
- Wang, X.; Hou, L.; Du, J.; Zhou, L.; Ge, X.; Guo, X.; Yang, H. Capsid, membrane and NS3 are the major viral proteins involved in autophagy induced by Japanese encephalitis virus. Vet. Microbiol. 2015, 178, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Rangarajan, P.N. Common host genes are activated in mouse brain by Japanese encephalitis and rabies viruses. J. Gen. Virol. 2003, 84, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Klotz, D.; Gerhauser, I. Interferon-stimulated genes—Mediators of the innate immune response during canine distemper virus infection. IJMS 2019, 20, 1620. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Wilkins, C.; Thomas, S.; Sekine, A.; Hendrick, D.M.; Voss, K.; Ireton, R.C.; Mooney, M.; Go, J.T.; Choonoo, G.; et al. Oas1b-dependent immune transcriptional profiles of West Nile virus infection in the collaborative cross. G3 (Bethesda) 2017, 7, 1665–1682. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hinson, E.R.; Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef]
- Nasr, N.; Maddocks, S.; Turville, S.G.; Harman, A.N.; Woolger, N.; Helbig, K.J.; Wilkinson, J.; Bye, C.R.; Wright, T.K.; Rambukwelle, D.; et al. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 2012, 120, 778–788. [Google Scholar] [CrossRef]
- Tan, K.S.; Olfat, F.; Phoon, M.C.; Hsu, J.P.; Howe, J.L.C.; Seet, J.E.; Chin, K.C.; Chow, V.T.K. In vivo and in vitro studies on the antiviral activities of viperin against influenza H1N1 virus infection. J. Gen. Virol. 2012, 93, 1269–1277. [Google Scholar] [CrossRef]
- Helbig, K.J.; Eyre, N.S.; Yip, E.; Narayana, S.; Li, K.; Fiches, G.; McCartney, E.M.; Jangra, R.K.; Lemon, S.M.; Beard, M.R. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology 2011, 54, 1506–1517. [Google Scholar] [CrossRef]
- Teng, T.-S.; Foo, S.-S.; Simamarta, D.; Lum, F.-M.; Teo, T.-H.; Lulla, A.; Yeo, N.K.W.; Koh, E.G.L.; Chow, A.; Leo, Y.S.; et al. Viperin restricts chikungunya virus replication and pathology. J. Clin. Investig. 2012, 122, 4447–4460. [Google Scholar] [CrossRef]
- Lindqvist, R.; Kurhade, C.; Gilthorpe, J.D.; Överby, A.K. Cell-type- and region-specific restriction of neurotropic flavivirus infection by viperin. J. NeuroInflamm. 2018, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Feng, L.; Chen, P.; Li, A.; Guo, S.; Jiao, X.; Zhang, C.; Zhao, Y.; Jin, X.; Zhong, K.; et al. Viperin inhibits classical swine fever virus replication by interacting with viral nonstructural 5A protein. J. Med. Virol. 2019, 92, e02054. [Google Scholar] [CrossRef]
- Bick, M.J.; Carroll, J.W.N.; Gao, G.; Goff, S.P.; Rice, C.M.; MacDonald, M.R. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 2003, 77, 11555–11562. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Möller, P.; Bick, M.J.; Wurr, S.; Becker, S.; Günther, S.; Kümmerer, B.M. Inhibition of filovirus replication by the zinc finger antiviral protein. J. Virol. 2007, 81, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, G.; Lv, F.; Wang, X.; Ji, X.; Xu, Y.; Sun, J.; Wu, L.; Zheng, Y.T.; Gao, G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 15834–15839. [Google Scholar] [CrossRef]
- Mao, R.; Nie, H.; Cai, D.; Zhang, J.; Liu, H.; Yan, R.; Cuconati, A.; Block, T.M.; Guo, J.-T.; Guo, H. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog. 2013, 9, e1003494-18. [Google Scholar] [CrossRef]
- Law, L.M.J.; Razooky, B.S.; Li, M.M.H.; You, S.; Jurado, A.; Rice, C.M.; MacDonald, M.R. ZAP’s stress granule localization is correlated with its antiviral activity and induced by virus replication. PLoS Pathog. 2019, 15, e1007798. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef]
- Hou, W.; Kang, H.S.; Kim, B.S. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J. Exp. Med. 2009, 206, 313–328. [Google Scholar] [CrossRef]
