Faecal Virome Analysis of Wild Animals from Brazil

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Viral Enrichment and Nucleic Acid Extraction
2.3. Sequence-Independent Amplification of Viral Nucleic Acids
2.4. Metagenomic Sequencing and Bioinformatics
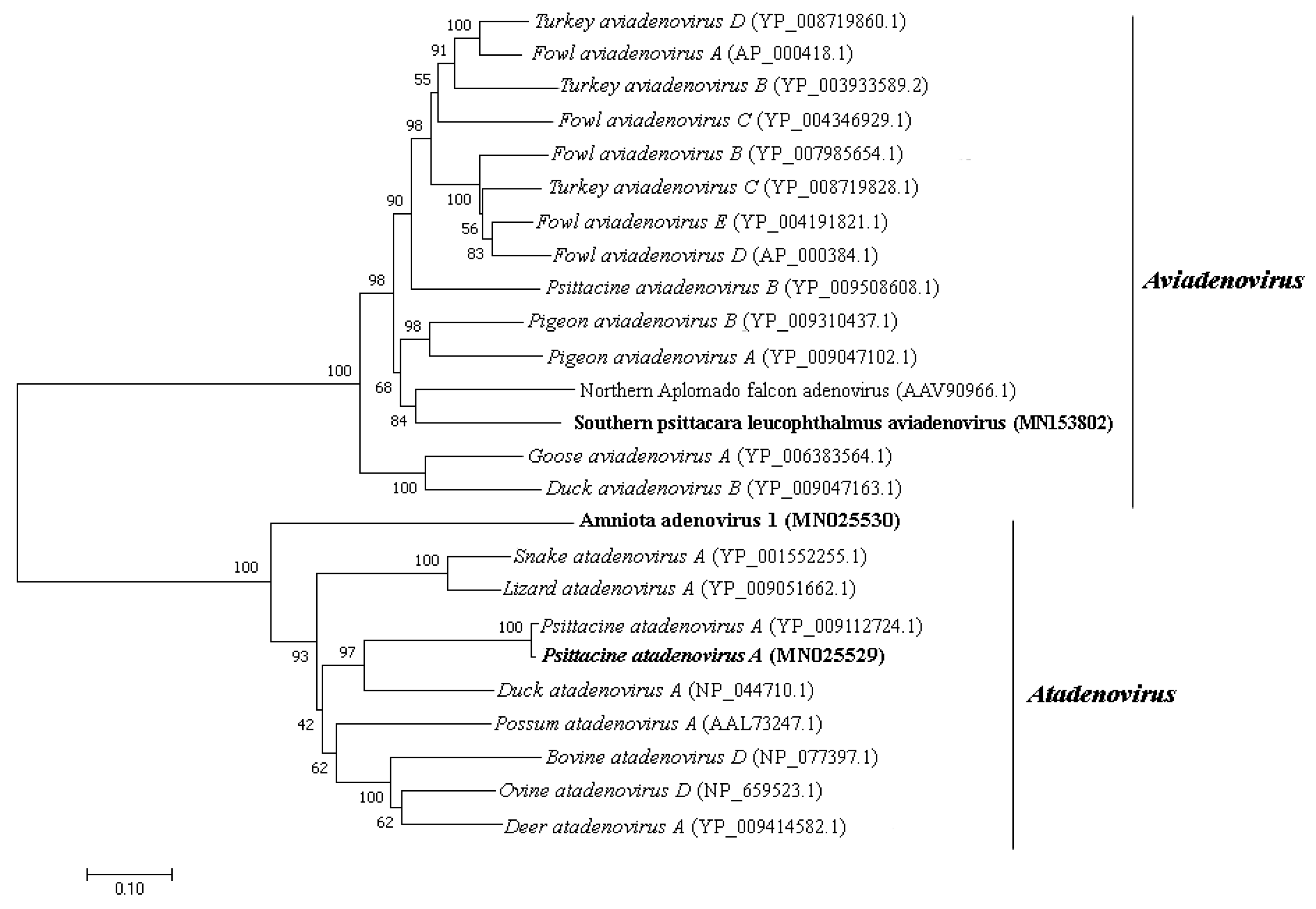
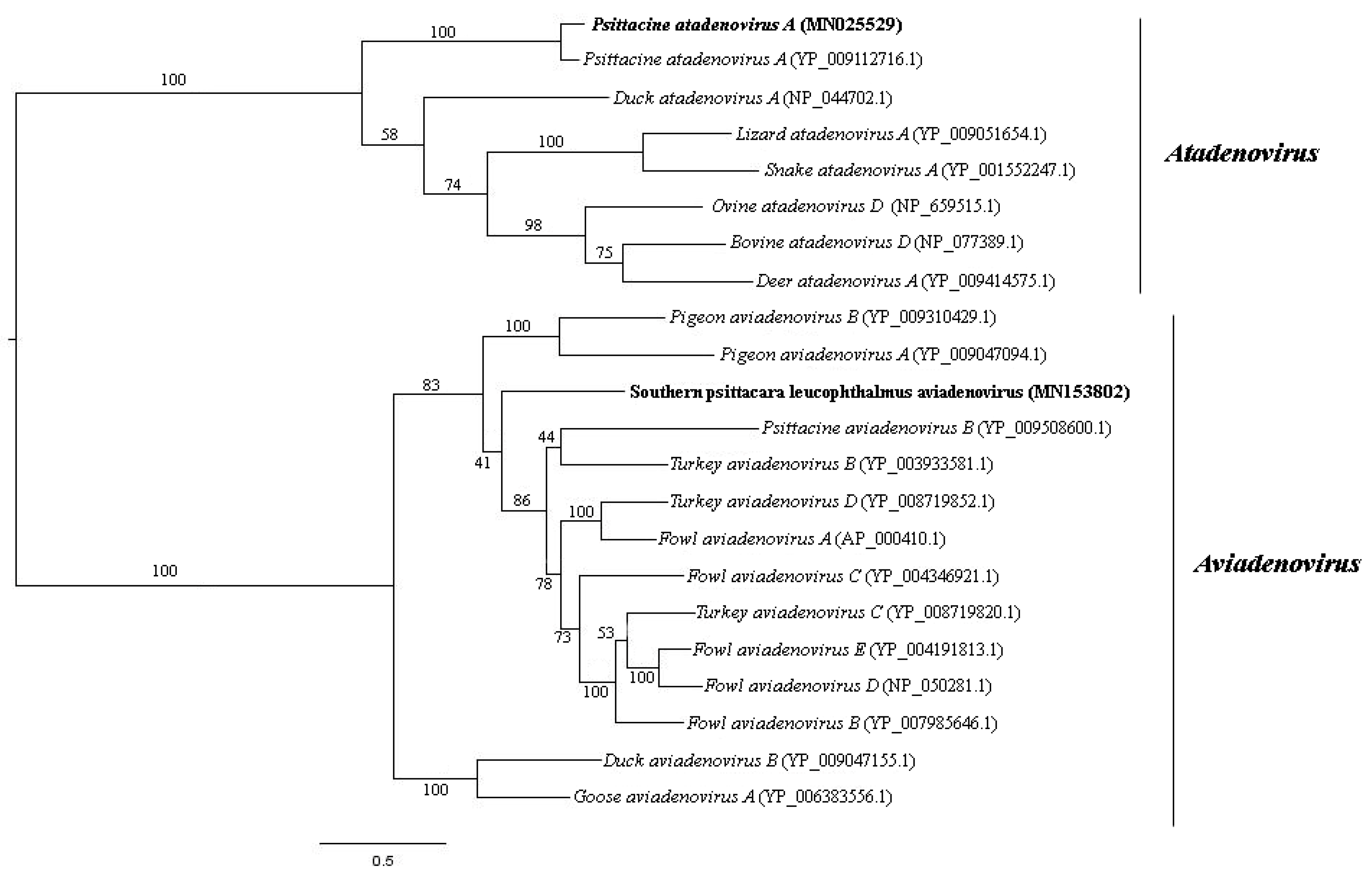
2.5. Phylogenetic Analysis
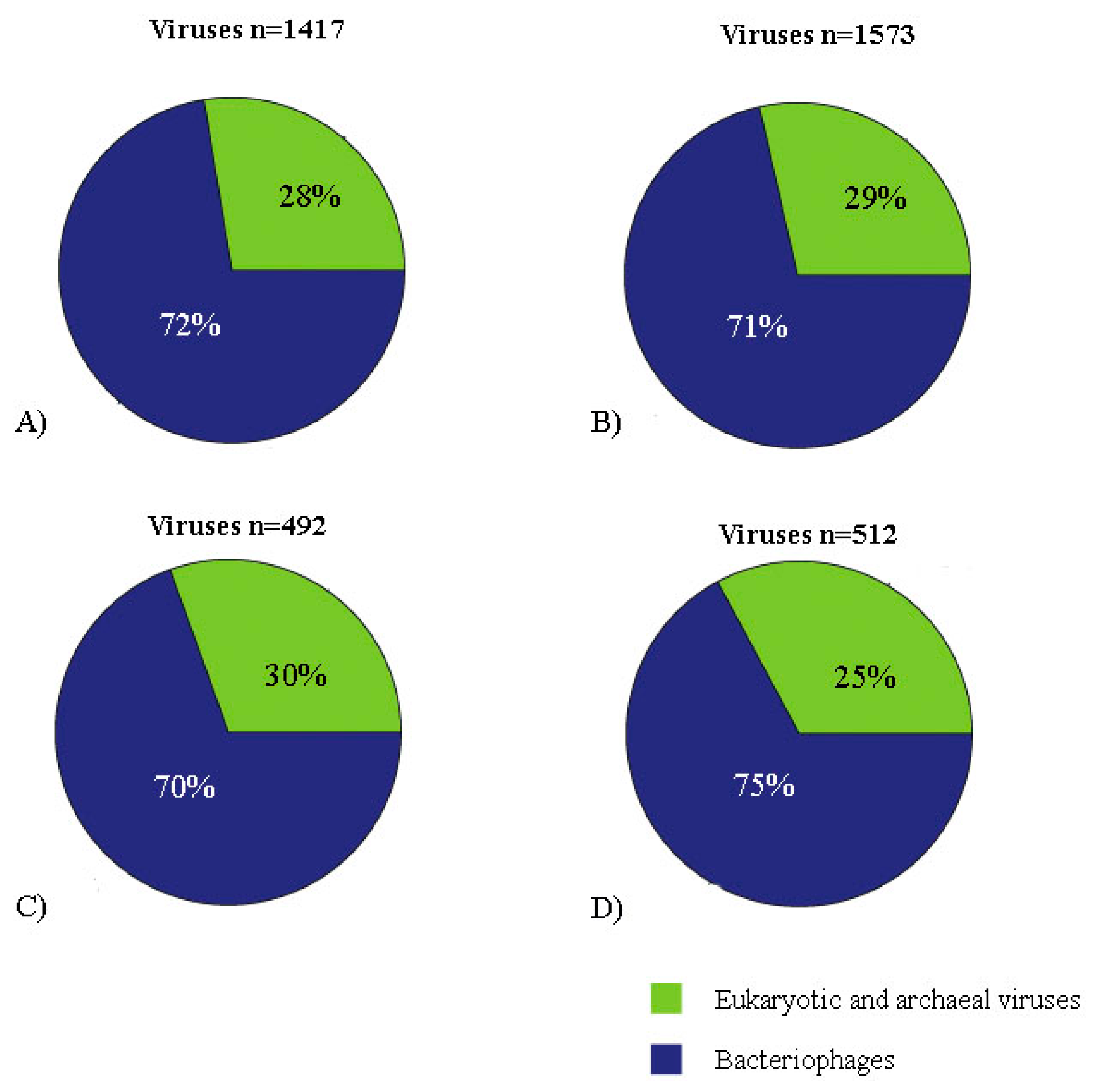
3. Results
3.1. Pool Information
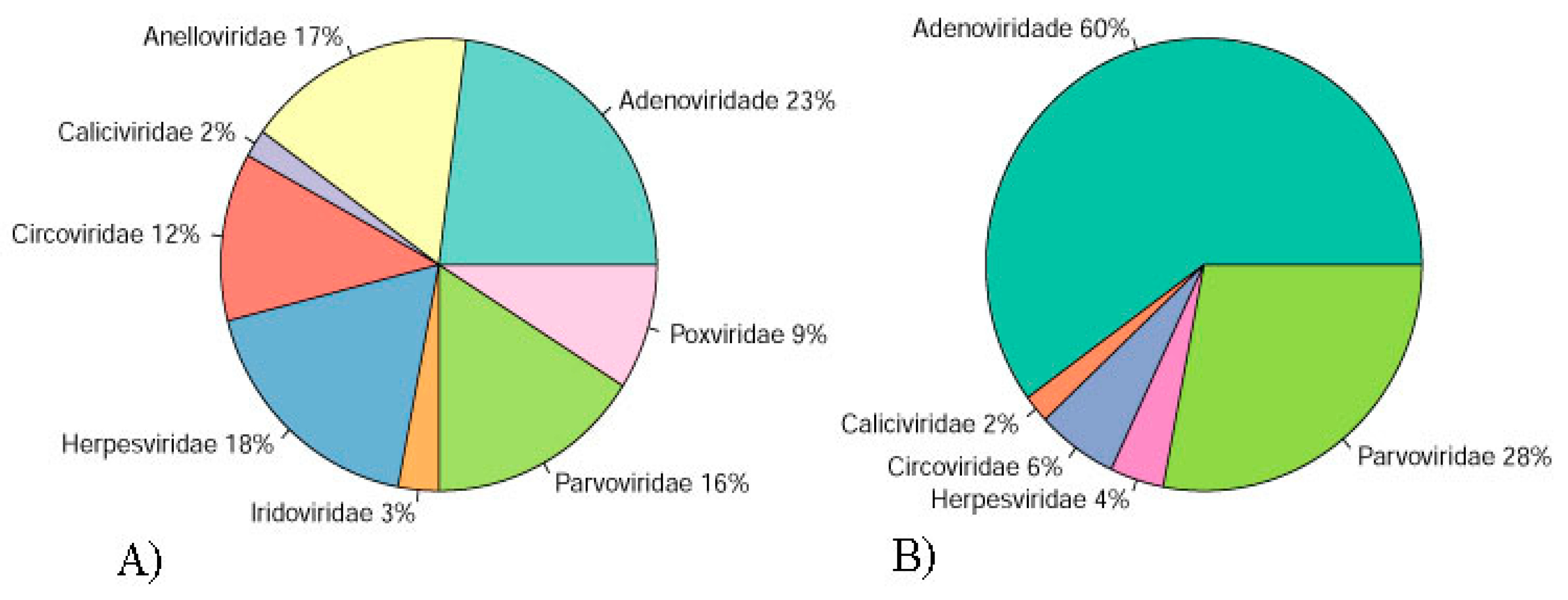
3.2. Pool 1
3.3. Pool 2
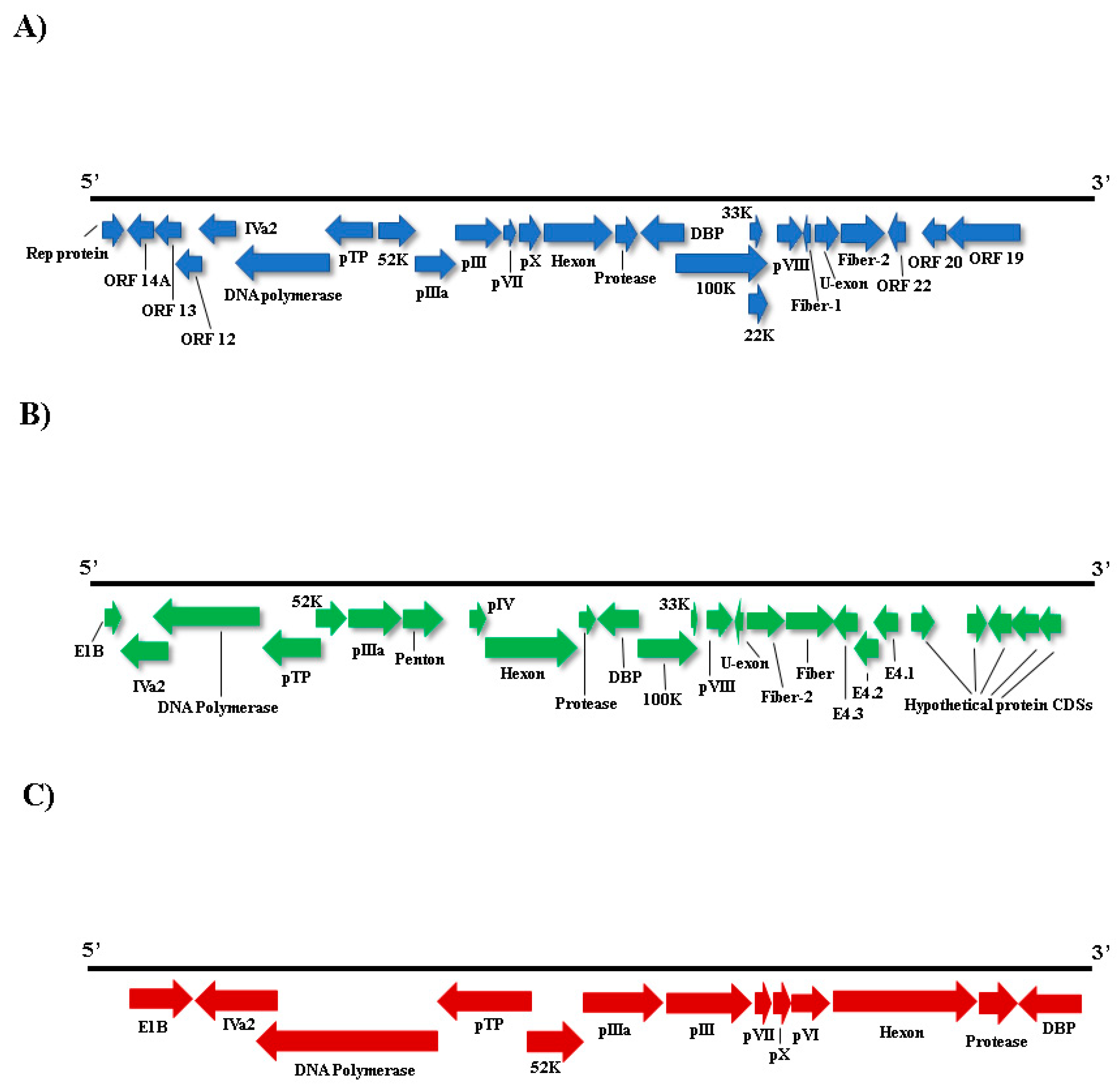
3.4. Adenoviridae
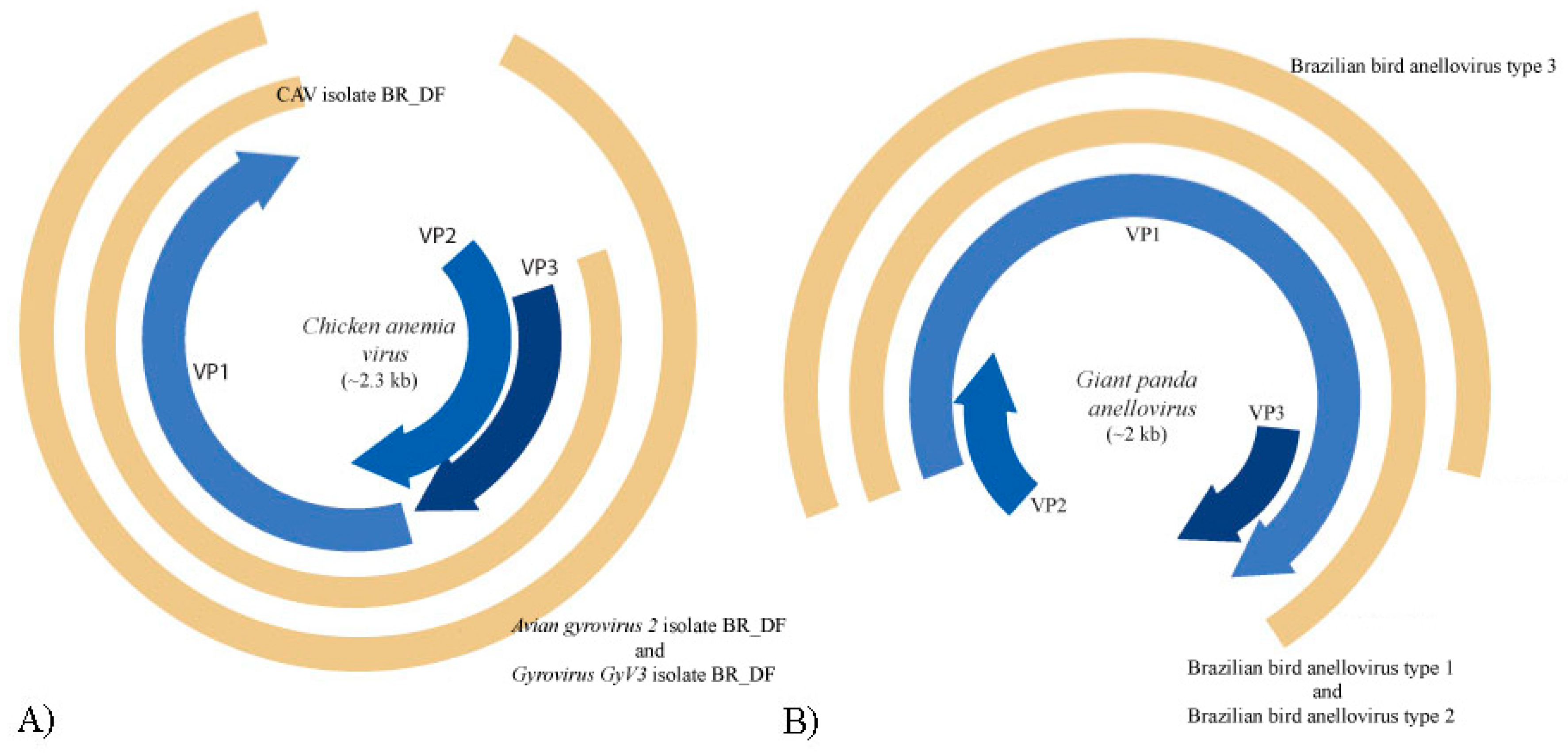
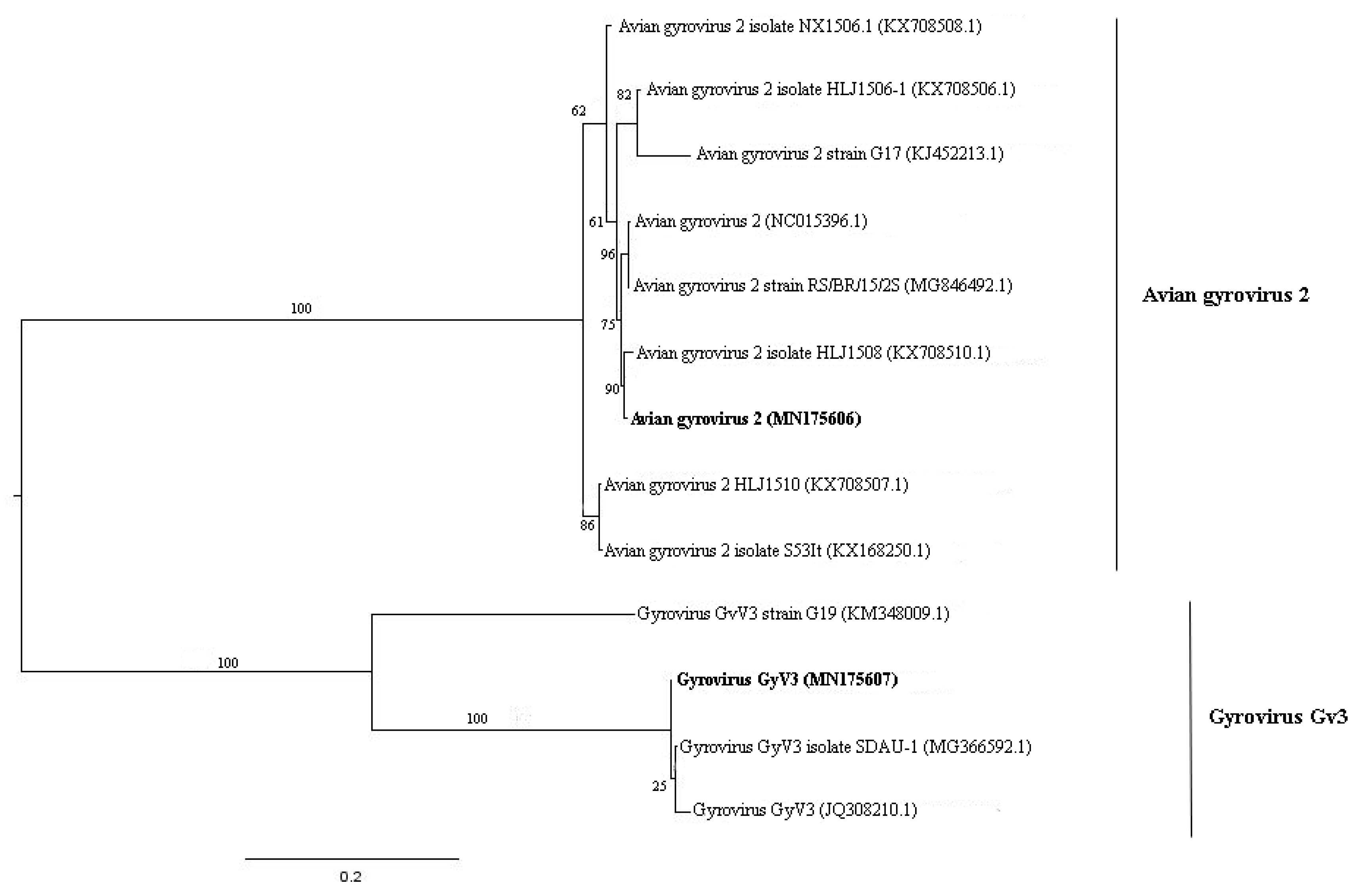
3.5. Anelloviridae
3.6. Caliciviridae
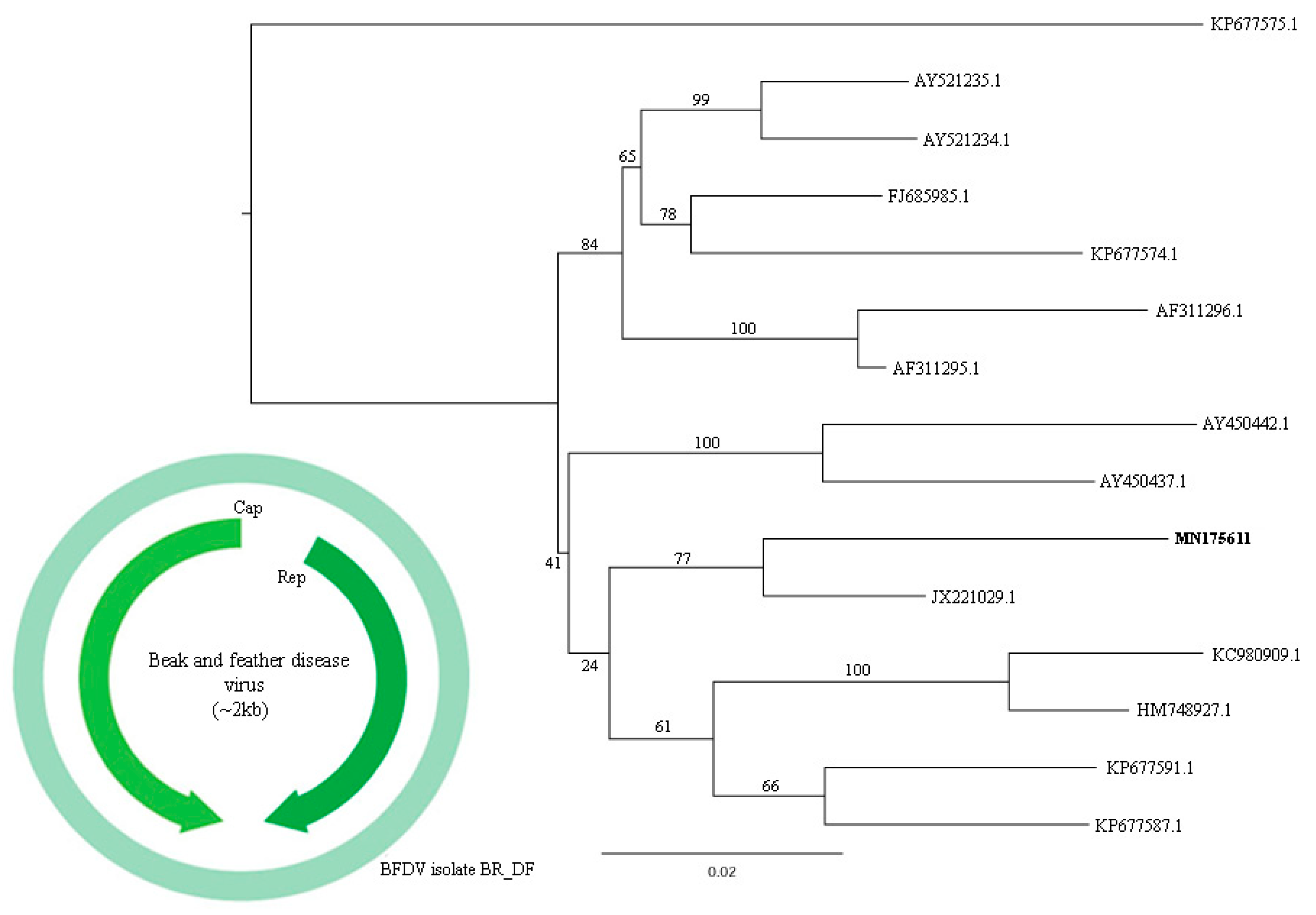
3.7. Circoviridae
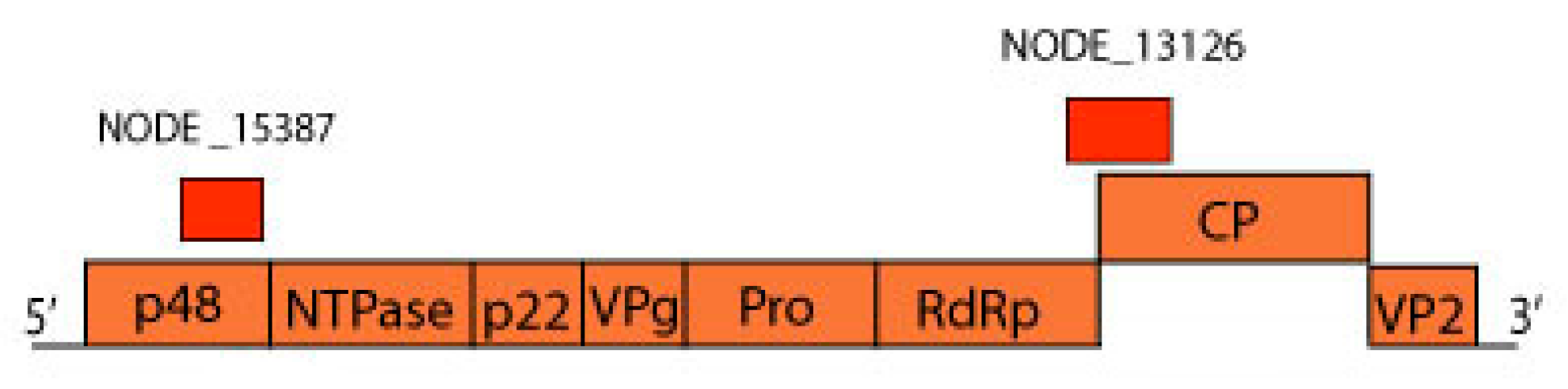
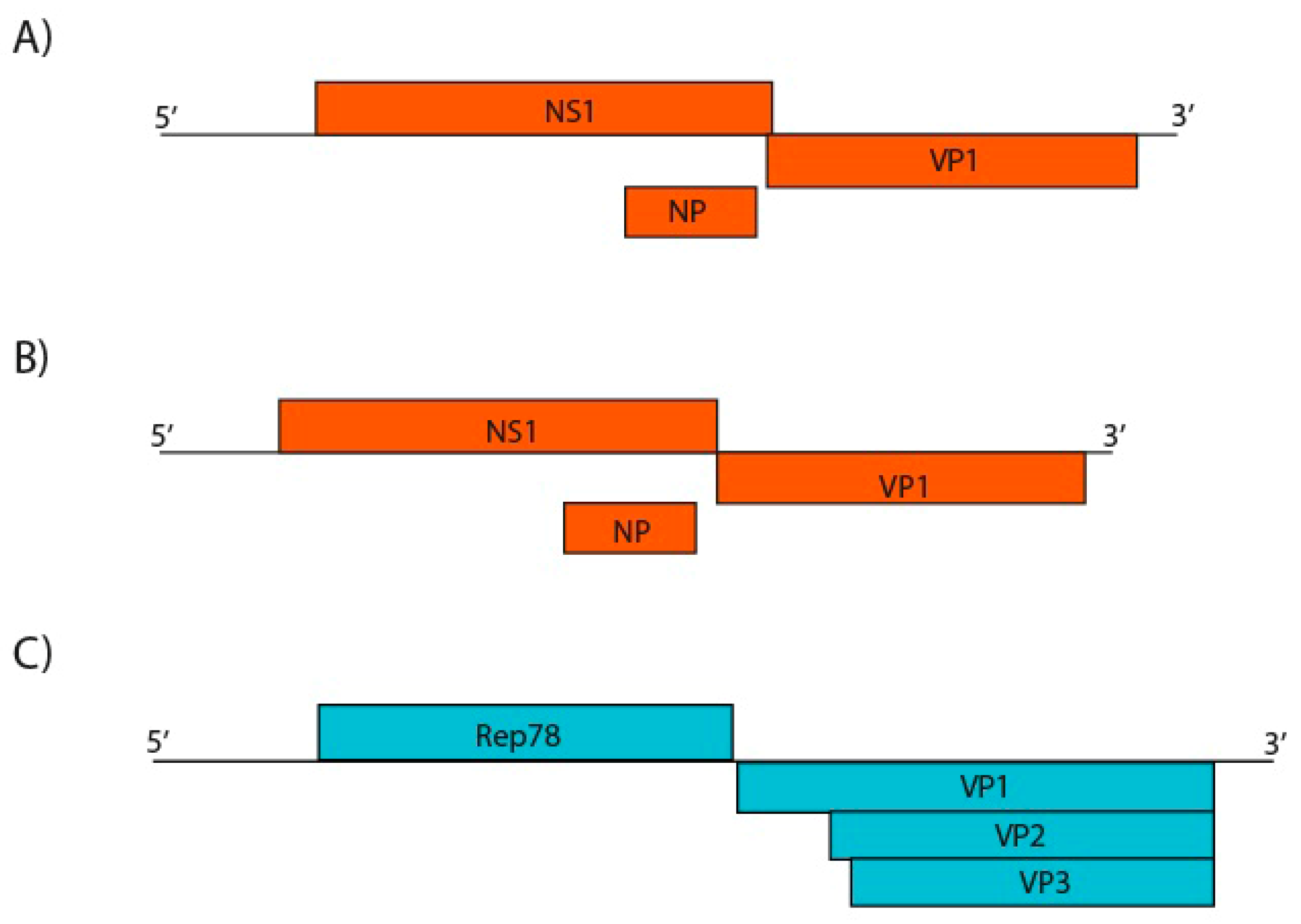
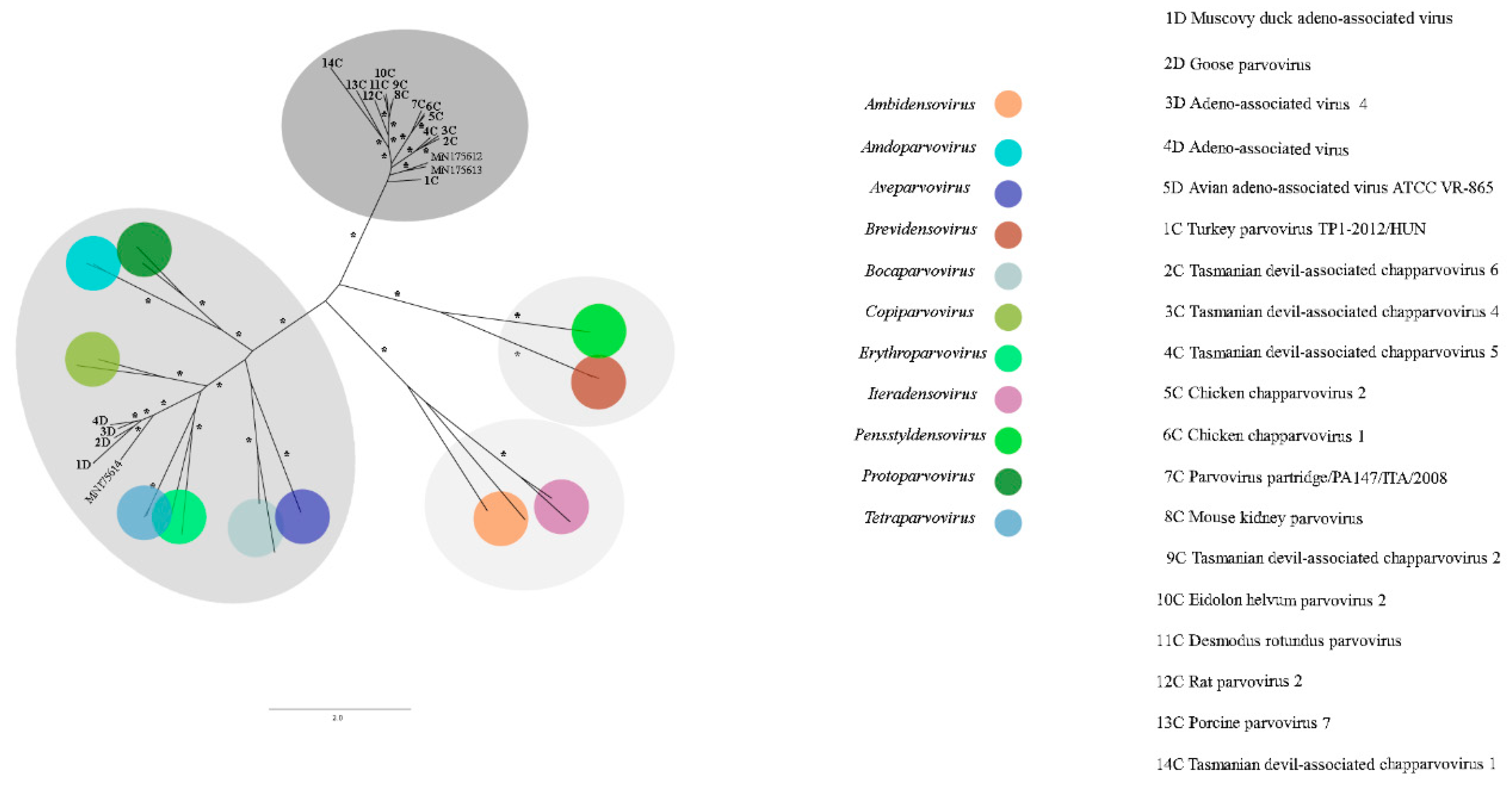
3.8. Parvoviridae
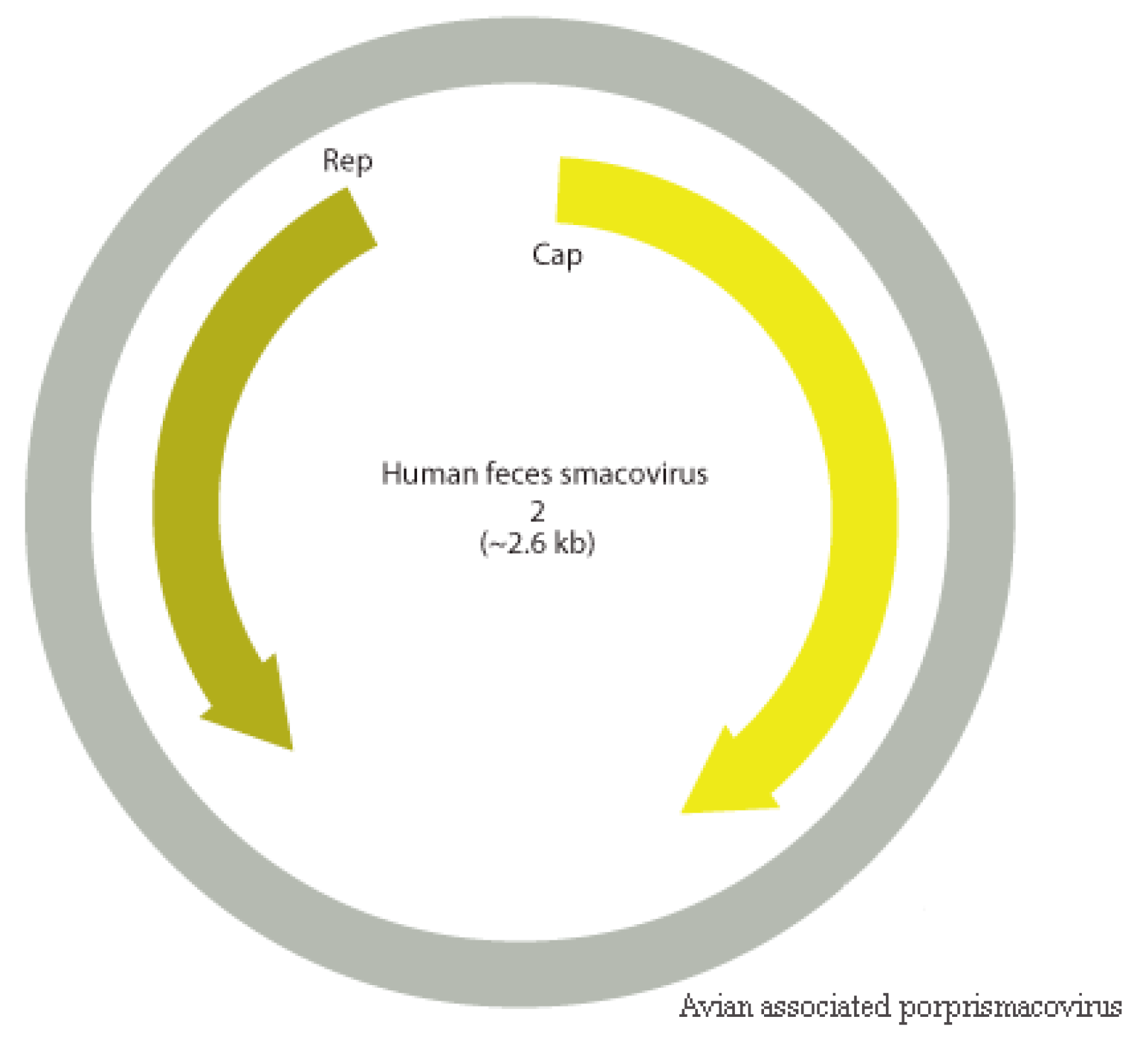
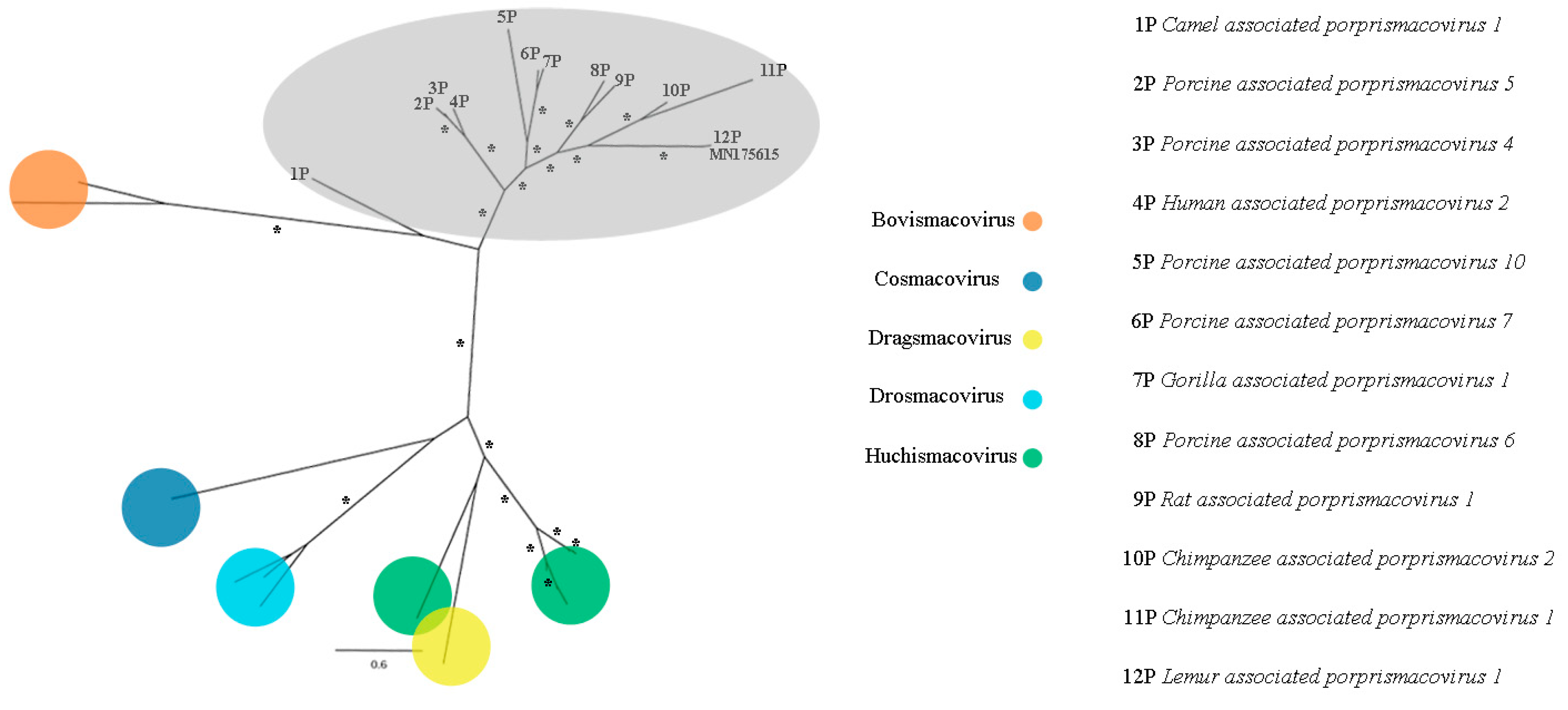
3.9. Smacoviridae
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Strassburg, B.; Brooks, T.; Feltran-Barbieri, R.; Iribarrem, A.; Crouzeilles, R.; Loyola, R.; Latawiec, A.; Oliveira Filho, F.J.; De Mattos Scaramuzza, C.; Scarano, F.; et al. Moment of truth for the Cerrado hotspot. Nat. Ecol. Evol. 2017, 1, 1–3. [Google Scholar] [CrossRef] [PubMed]
