Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans


Abstract
:1. Introduction
2. Historical Background
3. Molecular Basis of Phenotypic Variability and Disease Subtypes
3.1. Creutzfeldt–Jakob Disease (CJD) and Fatal Insomnia (FI)
3.2. Gerstmann–Sträussler–Scheinker (GSS) Disease and Variably Protease-Sensitive Prionopathy (VPSPr)
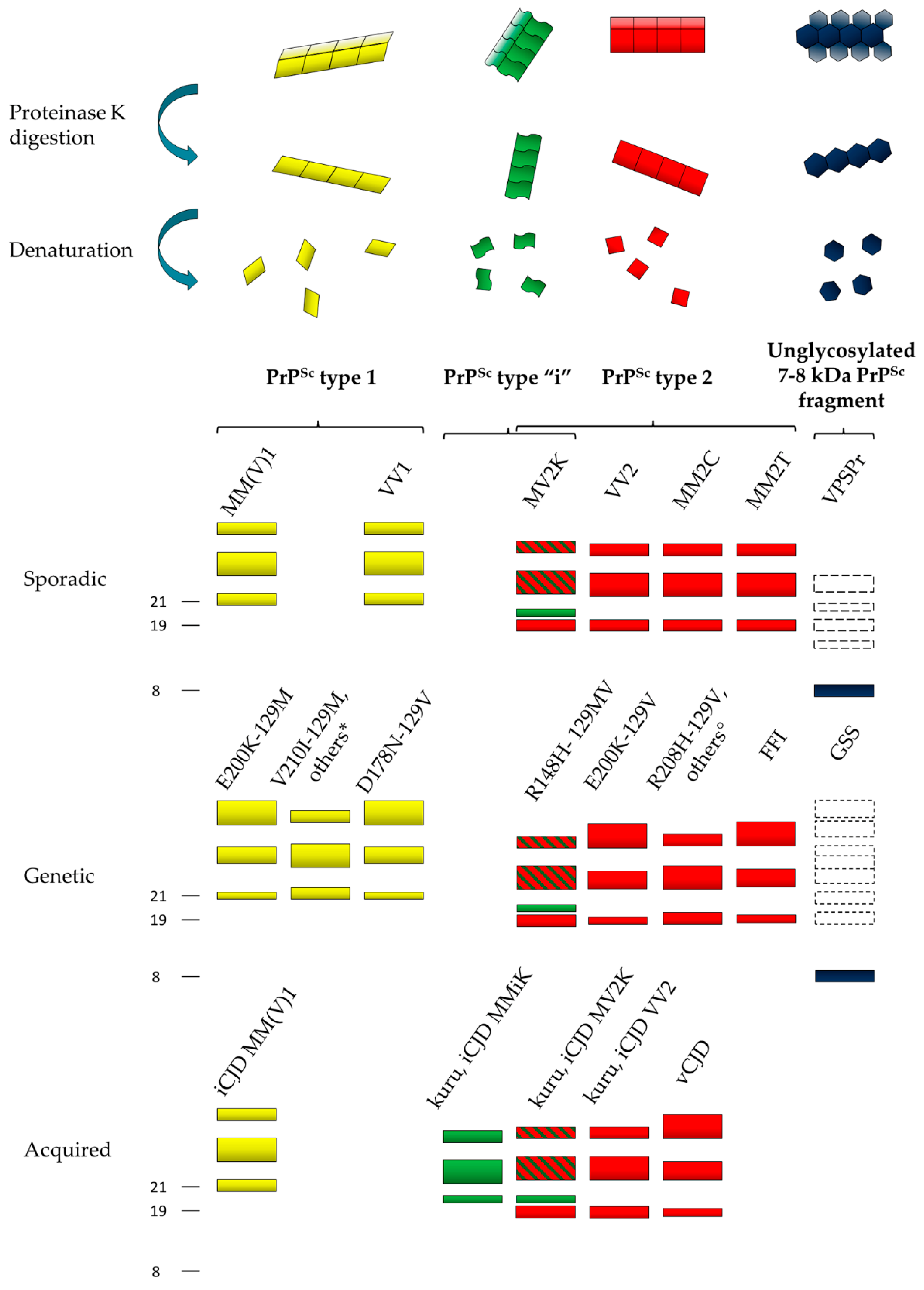
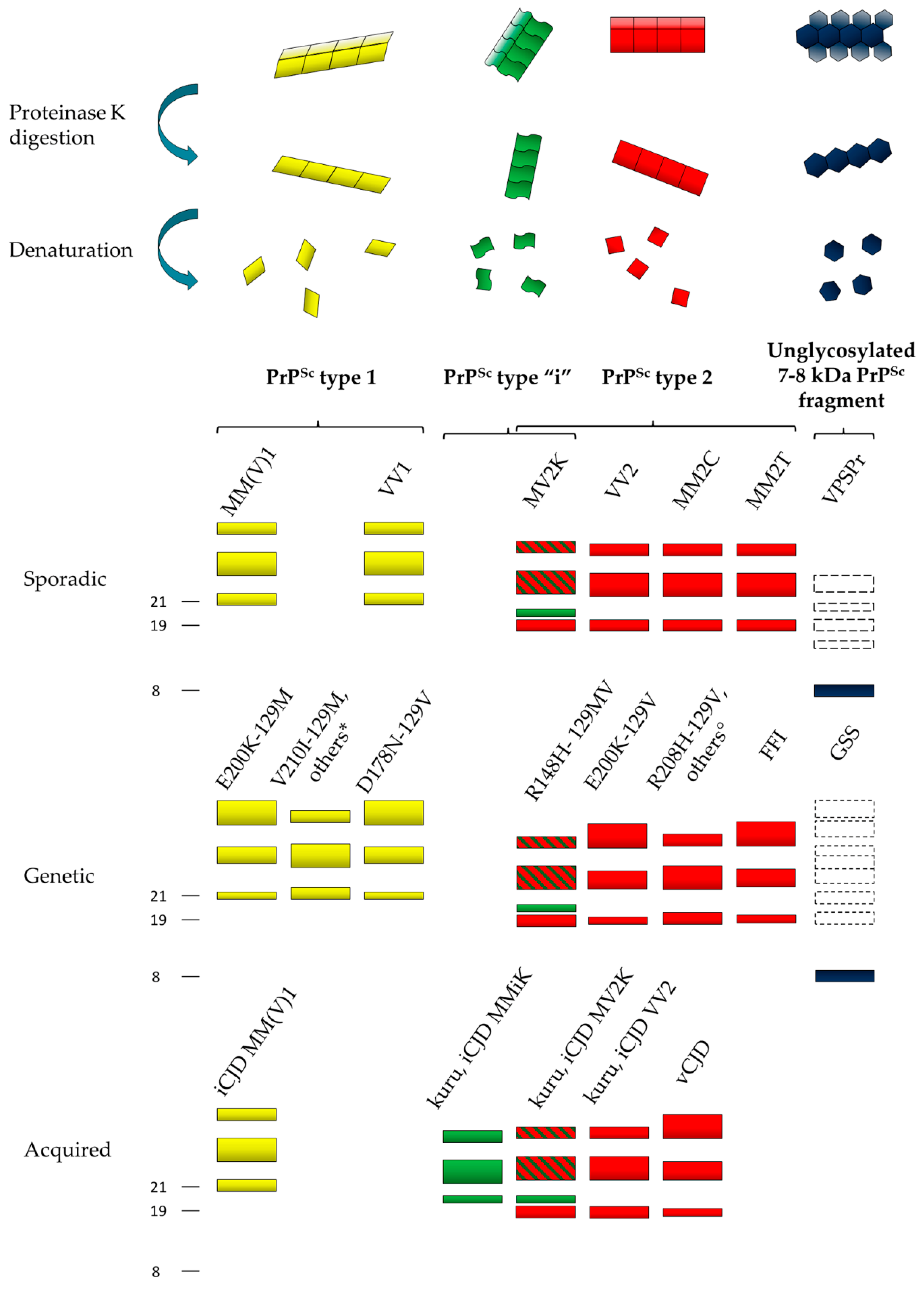
4. Characterization of Human Abnormal Prion Protein (PrPSc) by Conformational and Solubility Assays
5. Characterization of Human PrPSc Types Conversion and Seeding Activity by In Vitro Amplification Techniques
6. Characterization of Human Prion Strains by Experimental Transmission
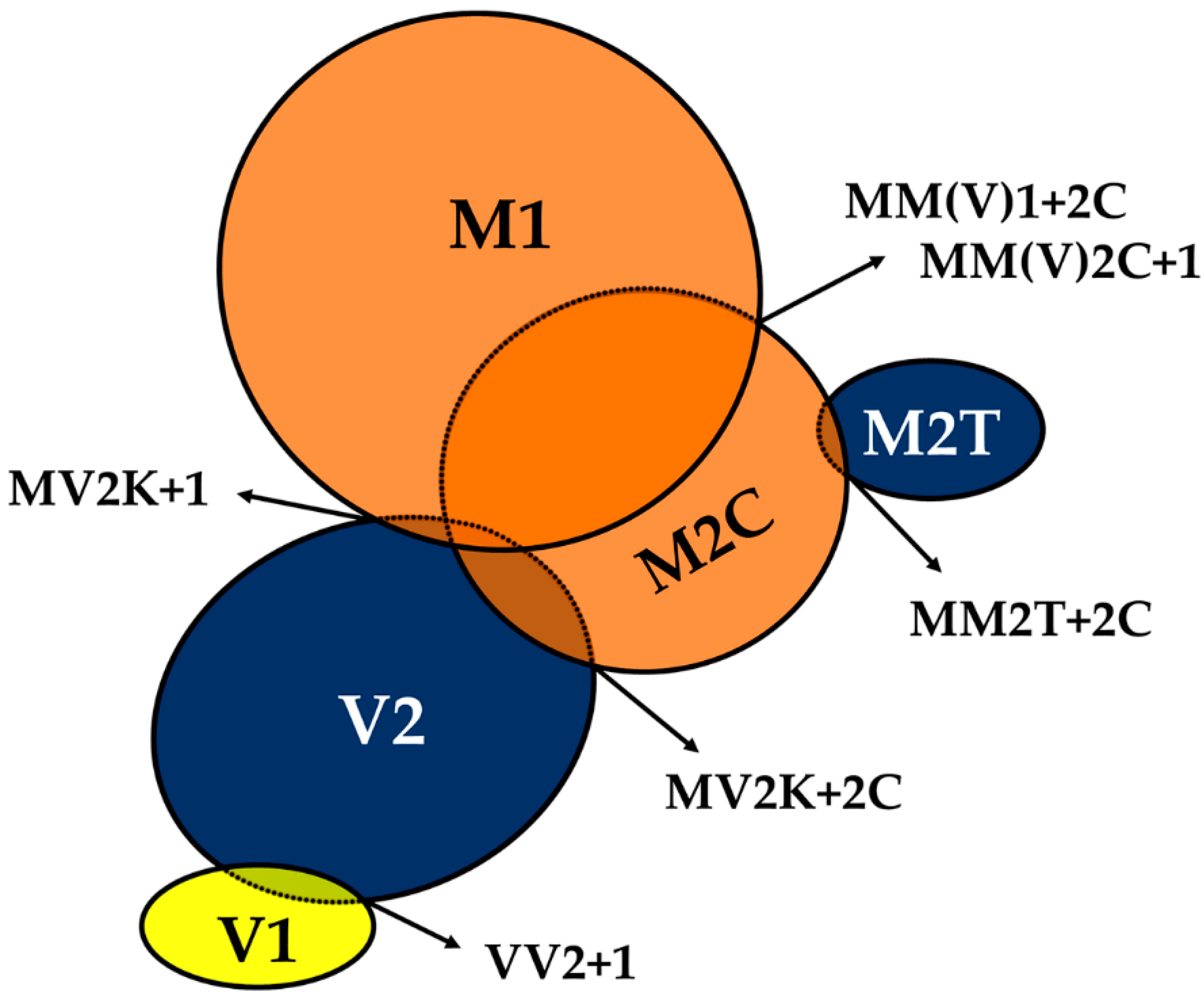
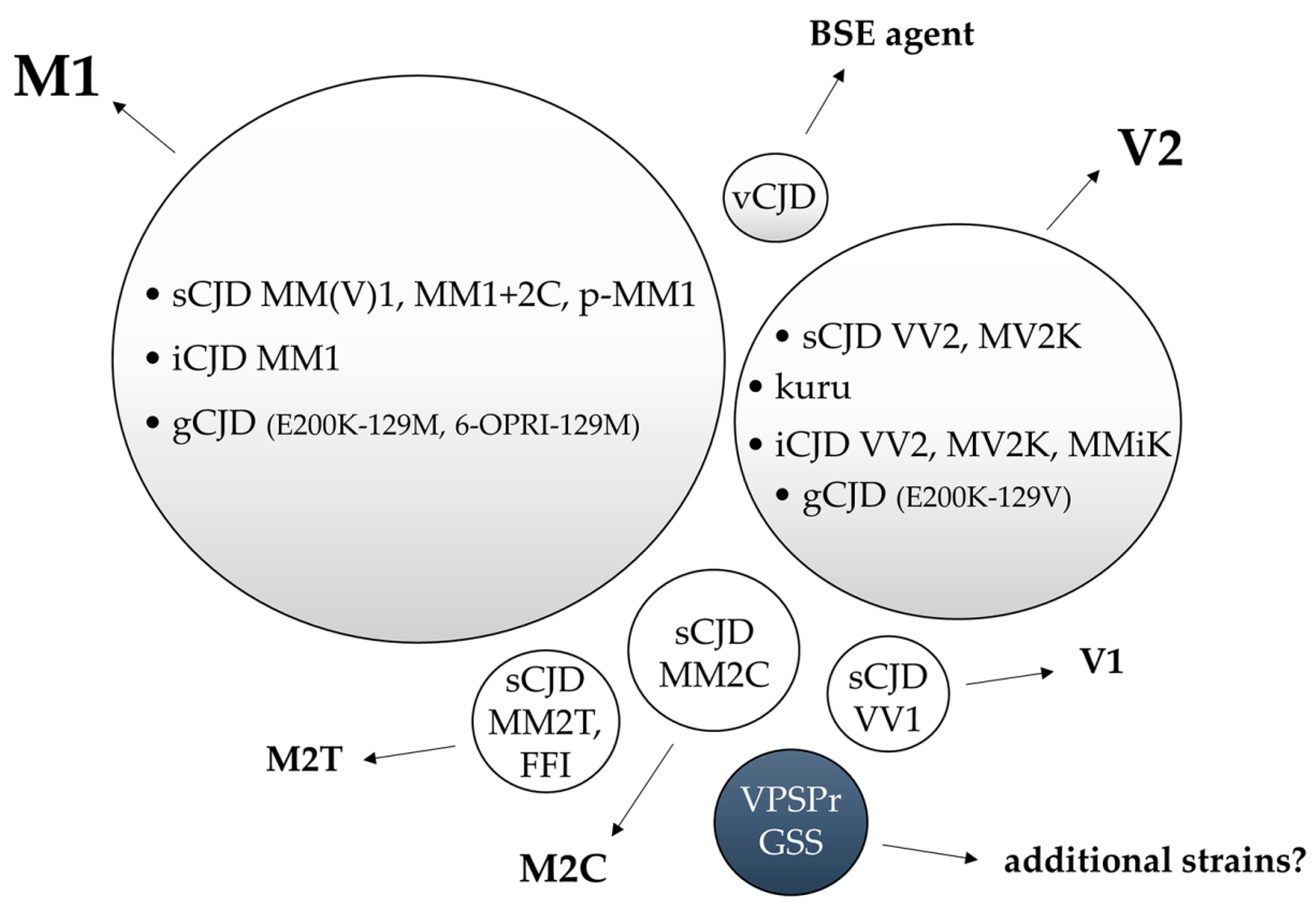
6.1. Strain M1
6.2. Strain V2
6.3. Other CJD Strains: M2T, M2C and V1
6.4. Variant CJD
6.5. Gerstmann–Straüssler–Scheinker (GSS) Disease
6.6. Other Inherited Prion Amyloidosis
6.7. Variably Protease-Sensitive Prionopathy (VPSPr)
7. Summary and Gaps in Our Understanding of Human Prion Strains
Author Contributions
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Caughey, B.; Baron, G.S.; Chesebro, B.; Jeffrey, M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 2009, 78, 177–204. [Google Scholar] [CrossRef]
- Bruce, M.E. TSE strain variation. Br. Med. Bull. 2003, 66, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.S.; Sawaya, M.R. Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Heikenwalder, M.; Polymenidou, M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007, 8, 552–561. [Google Scholar] [CrossRef]
- Baiardi, S.; Rossi, M.; Capellari, S.; Parchi, P. Recent advances in the histo-molecular pathology of human prion disease. Brain Pathol. 2019, 29, 278–300. [Google Scholar] [CrossRef]
- Pattison, I.H.; Millson, G.C. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J. Comp. Pathol. 1961, 71, 101–109. [Google Scholar] [CrossRef]
- Chandler, R.L.; Fisher, J. Experimental Transmission of Scrapie to Rats. Lancet 1963, 2, 1165. [Google Scholar] [CrossRef]
- Fraser, H.; Dickinson, A.G. Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J. Comp. Pathol. 1973, 83, 29–40. [Google Scholar] [CrossRef]
- Bruce, M.E.; Dickinson, A.G. Biological evidence that scrapie agent has an independent genome. J. Gen. Virol. 1987, 68, 79–89. [Google Scholar] [CrossRef]
- Gajdusek, D.C.; Gibbs, C.J.; Alpers, M. Transmission and passage of experimenal “kuru” to chimpanzees. Science 1967, 155, 212–214. [Google Scholar]
- Gibbs, C.J.; Gajdusek, D.C.; Asher, D.M.; Alpers, M.P.; Beck, E.; Daniel, P.M.; Matthews, W.B. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 1968, 161, 388–389. [Google Scholar] [CrossRef] [PubMed]
- Muramoto, T.; Kitamoto, T.; Hoque, M.Z.; Tateishi, J.; Goto, I. Species barrier prevents an abnormal isoform of prion protein from accumulating in follicular dendritic cells of mice with Creutzfeldt-Jakob disease. J. Virol. 1993, 67, 6808–6810. [Google Scholar] [PubMed]
- Bruce, M.; Chree, A.; McConnell, I.; Foster, J.; Pearson, G.; Fraser, H. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 1994, 343, 405–411. [Google Scholar]
- Parchi, P.; Saverioni, D. Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol 2012, 50, 20–45. [Google Scholar] [PubMed]
- Wadsworth, J.D.F.; Asante, E.A.; Collinge, J. Review: contribution of transgenic models to understanding human prion disease. Neuropathol. Appl. Neurobiol. 2010, 36, 576–597. [Google Scholar] [CrossRef]
- Nonno, R.; Di Bari, M.A.; Cardone, F.; Vaccari, G.; Fazzi, P.; Dell’Omo, G.; Cartoni, C.; Ingrosso, L.; Boyle, A.; Galeno, R.; et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006, 2, e12. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Cescatti, M.; Notari, S.; Schulz-Schaeffer, W.J.; Capellari, S.; Giese, A.; Zou, W.-Q.; Kretzschmar, H.; Ghetti, B.; Brown, P. Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain 2010, 133, 3030–3042. [Google Scholar] [CrossRef]
- Watts, J.C.; Giles, K.; Patel, S.; Oehler, A.; DeArmond, S.J.; Prusiner, S.B. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog. 2014, 10, e1003990. [Google Scholar] [CrossRef]
- Parchi, P.; Strammiello, R.; Giese, A.; Kretzschmar, H. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol. 2011, 121, 91–112. [Google Scholar] [CrossRef]
- Hsiao, K.; Baker, H.F.; Crow, T.J.; Poulter, M.; Owen, F.; Terwilliger, J.D.; Westaway, D.; Ott, J.; Prusiner, S.B. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature 1989, 338, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, L.G.; Petersen, R.B.; Tabaton, M.; Brown, P.; LeBlanc, A.C.; Montagna, P.; Cortelli, P.; Julien, J.; Vital, C.; Pendelbury, W.W. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science 1992, 258, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Deslys, J.P.; Lasmézas, C.; Dormont, D. Selection of specific strains in iatrogenic Creutzfeldt-Jakob disease. Lancet 1994, 343, 848–849. [Google Scholar] [CrossRef]
- De Silva, R.; Ironside, J.W.; McCardle, L.; Esmonde, T.; Bell, J.; Will, R.; Windl, O.; Dempster, M.; Estibeiro, P.; Lathe, R. Neuropathological phenotype and “prion protein” genotype correlation in sporadic Creutzfeldt-Jakob disease. Neurosci. Lett. 1994, 179, 50–52. [Google Scholar] [CrossRef]
- Miyazono, M.; Kitamoto, T.; Doh-ura, K.; Iwaki, T.; Tateishi, J. Creutzfeldt-Jakob disease with codon 129 polymorphism (valine): a comparative study of patients with codon 102 point mutation or without mutations. Acta Neuropathol. 1992, 84, 349–354. [Google Scholar] [CrossRef]