- Kim, B.S.; Jin, Y.H.; Meng, L.; Hou, W.; Kang, H.S.; Park, H.S.; Koh, C.S. IL-1 signal affects both protection and pathogenesis of virus-induced chronic CNS demyelinating disease. J. NeuroInflamm. 2012, 9, 217. [Google Scholar] [CrossRef]
- Savarin, C.; Stohlman, S.A.; Hinton, D.R.; Ransohoff, R.M.; Cua, D.J.; Bergmann, C.C. IFN-γ protects from lethal IL-17 mediated viral encephalomyelitis independent of neutrophils. J. Neuroinflamm. 2012, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.J.; Gaffen, S.L. Interleukin-17: A novel inflammatory cytokine that bridges innate and adaptive immunity. Front. Biosci. 2008, 13, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Duley, A.K.; Ploquin, M.J.Y.; Eksmond, U.; Ammann, C.G.; Messer, R.J.; Myers, L.; Hasenkrug, K.J.; Kassiotis, G. Negative impact of IFN-γ on early host immune responses to retroviral infection. J. Immunol. 2012, 189, 2521–2529. [Google Scholar] [CrossRef]
- Hirsch, R.L. Natural killer cells appear to play no role in the recovery of mice from Sindbis virus infection. Immunology 1981, 43, 81–89. [Google Scholar]
- Alsharifi, M.; Lobigs, M.; Simon, M.M.; Kersten, A.; Muller, K.; Koskinen, A.; Lee, E.; Mullbacher, A. NK cell-mediated immunopathology during an acute viral infection of the CNS. Eur. J. Immunol. 2006, 36, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Joly, E.; Mucke, L.; Oldstone, M.B. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science 1991, 253, 1283–1285. [Google Scholar] [CrossRef] [PubMed]
- Rall, G.F.; Mucke, L.; Oldstone, M.B. Consequences of cytotoxic T lymphocyte interaction with major histocompatibility complex class I-expressing neurons in vivo. J. Exp. Med. 1995, 182, 1201–1212. [Google Scholar] [CrossRef]
- Corriveau, R.A.; Huh, G.S.; Shatz, C.J. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 1998, 21, 505–520. [Google Scholar] [CrossRef]
- Horwitz, M.S.; Evans, C.F.; Klier, F.G.; Oldstone, M.B. Detailed in vivo analysis of interferon-gamma induced major histocompatibility complex expression in the central nervous system: Astrocytes fail to express major histocompatibility complex class I and II molecules. Lab. Investig. 1999, 79, 235–242. [Google Scholar]
- McDole, J.R.; Danzer, S.C.; Pun, R.Y.K.; Chen, Y.; Johnson, H.L.; Pirko, I.; Johnson, A.J. Rapid formation of extended processes and engagement of Theiler’s virus-infected neurons by CNS-infiltrating CD8 T cells. Am. J. Pathol. 2010, 177, 1823–1833. [Google Scholar] [CrossRef]
- Zajac, A.J.; Dye, J.M.; Quinn, D.G. Control of lymphocytic choriomeningitis virus infection in granzyme B deficient mice. Virology 2003, 305, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Samuel, M.A.; Diamond, M.S. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J. Virol. 2006, 80, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Larena, M.; Regner, M.; Lobigs, M. Cytolytic effector pathways and IFN-γ help protect against Japanese encephalitis. Eur. J. Immunol. 2013, 43, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.C.; Parra, B.; Hinton, D.R.; Chandran, R.; Morrison, M.; Stohlman, S.A. Perforin-mediated effector function within the central nervous system requires IFN-γ mediated MHC up-regulation. J. Immunol. 2003, 170, 3204–3213. [Google Scholar] [CrossRef] [PubMed]
- Hooks, J.J. The critical role of IFN-γ in experimental coronavirus retinopathy. IOVS 2003, 44, 3402–3408. [Google Scholar] [CrossRef]
- Kramer, M.D.; Simon, M.M. Are proteinases functional molecules of T lymphocytes? Immunol. Today 1987, 8, 140–142. [Google Scholar] [CrossRef]
- Metkar, S.S.; Menaa, C.; Pardo, J.; Wang, B.; Wallich, R.; Freudenberg, M.; Kim, S.; Raja, S.M.; Shi, L.; Simon, M.M.; et al. Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity 2008, 29, 720–733. [Google Scholar] [CrossRef]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Lüthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef]