- De Figueiredo, M.L.G.; Figueiredo, L.T.M. Emerging alphaviruses in the Americas: Chikungunya and mayaro. Rev. Soc. Bras. Med. Trop. 2014, 47, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Auguste, A.; Terzian, A.; Vedovello, D.; Riet-Correa, F.; Macário, V.; Mourão, M.P.G.; Ullmann, L.S.; Weaver, S.C.; Nogueira, M.L.; et al. Isolation and characterization of Madariaga Virus from a horse in Paraíba state, Brazil. Transbound. Emerg. Dis. 2015, 64, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, H.; Bozidis, P.; Franks, A.; Papadopoulou, C. Oropouche fever: A review. Viruses 2018, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Donalisio, M.R.; Freitas, A.R.R.; Zuben, A.P.B.V. Arboviruses emerging in Brazil: Challenges for clinic and implications for public health. Rev. Saude Publica 2017, 51, 30. [Google Scholar] [CrossRef] [PubMed]
- Hotez, P. Neglected Tropical Diseases in the Anthropocene: The Cases of Zika, Ebola, and Other Infections. PLoS Negl. Trop. Dis. 2016, 10, e0004648. [Google Scholar] [CrossRef] [PubMed]
- Gummow, B. Challenges posed by new and re-emerging infectious diseases in livestock production, wildlife and humans. Livest. Sci. 2010, 130, 41–46. [Google Scholar] [CrossRef]
- Chomel, B.B.; Belotto, A.; Meslin, F.X. Wildlife, exotic pets, and emerging zoonoses. Emerg. Infect. Dis. 2007, 13, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.J. Bacterial and parasitic zoonoses of exotic pets. Vet. Clin. N. Am. Exot. Anim. Pract. 2009, 12, 401–415. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A. The Global Virome Project. Science 2018, 359, 872–874. [Google Scholar] [CrossRef] [PubMed]