- Parchi, P.; Chen, S.G.; Brown, P.; Zou, W.; Capellari, S.; Budka, H.; Hainfellner, J.; Reyes, P.F.; Golden, G.T.; Hauw, J.J.; et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc. Natl. Acad. Sci. USA 1998, 95, 8322–8327. [Google Scholar] [CrossRef]
- Piccardo, P.; Dlouhy, S.R.; Lievens, P.M.; Young, K.; Bird, T.D.; Nochlin, D.; Dickson, D.W.; Vinters, H.V.; Zimmerman, T.R.; Mackenzie, I.R.; et al. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J. Neuropathol. Exp. Neurol. 1998, 57, 979–988. [Google Scholar] [CrossRef]
- Parchi, P.; Castellani, R.; Capellari, S.; Ghetti, B.; Young, K.; Chen, S.G.; Farlow, M.; Dickson, D.W.; Sima, A.A.; Trojanowski, J.Q.; et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 1996, 39, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Capellari, S.; Chen, S.G.; Petersen, R.B.; Gambetti, P.; Kopp, N.; Brown, P.; Kitamoto, T.; Tateishi, J.; Giese, A.; et al. Typing prion isoforms. Nature 1997, 386, 232–234. [Google Scholar] [CrossRef]
- Parchi, P.; Zou, W.; Wang, W.; Brown, P.; Capellari, S.; Ghetti, B.; Kopp, N.; Schulz-Schaeffer, W.J.; Kretzschmar, H.A.; Head, M.W.; et al. Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10168–10172. [Google Scholar] [CrossRef]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Parchi, P.; Strammiello, R.; Notari, S.; Giese, A.; Langeveld, J.P.M.; Ladogana, A.; Zerr, I.; Roncaroli, F.; Cras, P.; Ghetti, B.; et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 2009, 118, 659–671. [Google Scholar] [CrossRef]
- Parchi, P.; de Boni, L.; Saverioni, D.; Cohen, M.L.; Ferrer, I.; Gambetti, P.; Gelpi, E.; Giaccone, G.; Hauw, J.-J.; Höftberger, R.; et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012, 124, 517–529. [Google Scholar] [CrossRef]
- Kobayashi, A.; Matsuura, Y.; Mohri, S.; Kitamoto, T. Distinct origins of dura mater graft-associated Creutzfeldt-Jakob disease: past and future problems. Acta Neuropathol. Commun. 2014, 2, 32. [Google Scholar] [CrossRef]
- Notari, S.; Capellari, S.; Giese, A.; Westner, I.; Baruzzi, A.; Ghetti, B.; Gambetti, P.; Kretzschmar, H.A.; Parchi, P. Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. J. Biol. Chem. 2004, 279, 16797–16804. [Google Scholar] [CrossRef]
- Zou, W.-Q.; Capellari, S.; Parchi, P.; Sy, M.-S.; Gambetti, P.; Chen, S.G. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J. Biol. Chem. 2003, 278, 40429–40436. [Google Scholar] [CrossRef]
- Satoh, K.; Muramoto, T.; Tanaka, T.; Kitamoto, N.; Ironside, J.W.; Nagashima, K.; Yamada, M.; Sato, T.; Mohri, S.; Kitamoto, T. Association of an 11-12 kDa protease-resistant prion protein fragment with subtypes of dura graft-associated Creutzfeldt-Jakob disease and other prion diseases. J. Gen. Virol. 2003, 84, 2885–2893. [Google Scholar] [CrossRef]
- Pan, T.; Li, R.; Kang, S.-C.; Pastore, M.; Wong, B.-S.; Ironside, J.; Gambetti, P.; Sy, M.-S. Biochemical fingerprints of prion diseases: scrapie prion protein in human prion diseases that share prion genotype and type. J. Neurochem. 2005, 92, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Notari, S.; Strammiello, R.; Capellari, S.; Giese, A.; Cescatti, M.; Grassi, J.; Ghetti, B.; Langeveld, J.P.M.; Zou, W.-Q.; Gambetti, P.; et al. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. J. Biol. Chem. 2008, 283, 30557–30565. [Google Scholar] [CrossRef]
- Endo, T.; Groth, D.; Prusiner, S.B.; Kobata, A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 1989, 28, 8380–8388. [Google Scholar] [CrossRef]
- Monari, L.; Chen, S.G.; Brown, P.; Parchi, P.; Petersen, R.B.; Mikol, J.; Gray, F.; Cortelli, P.; Montagna, P.; Ghetti, B. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc. Natl. Acad. Sci. USA 1994, 91, 2839–2842. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Sidle, K.C.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Joiner, S.; Beck, J.A.; Campbell, T.A.; Dickinson, A.; Poulter, M.; Wadsworth, J.D.F.; Collinge, J. Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain 2006, 129, 676–685. [Google Scholar] [CrossRef]
- Pan, T.; Colucci, M.; Wong, B.S.; Li, R.; Liu, T.; Petersen, R.B.; Chen, S.; Gambetti, P.; Sy, M.S. Novel differences between two human prion strains revealed by two-dimensional gel electrophoresis. J. Biol. Chem. 2001, 276, 37284–37288. [Google Scholar] [CrossRef]
- Head, M.W.; Bunn, T.J.R.; Bishop, M.T.; McLoughlin, V.; Lowrie, S.; McKimmie, C.S.; Williams, M.C.; McCardle, L.; MacKenzie, J.; Knight, R.; et al. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991-2002. Ann. Neurol. 2004, 55, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Head, M.W.; Ironside, J.W. Sporadic Creutzfeldt-Jakob disease: discrete subtypes or a spectrum of disease? Brain 2009, 132, 2627–2629. [Google Scholar] [CrossRef]
- Lewis, V.; Hill, A.F.; Klug, G.M.; Boyd, A.; Masters, C.L.; Collins, S.J. Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology 2005, 65, 113–118. [Google Scholar] [CrossRef]
- Puoti, G.; Giaccone, G.; Rossi, G.; Canciani, B.; Bugiani, O.; Tagliavini, F. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology 1999, 53, 2173–2176. [Google Scholar] [CrossRef]
- Schoch, G.; Seeger, H.; Bogousslavsky, J.; Tolnay, M.; Janzer, R.C.; Aguzzi, A.; Glatzel, M. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med. 2006, 3, e14. [Google Scholar] [CrossRef]