- Decman, V.; Kinchington, P.R.; Harvey, S.A.K.; Hendricks, R.L. Gamma interferon can block herpes simplex virus type 1 reactivation from latency, even in the presence of late gene expression. J. Virol. 2005, 79, 10339–10347. [Google Scholar] [CrossRef]
- Verjans, G.M.G.M.; Hintzen, R.Q.; van Dun, J.M.; Poot, A.; Milikan, J.C.; Laman, J.D.; Langerak, A.W.; Kinchington, P.R.; Osterhaus, A.D.M.E. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc. Natl. Acad. Sci. USA 2007, 104, 3496–3501. [Google Scholar] [CrossRef]
- Knickelbein, J.E.; Khanna, K.M.; Yee, M.B.; Baty, C.J.; Kinchington, P.R.; Hendricks, R.L. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 2008, 322, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Joeckel, L.T.; Wallich, R.; Martin, P.; Sanchez-Martinez, D.; Weber, F.C.; Martin, S.F.; Borner, C.; Pardo, J.; Froelich, C.; Simon, M.M. Mouse granzyme K has pro-inflammatory potential. Cell Death Differ. 2011, 18, 1112–1119. [Google Scholar] [CrossRef]
- Simon, H.G.; Fruth, U.; Kramer, M.D.; Simon, M.M. A secretable serine proteinase with highly restricted specificity from cytolytic T lymphocytes inactivates retrovirus-associated reverse transcriptase. FEBS Lett. 1987, 223, 352–360. [Google Scholar] [CrossRef]
- Andrade, F.; Fellows, E.; Jenne, D.E.; Rosen, A.; Young, C.S.H. Granzyme H destroys the function of critical adenoviral proteins required for viral DNA replication and granzyme B inhibition. EMBO J. 2007, 26, 2148–2157. [Google Scholar] [CrossRef]
- Van Domselaar, R.; de Poot, S.A.H.; Remmerswaal, E.B.M.; Lai, K.W.; ten Berge, I.J.M.; Bovenschen, N. Granzyme M targets host cell hnRNP K that is essential for human cytomegalovirus replication. Cell Death Differ. 2012, 20, 419–429. [Google Scholar] [CrossRef]
- Ehlers, I.; Horke, S.; Reumann, K.; Rang, A.; Grosse, F.; Will, H.; Heise, T. Functional characterization of the interaction between human La and hepatitis B virus RNA. J. Biol. Chem. 2004, 279, 43437–43447. [Google Scholar] [CrossRef] [PubMed]
- Romero, V.; Fellows, E.; Jenne, D.E.; Andrade, F. Cleavage of La protein by granzyme H induces cytoplasmic translocation and interferes with La-mediated HCV-IRES translational activity. Cell Death Differ. 2009, 16, 340–348. [Google Scholar] [CrossRef]
- Vashist, S.; Anantpadma, M.; Sharma, H.; Vrati, S. La protein binds the predicted loop structures in the 3’ non-coding region of Japanese encephalitis virus genome: Role in virus replication. J. Gen. Virol. 2009, 90, 1343–1352. [Google Scholar] [CrossRef]
- Vashist, S.; Bhullar, D.; Vrati, S. La Protein can simultaneously bind to both 3′- and 5′-noncoding regions of Japanese encephalitis virus genome. DNA Cell Biol. 2011, 30, 339–346. [Google Scholar] [CrossRef]
- Lin, J.Y.; LI, M.L.; Huang, P.N.; Chien, K.Y.; Horng, J.T.; Shih, S.R. Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5’ untranslated region and participates in virus replication. J. Gen. Virol. 2008, 89, 2540–2549. [Google Scholar] [CrossRef]
- Wolf, D.; Witte, V.; Clark, P.; Blume, K.; Lichtenheld, M.G.; Baur, A.S. HIV Nef enhances Tat-mediated viral transcription through a hnRNP-K-nucleated signaling complex. Cell Host Microbe 2008, 4, 398–408. [Google Scholar] [CrossRef]
- Kanlaya, R.; Pattanakitsakul, S.-N.; Sinchaikul, S.; Chen, S.-T.; Thongboonkerd, V. Vimentin interacts with heterogeneous nuclear ribonucleoproteins and dengue nonstructural protein 1 and is important for viral replication and release. Mol. Biosyst. 2010, 6, 795–806. [Google Scholar] [CrossRef]
- Bouraï, M.; Lucas-Hourani, M.; Gad, H.H.; Drosten, C.; Jacob, Y.; Tafforeau, L.; Cassonnet, P.; Jones, L.M.; Judith, D.; Couderc, T.; et al. Mapping of chikungunya virus interactions with host proteins identified nsP2 as a highly connected viral component. J. Virol. 2012, 86, 3121–3134. [Google Scholar] [CrossRef] [PubMed]