- Stang, A.; Korn, K.; Wildner, O.; Uberla, K. Characterization of virus isolates by particle associated nucleic acid PCR. J. Clin. Microbiol. 2005, 43, 716–720. [Google Scholar] [CrossRef] [PubMed]
- FastQC: A quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 30 March 2019).
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinform 2015, 15, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.; Nikolenko, S.; Pham, S.; Prjibelski, A.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA7: Molecular evolutionary genetics analysis version 7.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Collar, N.; Boesman, P.; Sharpe, C.J. White-eyed Parakeet (Psittacara leucophthalmus). In Handbook of the Birds of the World Alive, 1st ed.; Del Hoyo, J., Elliott, A., Sargatal, J., Christie, D.A., de Juana, E., Eds.; Lynx Edicions: Barcelona, Spain, 2019. [Google Scholar]
- Rising, J.; Jaramillo, A. Saffron Finch (Sicalis flaveola). In Handbook of the Birds of the World Alive, 1st ed.; del Hoyo, J., Elliott, A., Sargatal, J., Christie, D.A., de Juana, E., Eds.; Lynx Edicions: Barcelona, Spain, 2019. [Google Scholar]
- Gardner, A.L. Order Didelphimorphia. In Mammal. Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, EUA, 2005. [Google Scholar]