- Uro-Coste, E.; Cassard, H.; Simon, S.; Lugan, S.; Bilheude, J.-M.; Perret-Liaudet, A.; Ironside, J.W.; Haik, S.; Basset-Leobon, C.; Lacroux, C.; et al. Beyond PrP res type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 2008, 4, e1000029. [Google Scholar] [CrossRef]
- Yull, H.M.; Ritchie, D.L.; Langeveld, J.P.M.; van Zijderveld, F.G.; Bruce, M.E.; Ironside, J.W.; Head, M.W. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am. J. Pathol. 2006, 168, 151–157. [Google Scholar] [CrossRef]
- Polymenidou, M.; Stoeck, K.; Glatzel, M.; Vey, M.; Bellon, A.; Aguzzi, A. Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol. 2005, 4, 805–814. [Google Scholar] [CrossRef]
- Kobayashi, A.; Mizukoshi, K.; Iwasaki, Y.; Miyata, H.; Yoshida, Y.; Kitamoto, T. Co-occurrence of types 1 and 2 PrP(res) in sporadic Creutzfeldt-Jakob disease MM1. Am. J. Pathol. 2011, 178, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Notari, S.; Capellari, S.; Langeveld, J.; Giese, A.; Strammiello, R.; Gambetti, P.; Kretzschmar, H.A.; Parchi, P. A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab. Invest. 2007, 87, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Cali, I.; Castellani, R.; Alshekhlee, A.; Cohen, Y.; Blevins, J.; Yuan, J.; Langeveld, J.P.M.; Parchi, P.; Safar, J.G.; Zou, W.-Q.; et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain 2009, 132, 2643–2658. [Google Scholar] [CrossRef] [PubMed]
- Piccardo, P.; Liepnieks, J.J.; William, A.; Dlouhy, S.R.; Farlow, M.R.; Young, K.; Nochlin, D.; Bird, T.D.; Nixon, R.R.; Ball, M.J.; et al. Prion proteins with different conformations accumulate in Gerstmann-Sträussler-Scheinker disease caused by A117V and F198S mutations. Am. J. Pathol. 2001, 158, 2201–2207. [Google Scholar] [CrossRef]
- Tagliavini, F.; Prelli, F.; Ghiso, J.; Bugiani, O.; Serban, D.; Prusiner, S.B.; Farlow, M.R.; Ghetti, B.; Frangione, B. Amyloid protein of Gerstmann-Sträussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58. EMBO J. 1991, 10, 513–519. [Google Scholar] [CrossRef]
- Tagliavini, F.; Lievens, P.M.; Tranchant, C.; Warter, J.M.; Mohr, M.; Giaccone, G.; Perini, F.; Rossi, G.; Salmona, M.; Piccardo, P.; et al. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Sträussler-Scheinker disease A117V. J. Biol. Chem. 2001, 276, 6009–6015. [Google Scholar] [CrossRef]
- Pirisinu, L.; Nonno, R.; Esposito, E.; Benestad, S.L.; Gambetti, P.; Agrimi, U.; Zou, W.-Q. Small ruminant nor98 prions share biochemical features with human gerstmann-sträussler-scheinker disease and variably protease-sensitive prionopathy. PLoS ONE 2013, 8, e66405. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.-Q.; Puoti, G.; Xiao, X.; Yuan, J.; Qing, L.; Cali, I.; Shimoji, M.; Langeveld, J.P.M.; Castellani, R.; Notari, S.; et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann. Neurol. 2010, 68, 162–172. [Google Scholar] [CrossRef]
- Peden, A.H.; Sarode, D.P.; Mulholland, C.R.; Barria, M.A.; Ritchie, D.L.; Ironside, J.W.; Head, M.W. The prion protein protease sensitivity, stability and seeding activity in variably protease sensitive prionopathy brain tissue suggests molecular overlaps with sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. Commun. 2014, 2, 152. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Pascual, M.; Rossi, M.; Gámez, J.; Lladó, A.; Valls, J.; Grau-Rivera, O.; Ávila Polo, R.; Llorens, F.; Zerr, I.; Ferrer, I.; et al. Variably protease-sensitive prionopathy presenting within ALS/FTD spectrum. Ann. Clin. Transl. Neurol. 2018, 5, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.G.; Teplow, D.B.; Parchi, P.; Teller, J.K.; Gambetti, P.; Autilio-Gambetti, L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995, 270, 19173–19180. [Google Scholar] [CrossRef]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996, 274, 2079–2082. [Google Scholar] [CrossRef]
- Bessen, R.A.; Marsh, R.F. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 1994, 68, 7859–7868. [Google Scholar] [PubMed]
- Wille, H.; Requena, J.R. The Structure of PrPSc Prions. Pathogens 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Saverioni, D.; Notari, S.; Capellari, S.; Poggiolini, I.; Giese, A.; Kretzschmar, H.A.; Parchi, P. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J. Biol. Chem. 2013, 288, 27972–27985. [Google Scholar] [CrossRef]
- Cescatti, M.; Saverioni, D.; Capellari, S.; Tagliavini, F.; Kitamoto, T.; Ironside, J.; Giese, A.; Parchi, P. Analysis of Conformational Stability of Abnormal Prion Protein Aggregates across the Spectrum of Creutzfeldt-Jakob Disease Prions. J. Virol. 2016, 90, 6244–6254. [Google Scholar] [CrossRef]
- Diack, A.B.; Head, M.W.; McCutcheon, S.; Boyle, A.; Knight, R.; Ironside, J.W.; Manson, J.C.; Will, R.G. Variant CJD. 18 years of research and surveillance. Prion 2014, 8, 286–295. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Redaelli, V.; Baiardi, S.; Mackenzie, G.; Windl, O.; Ritchie, D.L.; Didato, G.; Hernandez-Vara, J.; Rossi, M.; Capellari, S.; et al. Sporadic Fatal Insomnia in Europe: Phenotypic Features and Diagnostic Challenges. Ann. Neurol. 2018, 84, 347–360. [Google Scholar] [CrossRef]
- Bishop, M.T.; Will, R.G.; Manson, J.C. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc. Natl. Acad. Sci. USA 2010, 107, 12005–12010. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Sakuma, N.; Matsuura, Y.; Mohri, S.; Aguzzi, A.; Kitamoto, T. Experimental verification of a traceback phenomenon in prion infection. J. Virol. 2010, 84, 3230–3238. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Iwasaki, Y.; Otsuka, H.; Yamada, M.; Yoshida, M.; Matsuura, Y.; Mohri, S.; Kitamoto, T. Deciphering the pathogenesis of sporadic Creutzfeldt-Jakob disease with codon 129 M/V and type 2 abnormal prion protein. Acta Neuropathol. Commun. 2013, 1, 74. [Google Scholar] [CrossRef] [PubMed]