- Burnham, A.J.; Gong, L.; Hardy, R.W. Heterogeneous nuclear ribonuclear protein K interacts with Sindbis virus nonstructural proteins and viral subgenomic mRNA. Virology 2007, 367, 212–221. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, A.T.; Gebhart, N.N.; Meller, M.E.; Hardy, R.W.; Sokoloski, K.J. Identification and characterization of Sindbis virus RNA-host protein interactions. J. Virol. 2018, 92, e02171-27. [Google Scholar] [CrossRef] [PubMed]
- Varjak, M.; Saul, S.; Arike, L.; Lulla, A.; Peil, L.; Merits, A. Magnetic fractionation and proteomic dissection of cellular organelles occupied by the late replication complexes of Semliki Forest virus. J. Virol. 2013, 87, 10295–10312. [Google Scholar] [CrossRef]
- Tyor, W.R.; Wesselingh, S.; Levine, B. Long term intraparenchymal Ig secretion after acute viral encephalitis in mice. J. Immunol. 1992, 149, 4016–4020. [Google Scholar]
- Ravi, V.; Desai, A.S.; Shenoy, P.K.; Satishchandra, P.; Chandramuki, A.; Gourie-Devi, M. Persistence of Japanese encephalitis virus in the human nervous system. J. Med. Virol. 1993, 40, 326–329. [Google Scholar] [CrossRef]
- Donnelly, S.M.; Sheahan, B.J.; Atkins, G.J. Long-term effects of Semliki Forest virus infection in the mouse central nervous system. Neuropathol. Appl. Neurobiol. 1997, 23, 235–241. [Google Scholar] [CrossRef]
- Appler, K.K.; Brown, A.N.; Stewart, B.S.; Behr, M.J.; Demarest, V.L.; Wong, S.J.; Bernard, K.A. Persistence of West Nile virus in the central nervous system and periphery of mice. PLoS ONE 2010, 5, e10649. [Google Scholar] [CrossRef]
- Brooke, C.B.; Deming, D.J.; Whitmore, A.C.; White, L.J.; Johnston, R.E. T cells facilitate recovery from Venezuelan equine encephalitis virus-induced encephalomyelitis in the absence of antibody. J. Virol. 2010, 84, 4556–4568. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.S.; Demarest, V.L.; Wong, S.J.; Green, S.; Bernard, K.A. Persistence of virus-specific immune responses in the central nervous system of mice after West Nile virus infection. BMC Immunol. 2011, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, T.U.; Baxter, V.K.; Nilaratanakul, V.; Griffin, D.E. Recruitment and retention of B Cells in the central nervous system in response to alphavirus encephalomyelitis. J. Virol. 2013, 87, 2420–2429. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Waithman, J.; van Rooijen, N.; Heath, W.R.; Carbone, F.R. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 2008, 319, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Stock, A.T.; Ma, J.Z.; Jones, C.M.; Kent, S.J.; Mueller, S.N.; Heath, W.R.; Carbone, F.R.; Gebhardt, T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA 2012, 109, 7037–7042. [Google Scholar] [CrossRef]
- Steinbach, K.; Vincenti, I.; Kreutzfeldt, M.; Page, N.; Muschaweckh, A.; Wagner, I.; Drexler, I.; Pinschewer, D.; Korn, T.; Merkler, D. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J. Exp. Med. 2016, 213, 1571–1587. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Zhang, N.; Marshall, H.D.; Staron, M.M.; Guan, T.; Hu, Y.; Cauley, L.S.; Craft, J.; Kaech, S.M. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity 2014, 41, 633–645. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baxter, V.K.; Griffin, D.E. Interferon-Gamma Modulation of the Local T Cell Response to Alphavirus Encephalomyelitis. Viruses 2020, 12, 113. https://doi.org/10.3390/v12010113
Baxter VK, Griffin DE. Interferon-Gamma Modulation of the Local T Cell Response to Alphavirus Encephalomyelitis. Viruses. 2020; 12(1):113. https://doi.org/10.3390/v12010113
Chicago/Turabian StyleBaxter, Victoria K., and Diane E. Griffin. 2020. "Interferon-Gamma Modulation of the Local T Cell Response to Alphavirus Encephalomyelitis" Viruses 12, no. 1: 113. https://doi.org/10.3390/v12010113
APA StyleBaxter, V. K., & Griffin, D. E. (2020). Interferon-Gamma Modulation of the Local T Cell Response to Alphavirus Encephalomyelitis. Viruses, 12(1), 113. https://doi.org/10.3390/v12010113