- Groves, C.P. Order primates. In Mammal. Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, EUA, 2005. [Google Scholar]
- Favoretto, S.; Araújo, D.; Naylê, F.H.; Oliveira, D.; da Crus, N.G.; Mesquita, F.; Leal, F.; Machado, R.R.G.; Gaio, F.; Oliveira, W.F.; et al. Zika Virus in peridomestic neotropical primates, Northeast Brazil. Ecohealth 2019, 16, 61–69. [Google Scholar] [CrossRef]
- Wozencraft, W.C. Order Carnivora. In Mammal. Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, EUA, 2005. [Google Scholar]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Family Adenoviridae. In Virus Taxonomy: 9th Report of the International Committee on Taxonomy of Viruses; Elsevier: Oxford, UK, 2011. [Google Scholar]
- Binder, A.M.; Biggs, H.M.; Haynes, A.K.; Chommanard, C.; Lu, X.; Erdman, D.D.; Watson, J.T.; Gerber, S.I. Human adenovirus surveillance—United States, 2003–2016. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 1039–1042. [Google Scholar] [CrossRef]
- Li, Y.; Ge, X.; Zhang, H.; Zhou, P.; Zhu, Y.; Zhang, Y.; Yuan, J.; Wang, L.F.; Shi, Z. Host range, prevalence, and genetic diversity of adenoviruses in bats. J. Virol. 2010, 84, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Souza, W.; Fumagalli, M.; Araujo, J.; Sabino-Santos Jr, G.; Felipe, G.M.M.; Romeiro, M.; Modha, S.; Nardi, M.S.; Queiroz, L.; Durigon, E.L.; et al. Discovery of novel anelloviruses in small mammals expands the host