- Moda, F.; Suardi, S.; Di Fede, G.; Indaco, A.; Limido, L.; Vimercati, C.; Ruggerone, M.; Campagnani, I.; Langeveld, J.; Terruzzi, A.; et al. MM2-thalamic Creutzfeldt-Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathol. 2012, 22, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Kobayashi, A.; Ironside, J.W.; Mohri, S.; Kitamoto, T. Characterization of variant Creutzfeldt-Jakob disease prions in prion protein-humanized mice carrying distinct codon 129 genotypes. J. Biol. Chem. 2013, 288, 21659–21666. [Google Scholar] [CrossRef] [PubMed]
- Diack, A.B.; Ritchie, D.L.; Peden, A.H.; Brown, D.; Boyle, A.; Morabito, L.; Maclennan, D.; Burgoyne, P.; Jansen, C.; Knight, R.S.; et al. Variably protease-sensitive prionopathy, a unique prion variant with inefficient transmission properties. Emerging Infect. Dis. 2014, 20, 1969–1979. [Google Scholar] [CrossRef]
- Kobayashi, A.; Matsuura, Y.; Iwaki, T.; Iwasaki, Y.; Yoshida, M.; Takahashi, H.; Murayama, S.; Takao, M.; Kato, S.; Yamada, M.; et al. Sporadic Creutzfeldt-Jakob Disease MM1+2C and MM1 are Identical in Transmission Properties. Brain Pathol. 2016, 26, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Notari, S.; Xiao, X.; Espinosa, J.C.; Cohen, Y.; Qing, L.; Aguilar-Calvo, P.; Kofskey, D.; Cali, I.; Cracco, L.; Kong, Q.; et al. Transmission characteristics of variably protease-sensitive prionopathy. Emerging Infect. Dis. 2014, 20, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G.; Geschwind, M.D.; Deering, C.; Didorenko, S.; Sattavat, M.; Sanchez, H.; Serban, A.; Vey, M.; Baron, H.; Giles, K.; et al. Diagnosis of human prion disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3501–3506. [Google Scholar] [CrossRef]
- Choi, Y.P.; Gröner, A.; Ironside, J.W.; Head, M.W. Comparison of the level, distribution and form of disease-associated prion protein in variant and sporadic Creutzfeldt-Jakob diseased brain using conformation-dependent immunoassay and Western blot. J. Gen. Virol. 2011, 92, 727–732. [Google Scholar] [CrossRef]
- Kim, C.; Haldiman, T.; Cohen, Y.; Chen, W.; Blevins, J.; Sy, M.-S.; Cohen, M.; Safar, J.G. Protease-sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt-Jakob disease are indicator of progression rate. PLoS Pathog. 2011, 7, e1002242. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G.; Xiao, X.; Kabir, M.E.; Chen, S.; Kim, C.; Haldiman, T.; Cohen, Y.; Chen, W.; Cohen, M.L.; Surewicz, W.K. Structural determinants of phenotypic diversity and replication rate of human prions. PLoS Pathog. 2015, 11, e1004832. [Google Scholar] [CrossRef] [PubMed]
- Pirisinu, L.; Di Bari, M.; Marcon, S.; Vaccari, G.; D’Agostino, C.; Fazzi, P.; Esposito, E.; Galeno, R.; Langeveld, J.; Agrimi, U.; et al. A new method for the characterization of strain-specific conformational stability of protease-sensitive and protease-resistant PrPSc. PLoS ONE 2010, 5, e12723. [Google Scholar] [CrossRef] [PubMed]
- Cracco, L.; Notari, S.; Cali, I.; Sy, M.-S.; Chen, S.G.; Cohen, M.L.; Ghetti, B.; Appleby, B.S.; Zou, W.-Q.; Caughey, B.; et al. Novel strain properties distinguishing sporadic prion diseases sharing prion protein genotype and prion type. Sci. Rep. 2017, 7, 38280. [Google Scholar] [CrossRef]
- Bett, C.; Joshi-Barr, S.; Lucero, M.; Trejo, M.; Liberski, P.; Kelly, J.W.; Masliah, E.; Sigurdson, C.J. Biochemical properties of highly neuroinvasive prion strains. PLoS Pathog. 2012, 8, e1002522. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Anderes, L.; Suardi, S.; Cardone, F.; Castilla, J.; Frossard, M.-J.; Peano, S.; Saa, P.; Limido, L.; Carbonatto, M.; et al. Pre-symptomatic detection of prions by cyclic amplification of protein misfolding. FEBS Lett. 2005, 579, 638–642. [Google Scholar] [CrossRef]
- Rubenstein, R.; Chang, B. Re-assessment of PrP(Sc) distribution in sporadic and variant CJD. PLoS ONE 2013, 8, e66352. [Google Scholar] [CrossRef] [PubMed]
- Concha-Marambio, L.; Pritzkow, S.; Moda, F.; Tagliavini, F.; Ironside, J.W.; Schulz, P.E.; Soto, C. Detection of prions in blood from patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 2016, 8, 370ra183. [Google Scholar] [CrossRef]
- Bougard, D.; Brandel, J.-P.; Bélondrade, M.; Béringue, V.; Segarra, C.; Fleury, H.; Laplanche, J.-L.; Mayran, C.; Nicot, S.; Green, A.; et al. Detection of prions in the plasma of presymptomatic and symptomatic patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 2016, 8, 370ra182. [Google Scholar] [CrossRef]
- Jones, M.; Peden, A.H.; Prowse, C.V.; Gröner, A.; Manson, J.C.; Turner, M.L.; Ironside, J.W.; MacGregor, I.R.; Head, M.W. In vitro amplification and detection of variant Creutzfeldt-Jakob disease PrPSc. J. Pathol. 2007, 213, 21–26. [Google Scholar] [CrossRef]
- Yokoyama, T.; Takeuchi, A.; Yamamoto, M.; Kitamoto, T.; Ironside, J.W.; Morita, M. Heparin enhances the cell-protein misfolding cyclic amplification efficiency of variant Creutzfeldt-Jakob disease. Neurosci. Lett. 2011, 498, 119–123. [Google Scholar] [CrossRef]
- Atarashi, R.; Moore, R.A.; Sim, V.L.; Hughson, A.G.; Dorward, D.W.; Onwubiko, H.A.; Priola, S.A.; Caughey, B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 2007, 4, 645–650. [Google Scholar] [CrossRef]
- Jones, M.; Peden, A.H.; Yull, H.; Wight, D.; Bishop, M.T.; Prowse, C.V.; Turner, M.L.; Ironside, J.W.; MacGregor, I.R.; Head, M.W. Human platelets as a substrate source for the in vitro amplification of the abnormal prion protein (PrP) associated with variant Creutzfeldt-Jakob disease. Transfusion 2009, 49, 376–384. [Google Scholar] [CrossRef]
- Haldiman, T.; Kim, C.; Cohen, Y.; Chen, W.; Blevins, J.; Qing, L.; Cohen, M.L.; Langeveld, J.; Telling, G.C.; Kong, Q.; et al. Co-existence of distinct prion types enables conformational evolution of human PrPSc by competitive selection. J. Biol. Chem. 2013, 288, 29846–29861. [Google Scholar] [CrossRef]
- Xiao, X.; Yuan, J.; Qing, L.; Cali, I.; Mikol, J.; Delisle, M.-B.; Uro-Coste, E.; Zeng, L.; Abouelsaad, M.; Gazgalis, D.; et al. Comparative Study of Prions in Iatrogenic and Sporadic Creutzfeldt-Jakob Disease. J. Clin. Cell Immunol. 2014, 5, 240. [Google Scholar] [CrossRef]
- Moda, F.; Gambetti, P.; Notari, S.; Concha-Marambio, L.; Catania, M.; Park, K.-W.; Maderna, E.; Suardi, S.; Haïk, S.; Brandel, J.-P.; et al. Prions in the urine of patients with variant Creutzfeldt-Jakob disease. N. Engl. J. Med. 2014, 371, 530–539. [Google Scholar] [CrossRef]
- Takeuchi, A.; Kobayashi, A.; Parchi, P.; Yamada, M.; Morita, M.; Uno, S.; Kitamoto, T. Distinctive properties of plaque-type dura mater graft-associated Creutzfeldt-Jakob disease in cell-protein misfolding cyclic amplification. Lab. Invest. 2016, 96, 581–587. [Google Scholar] [CrossRef]
- Oshita, M.; Yokoyama, T.; Takei, Y.; Takeuchi, A.; Ironside, J.W.; Kitamoto, T.; Morita, M. Efficient propagation of variant Creutzfeldt-Jakob disease prion protein using the cell-protein misfolding cyclic amplification technique with samples containing plasma and heparin. Transfusion 2016, 56, 223–230. [Google Scholar] [CrossRef]
- Redaelli, V.; Bistaffa, E.; Zanusso, G.; Salzano, G.; Sacchetto, L.; Rossi, M.; De Luca, C.M.G.; Di Bari, M.; Portaleone, S.M.; Agrimi, U.; et al. Detection of prion seeding activity in the olfactory mucosa of patients with Fatal Familial Insomnia. Sci. Rep. 2017, 7, 46269. [Google Scholar] [CrossRef]
- Privat, N.; Levavasseur, E.; Yildirim, S.; Hannaoui, S.; Brandel, J.-P.; Laplanche, J.-L.; Béringue, V.; Seilhean, D.; Haïk, S. Region-specific protein misfolding cyclic amplification reproduces brain tropism of prion strains. J. Biol. Chem. 2017, 292, 16688–16696. [Google Scholar] [CrossRef]
- Ritchie, D.L.; Barria, M.A.; Peden, A.H.; Yull, H.M.; Kirkpatrick, J.; Adlard, P.; Ironside, J.W.; Head, M.W. UK Iatrogenic Creutzfeldt-Jakob disease: investigating human prion transmission across genotypic barriers using human tissue-based and molecular approaches. Acta Neuropathol. 2017, 133, 579–595. [Google Scholar] [CrossRef]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci. Rep. 2015, 5, 16912. [Google Scholar] [CrossRef]
- Imamura, M.; Tabeta, N.; Kato, N.; Matsuura, Y.; Iwamaru, Y.; Yokoyama, T.; Murayama, Y. Heparan Sulfate and Heparin Promote Faithful Prion Replication in Vitro by Binding to Normal and Abnormal Prion Proteins in Protein Misfolding Cyclic Amplification. J. Biol. Chem. 2016, 291, 26478–26486. [Google Scholar] [CrossRef]
- Atarashi, R.; Sano, K.; Satoh, K.; Nishida, N. Real-time quaking-induced conversion: a highly sensitive assay for prion detection. Prion 2011, 5, 150–153. [Google Scholar] [CrossRef]
- Orrú, C.D.; Bongianni, M.; Tonoli, G.; Ferrari, S.; Hughson, A.G.; Groveman, B.R.; Fiorini, M.; Pocchiari, M.; Monaco, S.; Caughey, B.; et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N. Engl. J. Med. 2014, 371, 519–529. [Google Scholar] [CrossRef]
- Cramm, M.; Schmitz, M.; Karch, A.; Mitrova, E.; Kuhn, F.; Schroeder, B.; Raeber, A.; Varges, D.; Kim, Y.-S.; Satoh, K.; et al. Stability and Reproducibility Underscore Utility of RT-QuIC for Diagnosis of Creutzfeldt-Jakob Disease. Mol. Neurobiol. 2016, 53, 1896–1904. [Google Scholar] [CrossRef]
- McGuire, L.I.; Poleggi, A.; Poggiolini, I.; Suardi, S.; Grznarova, K.; Shi, S.; de Vil, B.; Sarros, S.; Satoh, K.; Cheng, K.; et al. Cerebrospinal fluid real-time quaking-induced conversion is a robust and reliable test for sporadic creutzfeldt-jakob disease: An international study. Ann. Neurol. 2016, 80, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Orrú, C.D.; Groveman, B.R.; Hughson, A.G.; Zanusso, G.; Coulthart, M.B.; Caughey, B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio 2015, 6, e02451-14. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Baiardi, S.; Hughson, A.G.; McKenzie, N.; Moda, F.; Rossi, M.; Capellari, S.; Green, A.; Giaccone, G.; Caughey, B.; et al. High diagnostic value of second generation CSF RT-QuIC across the wide spectrum of CJD prions. Sci. Rep. 2017, 7, 10655. [Google Scholar] [CrossRef]
- Cramm, M.; Schmitz, M.; Karch, A.; Zafar, S.; Varges, D.; Mitrova, E.; Schroeder, B.; Raeber, A.; Kuhn, F.; Zerr, I. Characteristic CSF prion seeding efficiency in humans with prion diseases. Mol. Neurobiol. 2015, 51, 396–405. [Google Scholar] [CrossRef]
- Lattanzio, F.; Abu-Rumeileh, S.; Franceschini, A.; Kai, H.; Amore, G.; Poggiolini, I.; Rossi, M.; Baiardi, S.; McGuire, L.; Ladogana, A.; et al. Prion-specific and surrogate CSF biomarkers in Creutzfeldt-Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p-tau and Aβ42 levels. Acta Neuropathol. 2017, 133, 559–578. [Google Scholar] [CrossRef] [PubMed]