range and diversity of the Anelloviridae. Virology 2017, 514, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kemenesi, G.; Gellért, Á.; Dallos, B.; Görföl, T.; Boldogh, S.; Estók, P.; Marton, S.; Oldal, M.; Martella, V.; Bányai, K.; et al. Sequencing and molecular modeling identifies candidate members of Caliciviridae family in bats. Infect. Genet. Evol. 2016, 41, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Summa, M.; Henttonen, H.; Maunula, L. Human noroviruses in the faeces of wild birds and rodents-new potential transmission routes. Zoonoses Public Health 2018, 65, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segalés, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family circoviridae: Establishment of the genus cyclovirus and removal of the genus gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Ghorashi, S.A.; Forwood, J.K.; Bent, S.J.; Peters, A.; Raidal, S.R. Phylogeny of beak and feather disease virus in cockatoos demonstrates host generalism and multiple-variant infections within Psittaciformes. Virology 2014, 460, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, M.; Mortlock, M.; Weyer, J.; Bezuidt, O.; Seamark, E.C.J.; Kearney, T.; Gleasner, C.; Erkkila, T.H.; Cui, H.; Markotter, W. A metagenomic viral discovery approach identifies potential zoonotic and novel mammalian viruses in Neoromicia bats within South Africa. PLoS ONE 2018, 13, e0194527. [Google Scholar] [CrossRef]