- McGuire, L.I.; Peden, A.H.; Orrú, C.D.; Wilham, J.M.; Appleford, N.E.; Mallinson, G.; Andrews, M.; Head, M.W.; Caughey, B.; Will, R.G.; et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 2012, 72, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Bongianni, M.; Orrù, C.; Groveman, B.R.; Sacchetto, L.; Fiorini, M.; Tonoli, G.; Triva, G.; Capaldi, S.; Testi, S.; Ferrari, S.; et al. Diagnosis of Human Prion Disease Using Real-Time Quaking-Induced Conversion Testing of Olfactory Mucosa and Cerebrospinal Fluid Samples. JAMA Neurol. 2017, 74, 155–162. [Google Scholar] [CrossRef]
- Foutz, A.; Appleby, B.S.; Hamlin, C.; Liu, X.; Yang, S.; Cohen, Y.; Chen, W.; Blevins, J.; Fausett, C.; Wang, H.; et al. Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann. Neurol. 2017, 81, 79–92. [Google Scholar] [CrossRef]
- Groveman, B.R.; Orrú, C.D.; Hughson, A.G.; Bongianni, M.; Fiorini, M.; Imperiale, D.; Ladogana, A.; Pocchiari, M.; Zanusso, G.; Caughey, B. Extended and direct evaluation of RT-QuIC assays for Creutzfeldt-Jakob disease diagnosis. Ann. Clin. Transl. Neurol. 2017, 4, 139–144. [Google Scholar] [CrossRef]
- Baiardi, S.; Redaelli, V.; Ripellino, P.; Rossi, M.; Franceschini, A.; Moggio, M.; Sola, P.; Ladogana, A.; Fociani, P.; Magherini, A.; et al. Prion-related peripheral neuropathy in sporadic Creutzfeldt-Jakob disease. J. Neurol. Neurosurg. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Orrú, C.D.; Yuan, J.; Appleby, B.S.; Li, B.; Li, Y.; Winner, D.; Wang, Z.; Zhan, Y.-A.; Rodgers, M.; Rarick, J.; et al. Prion seeding activity and infectivity in skin samples from patients with sporadic Creutzfeldt-Jakob disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Orrù, C.D.; Soldau, K.; Cordano, C.; Llibre-Guerra, J.; Green, A.J.; Sanchez, H.; Groveman, B.R.; Edland, S.D.; Safar, J.G.; Lin, J.H.; et al. Prion Seeds Distribute throughout the Eyes of Sporadic Creutzfeldt-Jakob Disease Patients. MBio 2018, 9, e02095-18. [Google Scholar] [CrossRef] [PubMed]
- Peden, A.H.; McGuire, L.I.; Appleford, N.E.J.; Mallinson, G.; Wilham, J.M.; Orrú, C.D.; Caughey, B.; Ironside, J.W.; Knight, R.S.; Will, R.G.; et al. Sensitive and specific detection of sporadic Creutzfeldt-Jakob disease brain prion protein using real-time quaking-induced conversion. J. Gen. Virol. 2012, 93, 438–449. [Google Scholar] [CrossRef]
- Orrú, C.D.; Groveman, B.R.; Raymond, L.D.; Hughson, A.G.; Nonno, R.; Zou, W.; Ghetti, B.; Gambetti, P.; Caughey, B. Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog. 2015, 11, e1004983. [Google Scholar]
- Meiner, Z.; Gabizon, R.; Prusiner, S.B. Familial Creutzfeldt-Jakob disease. Codon 200 prion disease in Libyan Jews. Medicine (Baltimore) 1997, 76, 227–237. [Google Scholar] [CrossRef]
- Bartz, J.C. Prion Strain Diversity. Cold Spring Harb Perspect Med. 2016, 6. [Google Scholar] [CrossRef]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, e1006323. [Google Scholar] [CrossRef] [PubMed]
- Korth, C.; Kaneko, K.; Groth, D.; Heye, N.; Telling, G.; Mastrianni, J.; Parchi, P.; Gambetti, P.; Will, R.; Ironside, J.; et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc. Natl. Acad. Sci. USA 2003, 100, 4784–4789. [Google Scholar] [CrossRef] [PubMed]
- Jaumain, E.; Quadrio, I.; Herzog, L.; Reine, F.; Rezaei, H.; Andréoletti, O.; Laude, H.; Perret-Liaudet, A.; Haïk, S.; Béringue, V. Absence of Evidence for a Causal Link between Bovine Spongiform Encephalopathy Strain Variant L-BSE and Known Forms of Sporadic Creutzfeldt-Jakob Disease in Human PrP Transgenic Mice. J. Virol. 2016, 90, 10867–10874. [Google Scholar] [CrossRef]
- Watts, J.C.; Giles, K.; Serban, A.; Patel, S.; Oehler, A.; Bhardwaj, S.; Guan, S.; Greicius, M.D.; Miller, B.L.; DeArmond, S.J.; et al. Modulation of Creutzfeldt-Jakob disease prion propagation by the A224V mutation. Ann. Neurol. 2015, 78, 540–553. [Google Scholar] [CrossRef]
- Beck, E.; Daniel, P.M.; Asher, D.M.; Gajdusek, D.C.; Gibbs, C.J. Experimental kuru in the chimpanzee. A neuropathological study. Brain 1973, 96, 441–462. [Google Scholar] [CrossRef]
- Brown, P.; Cathala, F.; Castaigne, P.; Gajdusek, D.C. Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann. Neurol. 1986, 20, 597–602. [Google Scholar] [CrossRef]
- Brown, P.; Gibbs, C.J.; Rodgers-Johnson, P.; Asher, D.M.; Sulima, M.P.; Bacote, A.; Goldfarb, L.G.; Gajdusek, D.C. Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann. Neurol. 1994, 35, 513–529. [Google Scholar] [CrossRef]
- Cervenáková, L.; Goldfarb, L.G.; Garruto, R.; Lee, H.S.; Gajdusek, D.C.; Brown, P. Phenotype-genotype studies in kuru: implications for new variant Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. USA 1998, 95, 13239–13241. [Google Scholar] [CrossRef]
- Kobayashi, A.; Arima, K.; Ogawa, M.; Murata, M.; Fukuda, T.; Kitamoto, T. Plaque-type deposition of prion protein in the damaged white matter of sporadic Creutzfeldt-Jakob disease MM1 patients. Acta Neuropathol. 2008, 116, 561–566. [Google Scholar] [CrossRef]
- Rossi, M.; Saverioni, D.; Di Bari, M.; Baiardi, S.; Lemstra, A.W.; Pirisinu, L.; Capellari, S.; Rozemuller, A.; Nonno, R.; Parchi, P. Atypical Creutzfeldt-Jakob disease with PrP-amyloid plaques in white matter: molecular characterization and transmission to bank voles show the M1 strain signature. Acta Neuropathol. Commun. 2017, 5, 87. [Google Scholar] [CrossRef]
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.; Gowland, I.; Collinge, J.; Doey, L.J.; Lantos, P. The same prion strain causes vCJD and BSE. Nature 1997, 389, 448–450, 526. [Google Scholar] [CrossRef]