- François, S.; Filloux, D.; Roumagnac, P.; Bigot, D.; Gayral, P.; Martin, D.P.; Froissart, R.; Ogliastro, M. Discovery of parvovirus-related-sequences in an unexpected broad range of animals. Sci. Rep. 2016, 6, 30880. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; To, K.K.-W.; Chen, H.; Yuen, K.-Y. Cross-species transmission and emergence of novel viruses from birds. Curr. Opin. Virol. 2015, 10, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, R. Novel viruses in birds: Flying through the roof or is a cage needed? Vet. J. 2018, 233, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Fagbo, S.F.; Alagaili, A.n.; Nitido, A.; Williams, S.H.; Lee, J.B.; Durosinlorun, A.; Garcia, J.E.; Jain, K.; Kapoor, V.; et al. A viral metagenomic survey identifies known and novel mammalian viruses in bats from Saudi Arabia. PLoS ONE 2019, 14, e0214227. [Google Scholar] [CrossRef] [PubMed]
- Conceição-Neto, N.; Godinho, R.; Álvares, F.; Yinda, K.; Deboutte, W.; Zeller, M.; Laenen, L.; Heylen, E.; Roque, S.; Petrucci-Fonseca, F.; et al. Viral gut metagenomics of sympatric wild and domestic canids, and monitoring of viruses: Insights from an endangered wolf population. Ecol. Evol. 2017, 7, 4135–4146. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses Virus Taxonomy: 2018b Release. Available online: talk.ictvonline:ictv-reports/ictv_9th_report (accessed on 18 August 2019).
- To, K.K.W.; Tse, H.; Chan, W.-M.; Choi, G.K.Y.; Anna, J.X.Z.; Sridhar, S.; Wong, S.; Chan, J.; Chan, A.S.F.; Woo, P.C.Y.; et al. A novel psittacine adenovirus identified during an outbreak of avian chlamydiosis and human psittacosis: Zoonosis associated with virus-bacterium coinfection in birds. PLoS Negl. Trop. Dis. 2014, 8, e3318. [Google Scholar] [CrossRef]
- Hess, M.; Blöcker, H.; Brandt, P. The complete nucleotide sequence of the egg drop syndrome Virus: An intermediate between Mastadenoviruses and Aviadenoviruses. Virology 1997, 238, 145–156. [Google Scholar] [CrossRef]
- Schrenzel, M.; Oaks, J.L.; & Rotstein, D.; Maalouf, G.; Snook, E.; Sandfort, C.; Rideout, B. Characterization of a new species of adenovirus in falcons. J. Clin. Microbiol. 2005, 43, 3402–3413. [Google Scholar] [CrossRef]
- Schachner, A.; Matos, M.; Grafl, B.; Hess, M. Fowl adenovirus-induced diseases and strategies for their control—A review on the current global situation. Avian Pathol. 2017, 47, 111–126. [Google Scholar] [CrossRef]
- Chiocca, S.; Kurzbauer, R.; Schaffner, G.; Baker, A.; Mautner, V.; Cotten, M. The complete DNA sequence and genomic organization of the avian adenovirus CELO. J. Virol. 1996, 70, 2939–2949. [Google Scholar] [PubMed]
- Phan, T.G.; Li, L.; O’Ryan, M.G.; Cortes, H.; Mamani, N.; Bonkoungou, I.J.O.; Wang, C.; Leutenegger, C.M.; Delwartcorresponding, E. A third gyrovirus species in human faeces. J. Gen. Virol. 2012, 93, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Smuts, H. Novel gyroviruses, including chicken anaemia virus, in clinical and chicken samples from South Africa. Adv. Virol. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pesavento, P.A.; Gaynor, A.M.; Duerr, R.S.; Phan, T.G.; Zhang, W.; Deng, X.; Delwart, E. A gyrovirus infecting a sea bird. Arch. Virol. 2015, 160, 2105–2109. [Google Scholar] [CrossRef] [PubMed]
- Shuai, Y.; Tianbei, T.; Xiang, G.; Chunyan, H.; Nana, Y.; Aijing, L.; Honglei, G.; Yulong, G.; Hongyu, C.; Changjun, L.; et al. Molecular epidemiology of chicken anaemia virus in sick chickens in China from 2014 to 2015. PLoS ONE 2019, 14, e0210696. [Google Scholar] [CrossRef]
- Farkas, T.; Maeda, K.; Sugiura, H.; Kai, K.; Hirai, K.; Otsuki, K.; Hayashi, T. A serological survey of chickens, Japanese quail, pigeons, ducks and crows for antibodies to chicken anaemia virus (CAV) in Japan. Avian Pathol. 1998, 27, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.-B.; Chang, H.-M.; Wu, W.-C.; Chou, Y.-C.; Yu, C.-P. First detection of chicken anemia virus and norovirus genogrupo II in stoll of children with acute gastroenteritis in Taiwan. Southeast Asian J. Trop. Med. Public Health 2016, 47, 416–423. [Google Scholar]
- Lichun, F.; Yang, L.; Yixin, W.; Jiayuan, F.; Shuai, C.; Xiaohan, L.; Shuang, C.; Peng, Z. Genetic analysis of two chicken infectious anemia virus variants-related gyrovirus in stray mice and dogs: The first report in China, 2015. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Xinheng, Z.; Yuanjia, L.; Jun, J.; Feng, C.; Baoli, S.; Chunyi, X.; Jingyun, M.; Yingzuo, B.; Qingmei, X. Identification of a chicken anemia virus variant-related Gyrovirus in stray cats in China, 2012. Biomed. Res. Int. 2012, 2014. [Google Scholar] [CrossRef]
- Maggi, F.; Macera, L.; Focosi, D.; Vatteroni, M.L.; Boggi, U.; Antonelli, G.; Eloit, M.; Pistello, M. Human gyrovirus DNA in human blood, Italy. Emerg. Infect. Dis. 2012, 18, 956–959. [Google Scholar] [CrossRef]
- Chu, D.K.; Poon, L.L.; Chiu, S.S.; Chan, K.H.; Ng, E.M.; Bauer, I.; Cheung, T.K.; Ng, I.H.; Guan, Y.; Wang, D.; et al. Characterization of a novel gyrovirus in human stool and chicken meat. J. Clin. Virol. 2012, 55, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Rijsewijk, F.A.; Dos Santos, H.F.; Teixeira, T.F.; Cibulski, S.P.; Varela, A.P.; Dezen, D.; Franco, A.C.; Roehe, P.M. Discovery of a genome of a distant relative of chicken anemia virus reveals a new member of the genus Gyrovirus. Arch. Virol. 2011, 156, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Abolnik, C.; Wandrag, D. Avian gyrovirus 2 and avirulent Newcastle disease virus coinfection in a chicken flock with neurologic symptoms and high Mortalities. Avian Dis. 2014, 58, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Biagini, P.; Bedarida, S.; Touinssi, M.; Galicher, V.; Philippe de, M. Human gyrovirus in healthy blood donors, France. Emerg. Infect. Dis. 2013, 19, 1014–1015. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Gao, X.; Tuo, T.; Han, C.; Gao, Y.; Qi, X.; Zhang, Y.; Liu, C.; Gao, H.; Wang, Y.; et al. Novel characteristics of the avian gyrovirus 2 genome. Sci. Rep. 2017, 7, 41068. [Google Scholar] [CrossRef] [PubMed]
- Fehér, E.; Pazár, P.; Mihalov-Kovács, E.; Farkas, S.; Lengyel, G.; Jakab, F.; Martella, V.; Banyai, K. Molecular detection and characterization of human gyroviruses identified in the ferret fecal virome. Arch. Virol. 2014, 159, 3401–3406. [Google Scholar] [CrossRef]
- Li, G.; Yuan, S.; He, M.; Zhao, M.; Hao, X.; Song, M.; Zhang, L.; Qiao, C.; Huang, L.; Zhang, L.; et al. Emergence of gyrovirus 3 in commercial broiler chickens with transmissible viral proventriculitis. Transbound. Emerg. Dis. 2018, 65, 1170–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, S.; Shan, T.; Hou, R.; Liu, Z.; Li, W.; Guo, L.; Wang, Y.; Chen, P.; Wang, X.; et al. Virome comparisons in wild-diseased and healthy captive giant pandas. Microbiome 2017, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, H.; Wang, Y.; Liu, Z.; Li, J.; Guo, L.; Yang, S.; Shen, Q.; Zhao, X.; Cui, L.; et al. Identification and genomic characterization of a novel species of feline anellovirus. Virol. J. 2016, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Takahashi, M.; Nishizawa, T.; Tawara, A.; Fukai, K.; Muramatsu, U.; Naito, Y.; Yoshikawa, A.J. Genomic characterization of TT viruses (TTVs) in pigs, cats and dogs and their relatedness with species-specific TTVs in primates and tupaias. Gen. Virol. 2002, 83, 1291–1297. [Google Scholar] [CrossRef]
- Pass, D.A.; Perry, R.A. The pathology of psittacine beak and feather disease. Aust. Vet. J. 1984, 61, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, B.W.; Niagro, F.D.; Lukert, P.D.; Steffens, W.L.; Latimer, K.S. Characterization of a new virus from cockatoos with psittacine beak and feather disease. Virology 1989, 171, 83–88. [Google Scholar] [CrossRef]
- Todd, D. Circoviruses: Immunosuppressive threats to avian species: A review. Avian Pathol. 2000, 29, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Catedral, L.; McInnes, K.; Hauber, M.E.; Brunton, D.H. First report of beak and feather disease virus (BFDV) in wild Red-fronted Parakeets (Cyanoramphus novaezelandiae) in New Zealand. Emu 2009, 109, 244–247. [Google Scholar] [CrossRef]
- Werther, K.; Raso, T.F.; Durigon, E.L.; Latimer, K.S.; Campagnoli, R.P. Description of the first case of psittacine beak and feather disease in Brazil. In Proceedings of the International Virtual Conferences in Veterinary Medicine: Diseases of Psittacine Birds, Athens, GA, USA, 15–30 June 1998. [Google Scholar]
- Weizhong, L.; Adam, G. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Todd, K.; Tripp, R. Human Norovirus: Experimental Models of Infection. Viruses 2019, 11, 151. [Google Scholar] [CrossRef]
- Siebenga, J.J.; Vennema, H.; Zheng, D.P.; Vinje’, J.; Lee, B.E.; Pang, X.L.; Ho, E.C.; Lim, W.; Choudekar, A.; Broor, S.; et al. Norovirus illness is a global problem: Emergence and spread of norovirus GII.4 variants. J. Infect. Dis. 2009, 200, 802–812. [Google Scholar] [CrossRef]
- Mesquita, J.R.; Costantini, V.P.; Cannon, J.L.; Lin, S.C.; Nascimento, M.S.; Vinjé, J. Presence of antibodies against genogroup VI norovirus in humans. Virol. J. 2013, 10, 176. [Google Scholar] [CrossRef]
- Wang, Q.H.; Han, M.; Cheetham, S.; Souza, M.; Funk, J.; Saif, L. Porcine noroviruses related to human noroviruses. Emerg. Infect. Dis. 2005, 11, 1874–1881. [Google Scholar] [CrossRef]