- Mead, S.; Poulter, M.; Beck, J.; Webb, T.E.F.; Campbell, T.A.; Linehan, J.M.; Desbruslais, M.; Joiner, S.; Wadsworth, J.D.F.; King, A.; et al. Inherited prion disease with six octapeptide repeat insertional mutation--molecular analysis of phenotypic heterogeneity. Brain 2006, 129, 2297–2317. [Google Scholar] [CrossRef]
- Kobayashi, A.; Asano, M.; Mohri, S.; Kitamoto, T. A traceback phenomenon can reveal the origin of prion infection. Neuropathology 2009, 29, 619–624. [Google Scholar] [CrossRef]
- Kobayashi, A.; Asano, M.; Mohri, S.; Kitamoto, T. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J. Biol. Chem. 2007, 282, 30022–30028. [Google Scholar] [CrossRef]
- Wadsworth, J.D.F.; Joiner, S.; Linehan, J.M.; Desbruslais, M.; Fox, K.; Cooper, S.; Cronier, S.; Asante, E.A.; Mead, S.; Brandner, S.; et al. Kuru prions and sporadic Creutzfeldt-Jakob disease prions have equivalent transmission properties in transgenic and wild-type mice. Proc. Natl. Acad. Sci. USA 2008, 105, 3885–3890. [Google Scholar] [CrossRef]
- Asante, E.A.; Gowland, I.; Grimshaw, A.; Linehan, J.M.; Smidak, M.; Houghton, R.; Osiguwa, O.; Tomlinson, A.; Joiner, S.; Brandner, S.; et al. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J. Gen. Virol. 2009, 90, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, J.; Brown, P.; Kitamoto, T.; Hoque, Z.M.; Roos, R.; Wollman, R.; Cervenáková, L.; Gajdusek, D.C. First experimental transmission of fatal familial insomnia. Nature 1995, 376, 434–435. [Google Scholar] [CrossRef]
- Collinge, J.; Palmer, M.S.; Sidle, K.C.; Gowland, I.; Medori, R.; Ironside, J.; Lantos, P. Transmission of fatal familial insomnia to laboratory animals. Lancet 1995, 346, 569–570. [Google Scholar] [CrossRef]
- Mastrianni, J.A.; Nixon, R.; Layzer, R.; Telling, G.C.; Han, D.; DeArmond, S.J.; Prusiner, S.B. Prion protein conformation in a patient with sporadic fatal insomnia. N. Engl. J. Med. 1999, 340, 1630–1638. [Google Scholar] [CrossRef]
- Chapuis, J.; Moudjou, M.; Reine, F.; Herzog, L.; Jaumain, E.; Chapuis, C.; Quadrio, I.; Boulliat, J.; Perret-Liaudet, A.; Dron, M.; et al. Emergence of two prion subtypes in ovine PrP transgenic mice infected with human MM2-cortical Creutzfeldt-Jakob disease prions. Acta Neuropathol. Commun. 2016, 4, 10. [Google Scholar] [CrossRef]
- Kobayashi, A.; Matsuura, Y.; Takeuchi, A.; Yamada, M.; Miyoshi, I.; Mohri, S.; Kitamoto, T. A domain responsible for spontaneous conversion of bank vole prion protein. Brain Pathol. 2019, 29, 155–163. [Google Scholar] [CrossRef]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef]
- Lasmézas, C.I.; Deslys, J.P.; Robain, O.; Jaegly, A.; Beringue, V.; Peyrin, J.M.; Fournier, J.G.; Hauw, J.J.; Rossier, J.; Dormont, D. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 1997, 275, 402–405. [Google Scholar] [CrossRef]
- Bishop, M.T.; Hart, P.; Aitchison, L.; Baybutt, H.N.; Plinston, C.; Thomson, V.; Tuzi, N.L.; Head, M.W.; Ironside, J.W.; Will, R.G.; et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006, 5, 393–398. [Google Scholar] [CrossRef]
- Asano, M.; Mohri, S.; Ironside, J.W.; Ito, M.; Tamaoki, N.; Kitamoto, T. vCJD prion acquires altered virulence through trans-species infection. Biochem. Biophys. Res. Commun. 2006, 342, 293–299. [Google Scholar] [CrossRef]
- Wadsworth, J.D.F.; Asante, E.A.; Desbruslais, M.; Linehan, J.M.; Joiner, S.; Gowland, I.; Welch, J.; Stone, L.; Lloyd, S.E.; Hill, A.F.; et al. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 2004, 306, 1793–1796. [Google Scholar] [CrossRef]
- Asante, E.A.; Linehan, J.M.; Desbruslais, M.; Joiner, S.; Gowland, I.; Wood, A.L.; Welch, J.; Hill, A.F.; Lloyd, S.E.; Wadsworth, J.D.F.; et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002, 21, 6358–6366. [Google Scholar] [CrossRef]
- Scott, M.R.; Will, R.; Ironside, J.; Nguyen, H.O.; Tremblay, P.; DeArmond, S.J.; Prusiner, S.B. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc. Natl. Acad. Sci. USA 1999, 96, 15137–15142. [Google Scholar] [CrossRef]
- Bian, J.; Khaychuk, V.; Angers, R.C.; Fernández-Borges, N.; Vidal, E.; Meyerett-Reid, C.; Kim, S.; Calvi, C.L.; Bartz, J.C.; Hoover, E.A.; et al. Prion replication without host adaptation during interspecies transmissions. Proc. Natl. Acad. Sci. USA 2017, 114, 1141–1146. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef]
- Mok, T.; Jaunmuktane, Z.; Joiner, S.; Campbell, T.; Morgan, C.; Wakerley, B.; Golestani, F.; Rudge, P.; Mead, S.; Jäger, H.R.; et al. Variant Creutzfeldt-Jakob Disease in a Patient with Heterozygosity at PRNP Codon 129. N. Engl. J. Med. 2017, 376, 292–294. [Google Scholar] [CrossRef]
- Cooper, J.D.; Bird, S.M. Predicting incidence of variant Creutzfeldt-Jakob disease from UK dietary exposure to bovine spongiform encephalopathy for the 1940 to 1969 and post-1969 birth cohorts. Int. J. Epidemiol. 2003, 32, 784–791. [Google Scholar] [CrossRef]
- Ritchie, D.L.; Boyle, A.; McConnell, I.; Head, M.W.; Ironside, J.W.; Bruce, M.E. Transmissions of variant Creutzfeldt-Jakob disease from brain and lymphoreticular tissue show uniform and conserved bovine spongiform encephalopathy-related phenotypic properties on primary and secondary passage in wild-type mice. J. Gen. Virol. 2009, 90, 3075–3082. [Google Scholar] [CrossRef]
- Bruce, M.E.; McConnell, I.; Will, R.G.; Ironside, J.W. Detection of variant Creutzfeldt-Jakob disease infectivity in extraneural tissues. Lancet 2001, 358, 208–209. [Google Scholar] [CrossRef]