- Martella, V.; Lorusso, E.; Decaro, N.; Elia, G.; Radogna, A.; D’Abramo, M.; Buonavoglia, C. Detection and molecular characterization of a canine norovirus. Emerg. Infect. Dis. 2008, 14, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Pinto, P.; Wang, Q.; Chen, N.; Dubovi, E.; Daniels, J.B.; Millward, L.M.; Buonavoglia, C.; Martella, V.; Saif, L. Discovery and Genomic Characterization of noroviruses from a gastroenteritis outbreak in domestic cats in the US. PLoS ONE 2012, 7, e32739. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Campolo, M.; Lorusso, E.; Cavicchio, P.; Camero, M.; Bellacicco, A.L.; Decaro, N.; Elia, G.; Greco, G.; Corrente, M.; et al. Norovirus in captive lion cub (Panthera leo). Emerg. Infect. Dis. 2007, 13, 1071–1073. [Google Scholar] [CrossRef]
- Caddy, S.; Emmott, E.; El-Attar, L.; Mitchell, J.; Rougemont, A.; Brownlie, J.; Goodfellow, I. Serological evidence for multiple strains of canine norovirus in the UK dog population. PLoS ONE 2013, 8, e81596. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.P.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.S.; Leggett, R.M.; Bexfield, N.H.; Alston, M.; Daly, G.; Todd, S.; Tachedjian, M.; Holmes, C.; Crameri, S.; Wang, L.F.; et al. Metagenomic study of the viruses of African straw-coloured fruit bats: Detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology 2013, 441, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Lai, T.T.; Chen, Q.Y.; Wu, X.M.; Che, Y.L.; Yan, S.; Wang, C.Y.; Wang, L.B.; Zhou, L.J. Classification tree method for determining factors that affecting hatchability in chukar partridge (Alectoris chukar) eggs. Kafkas Univ. Vet. Fak. Derg 2018, 24, 473–477. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Delwart, E.; Pankovics, P. Novel circular single-stranded DNA virus from turkey faeces. Arch. Virol. 2014, 159, 2161–2164. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Z.; Wang, Y.; Li, W.; Fu, X.; Lin, Y.; Shen, Q.; Wang, X.; Wang, H.; Zhang, W. A novel rodent Chapparvovirus in feces of wild rats. Virol. J. 2016, 13, 133. [Google Scholar] [CrossRef]
- Chong, R.; Shi, M.; Grueber, C.; Holmes, E.; Hogg, C.; Belov, K.; Barrs, V. Characterisation of the faecal virome of captive and wild Tasmanian devils using virus-like particles metagenomics and meta-transcriptomics. bioRxivorg 2018, 443457. [Google Scholar] [CrossRef]
- Lima, D.; Cibulski, S.; Tochetto, C.; Varela, A.P.M.; Finkler, F.; Teixeira, T.F.; Loiko, M.; Cerva, C.; Junqueira, D.; Mayer, F.; et al. The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res. 2018, 261, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, S.; Liu, D.; Zhou, C.; Li, W.; Lin, Y.; Wang, X.; Shen, Q.; Wang, H.; Li, C.; et al. The fecal virome of red-crowned cranes. Arch. Virol. 2018, 164, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral Diversity of House Mice in New York City. MBio 2018, 9, e01354–17. [Google Scholar] [CrossRef] [PubMed]
- Palinski, R.M.; Mitra, N.; Hause, B.M. Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs. Virus Genes 2016, 52, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Souza, W.; Romeiro, M.; Fumagalli, M.; Modha, S.; Araujo, J.; Queiroz, L.; Durigon, E.L.; Figueiredo, L.T.M.; Murcia, P.; Gifford, R. Chapparvoviruses occur in at least three vertebrate classes and have a broad biogeographic distribution. J. Gen. Virol. 2017, 98, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.; Agbandje-McKenna, M.; Souza, W.; Pénzes, J. An ancient lineage of highly divergent parvoviruses infects both vertebrate and invertebrate hosts. bioRxivorg 2019, 11, 525. [Google Scholar] [CrossRef]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.; Henderson, J.; Jormakka, M.; O’Rourke, M.; Padula, M.; Pinello, N.; Henry, M.; et al. An atypical parvovirus drives chronic tubulointerstitial nephropathy and kidney fibrosis. Cell 2018, 175, 530–543. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. Viruses and Human Disease, 2nd ed.; Academic Press: Cambridge, MA, USA, 2008; pp. 261–323. [Google Scholar]
- Berns, K.I.; Muzyczka, N. AAV: An Overview of unanswered questions. Hum. Gene. Ther. 2017, 28, 308–313. [Google Scholar] [CrossRef]
- Díez-Villaseñor, C.; Rodríguez-Valera, F. CRISPR analysis suggests that small circular single-stranded DNA smacoviruses infect Archaea instead of humans. Nat. Commun. 2018, 10, 294. [Google Scholar] [CrossRef]
- Fontenele, R.S.; Lacorte, C.; Lamas, N.S.; Schmidlin, K.; Varsani, A.; Ribeiro, S.G. Single stranded DNA viruses associated with capybara faeces sampled in Brazil. Viruses 2019, 11, 710. [Google Scholar] [CrossRef]
- Kluge, M.; Campos, F.; Tavares, M.; Amorim, D.; Pedone Valdez, F.; Giongo, A.; Roehe, P.; Franco, A. Metagenomic survey of viral diversity obtained from feces of subantarctic and south american fur seals. PLoS ONE 2016, 11, e0151921. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, L.O.; Larsson, J.; Hayer, J.; Berg, M.; Jacobson, M. The intestinal eukaryotic virome in healthy and diarrhoeic neonatal piglets. PLoS ONE 2016, 11, e0151481. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pool Number | Contig ID | GenBank Accession Number | Virus Name | Closely Related Virus Type | Pairwise Identity (Hexon aa) | Pairwise Identity (DNA Polymerase aa) |
|---|---|---|---|---|---|---|
| 2 | k119 1050 | MN025530 | Amniota adenovirus 1 | Duck atadenovirus A (NP_044710.1) | 64.5 %. | 41.9 % |
| 2 | k119 2350 | MN025529 | Psittacine adenovirus 3 | Psittacine adenovirus 3 (YP_009112724.1) | 97.6 % | --- |
| 2 | k119 2155 | MN025529 | Psittacine adenovirus 3 | Psittacine adenovirus 3 (YP009112716.1) | ---- | 90.2 % |
| 2 | k119 380 | MN153802 | Southern psittacara leucophthalmus aviadenovirus | Fowl aviadenovirus A (AP_000410.1) | --- | 61.7 % |
| 2 | NODE 39 | MN153802 | Southern psittacara leucophthalmus aviadenovirus | Northern Aplomado falcon adenovirus (AAV90966.1) | 73.9 % | --- |
| Pool Number | Contig ID | GenBank Accession Number | Virus Name | Closely Related Virus Type | Pairwise Identity (ORF nt) |
|---|---|---|---|---|---|
| 1 | k119 16753 | MN175605 | Chicken anemia virus | Chicken anemia virus (JQ308213.1) | 98.8%. |
| 1 | k119 6992 | MN175606 | Avian gyrovirus 2 | Avian gyrovirus 2 (KX708510.1) | 98.8%. |
| 1 | k119 6843 | MN175607 | Gyrovirus GyV3 | Gyrovirus GyV3 (MG366592.1) | 99.4% |
| 1 | NODE 177 | MN175608 | Brazilian bird anellovirus type 1 | Giant panda anellovirus (MF327552.1) | 54.6%, |
| 1 | NODE 986 | MN175609 | Brazilian bird anellovirus type 2 | Giant panda anellovirus (MF327552.1) | 50.7% |
| 1 | NODE 1090 | MN175610 | Brazilian bird anellovirus type 3 | Giant panda anellovirus (MF327552.1) | 51.7% |
| Pool Number | Contig ID | GenBank Accession Number | Virus Name | Closely Related Virus Type | Pairwise Identity (NS1 aa) |
|---|---|---|---|---|---|
| 1 | k119 1463 | MN175612 | Avian chapparvovirus | Turkey parvovirus TP1-2012/HUN (AHF54687.1) | 45.3% |
| 2 | k119 15398 | MN175613 | Psittacara leucophthalmus chapparvovirus | Turkey parvovirus TP1-2012/HUN (AHF54687.1) | 44.9% |
| 1 | k119 1997 | MN175614 | Avian adeno-associated virus isolate BR_DF | Adeno-associated virus (YP_009552823.1)) | 42.70% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
A. Duarte, M.; F. Silva, J.M.; R. Brito, C.; S. Teixeira, D.; L. Melo, F.; M. Ribeiro, B.; Nagata, T.; S. Campos, F. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. https://doi.org/10.3390/v11090803
A. Duarte M, F. Silva JM, R. Brito C, S. Teixeira D, L. Melo F, M. Ribeiro B, Nagata T, S. Campos F. Faecal Virome Analysis of Wild Animals from Brazil. Viruses. 2019; 11(9):803. https://doi.org/10.3390/v11090803
Chicago/Turabian StyleA. Duarte, Matheus, João M. F. Silva, Clara R. Brito, Danilo S. Teixeira, Fernando L. Melo, Bergmann M. Ribeiro, Tatsuya Nagata, and Fabrício S. Campos. 2019. "Faecal Virome Analysis of Wild Animals from Brazil" Viruses 11, no. 9: 803. https://doi.org/10.3390/v11090803
APA StyleA. Duarte, M., F. Silva, J. M., R. Brito, C., S. Teixeira, D., L. Melo, F., M. Ribeiro, B., Nagata, T., & S. Campos, F. (2019). Faecal Virome Analysis of Wild Animals from Brazil. Viruses, 11(9), 803. https://doi.org/10.3390/v11090803