- Cervenakova, L.; Yakovleva, O.; McKenzie, C.; Kolchinsky, S.; McShane, L.; Drohan, W.N.; Brown, P. Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 2003, 43, 1687–1694. [Google Scholar] [CrossRef]
- Wadsworth, J.D.F.; Joiner, S.; Fox, K.; Linehan, J.M.; Desbruslais, M.; Brandner, S.; Asante, E.A.; Collinge, J. Prion infectivity in variant Creutzfeldt-Jakob disease rectum. Gut 2007, 56, 90–94. [Google Scholar] [CrossRef]
- Béringue, V.; Le Dur, A.; Tixador, P.; Reine, F.; Lepourry, L.; Perret-Liaudet, A.; Haïk, S.; Vilotte, J.-L.; Fontés, M.; Laude, H. Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS ONE 2008, 3, e1419. [Google Scholar] [CrossRef]
- Herzog, C.; Salès, N.; Etchegaray, N.; Charbonnier, A.; Freire, S.; Dormont, D.; Deslys, J.-P.; Lasmézas, C.I. Tissue distribution of bovine spongiform encephalopathy agent in primates after intravenous or oral infection. Lancet 2004, 363, 422–428. [Google Scholar] [CrossRef]
- Holznagel, E.; Yutzy, B.; Schulz-Schaeffer, W.; Kruip, C.; Hahmann, U.; Bierke, P.; Torres, J.-M.; Kim, Y.-S.; Thomzig, A.; Beekes, M.; et al. Foodborne transmission of bovine spongiform encephalopathy to nonhuman primates. Emerging Infect. Dis. 2013, 19, 712–720. [Google Scholar] [CrossRef]
- Asante, E.A.; Linehan, J.M.; Gowland, I.; Joiner, S.; Fox, K.; Cooper, S.; Osiguwa, O.; Gorry, M.; Welch, J.; Houghton, R.; et al. Dissociation of pathological and molecular phenotype of variant Creutzfeldt-Jakob disease in transgenic human prion protein 129 heterozygous mice. Proc. Natl. Acad. Sci. USA 2006, 103, 10759–10764. [Google Scholar] [CrossRef]
- Fernández-Borges, N.; Espinosa, J.C.; Marín-Moreno, A.; Aguilar-Calvo, P.; Asante, E.A.; Kitamoto, T.; Mohri, S.; Andréoletti, O.; Torres, J.M. Protective Effect of Val129-PrP against Bovine Spongiform Encephalopathy but not Variant Creutzfeldt-Jakob Disease. Emerging Infect. Dis. 2017, 23, 1522–1530. [Google Scholar] [CrossRef]
- Comoy, E.E.; Mikol, J.; Jaffré, N.; Lebon, V.; Levavasseur, E.; Streichenberger, N.; Sumian, C.; Perret-Liaudet, A.; Eloit, M.; Andreoletti, O.; et al. Experimental transfusion of variant CJD-infected blood reveals previously uncharacterised prion disorder in mice and macaque. Nat. Commun. 2017, 8, 1268. [Google Scholar] [CrossRef]
- Tateishi, J.; Kitamoto, T. Inherited prion diseases and transmission to rodents. Brain Pathol. 1995, 5, 53–59. [Google Scholar] [CrossRef]
- Masters, C.L.; Gajdusek, D.C.; Gibbs, C.J. Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Sträussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain 1981, 104, 559–588. [Google Scholar] [CrossRef]
- Manson, J.C.; Jamieson, E.; Baybutt, H.; Tuzi, N.L.; Barron, R.; McConnell, I.; Somerville, R.; Ironside, J.; Will, R.; Sy, M.S.; et al. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 1999, 18, 6855–6864. [Google Scholar] [CrossRef]
- Piccardo, P.; Manson, J.C.; King, D.; Ghetti, B.; Barron, R.M. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc. Natl. Acad. Sci. USA 2007, 104, 4712–4717. [Google Scholar] [CrossRef]
- Piccardo, P.; King, D.; Telling, G.; Manson, J.C.; Barron, R.M. Dissociation of prion protein amyloid seeding from transmission of a spongiform encephalopathy. J. Virol. 2013, 87, 12349–12356. [Google Scholar] [CrossRef]
- Asante, E.A.; Grimshaw, A.; Smidak, M.; Jakubcova, T.; Tomlinson, A.; Jeelani, A.; Hamdan, S.; Powell, C.; Joiner, S.; Linehan, J.M.; et al. Transmission Properties of Human PrP 102L Prions Challenge the Relevance of Mouse Models of GSS. PLoS Pathog. 2015, 11, e1004953. [Google Scholar] [CrossRef]
- Asante, E.A.; Linehan, J.M.; Smidak, M.; Tomlinson, A.; Grimshaw, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Powell, C.; Brandner, S.; et al. Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog. 2013, 9, e1003643. [Google Scholar] [CrossRef]
- Race, B.; Williams, K.; Hughson, A.G.; Jansen, C.; Parchi, P.; Rozemuller, A.J.M.; Chesebro, B. Familial human prion diseases associated with prion protein mutations Y226X and G131V are transmissible to transgenic mice expressing human prion protein. Acta Neuropathol. Commun. 2018, 6, 13. [Google Scholar] [CrossRef]
- Pirisinu, L.; Di Bari, M.A.; D’Agostino, C.; Marcon, S.; Riccardi, G.; Poleggi, A.; Cohen, M.L.; Appleby, B.S.; Gambetti, P.; Ghetti, B.; et al. Gerstmann-Sträussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci. Rep. 2016, 6, 20443. [Google Scholar] [CrossRef]
- Mead, S.; Gandhi, S.; Beck, J.; Caine, D.; Gallujipali, D.; Carswell, C.; Hyare, H.; Joiner, S.; Ayling, H.; Lashley, T.; et al. A novel prion disease associated with diarrhea and autonomic neuropathy. N. Engl. J. Med. 2013, 369, 1904–1914. [Google Scholar] [CrossRef]
- Nonno, R.; Notari, S.; Di Bari, M.A.; Cali, I.; Pirisinu, L.; d’Agostino, C.; Cracco, L.; Kofskey, D.; Vanni, I.; Lavrich, J.; et al. Variable Protease-Sensitive Prionopathy Transmission to Bank Voles. Emerging Infect. Dis. 2019, 25, 73–81. [Google Scholar] [CrossRef]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef]

{kind=link}
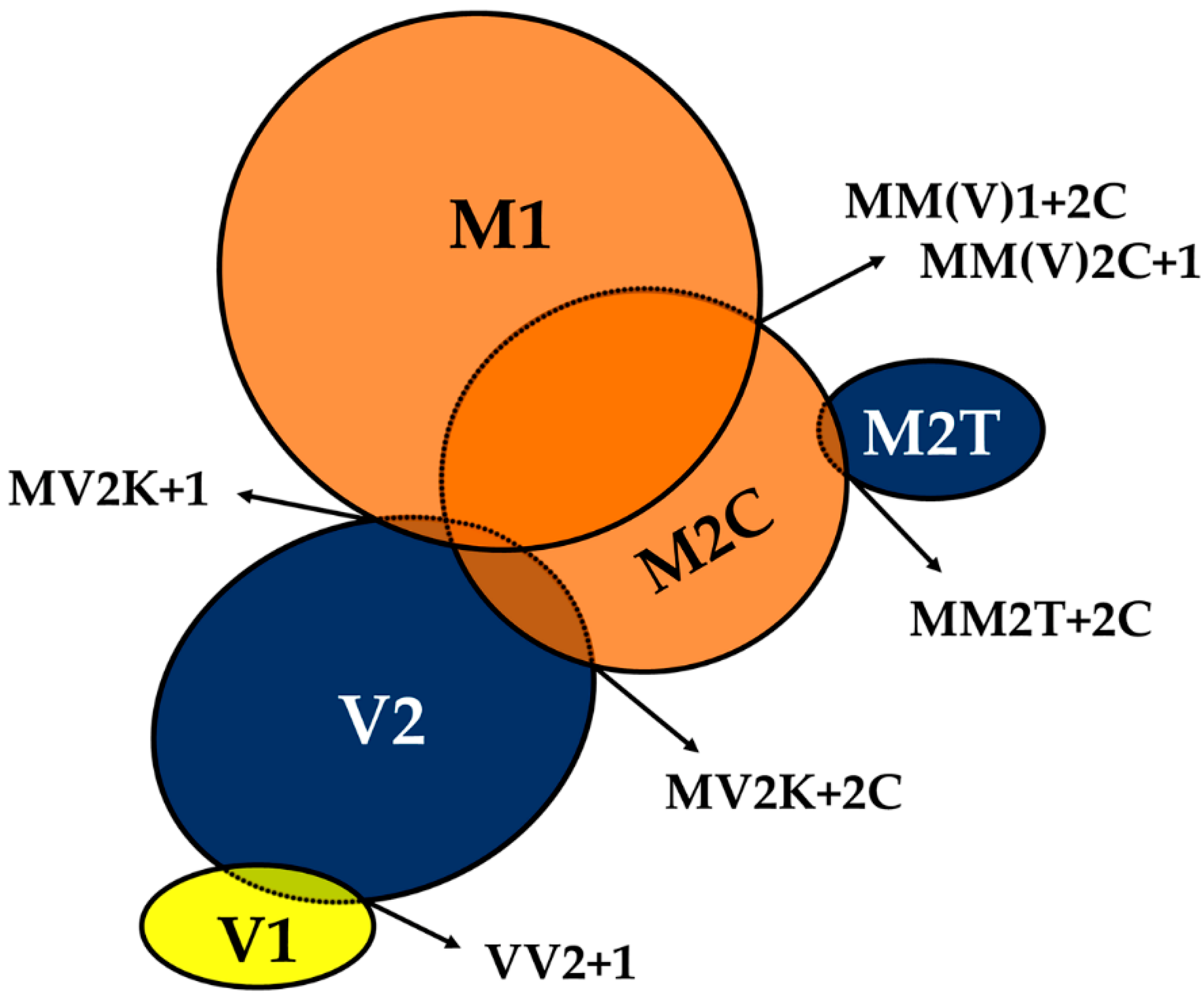{kind=link}
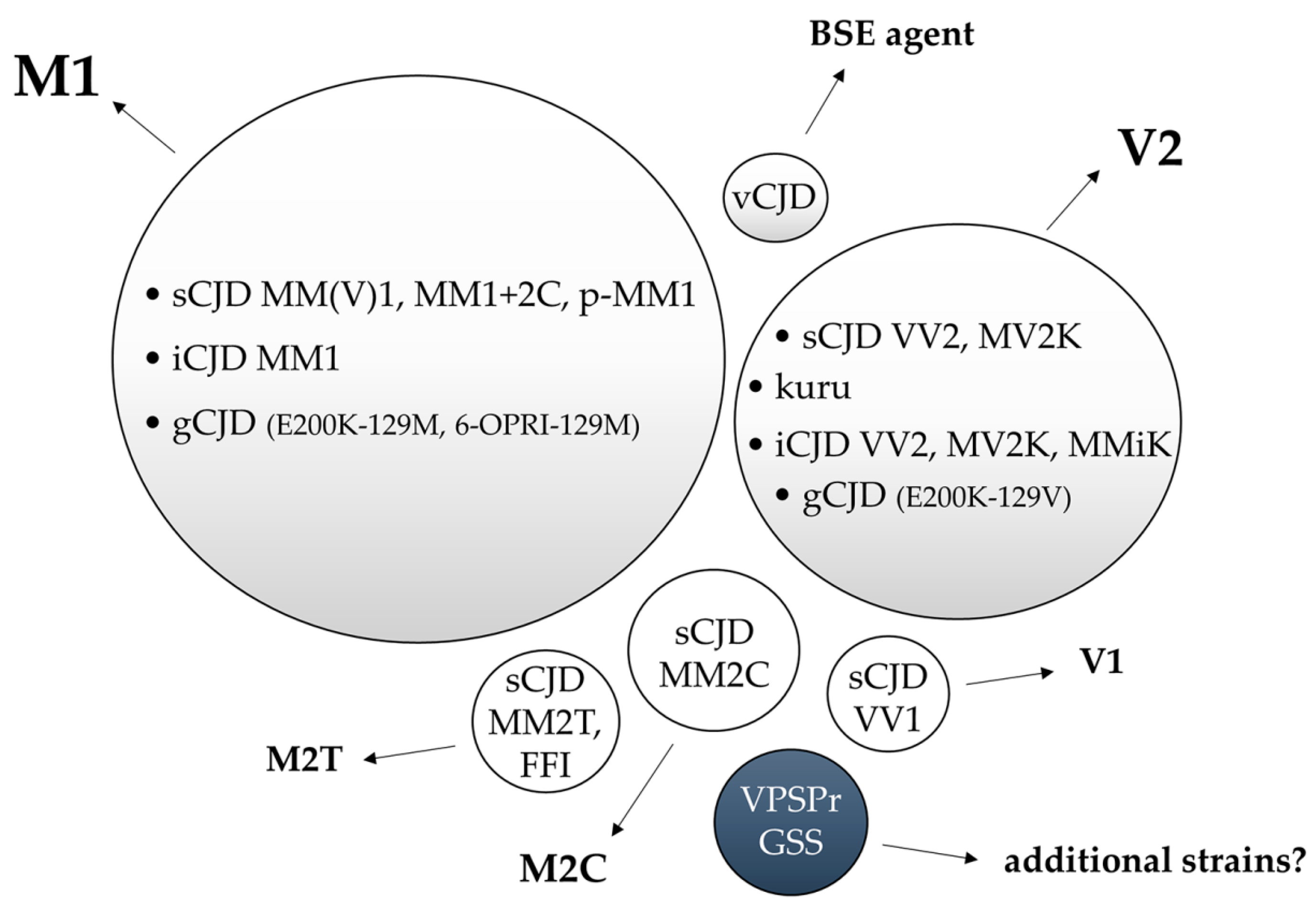{kind=link}
| Human Prion Disorders | Phenotype | VV2 | MV2K | MM(V)1 | vCJD | MM2T | MM2C | VV1 | VPSPr |
| Frequency (% of Cases) | 15 | 8 | 40 | Rare | <1 | <1 | <1 | ~1 | |
| Mean Duration (Months) | 6.3 | 15.8 | 4.0 | 14.0 | 30.2 | 20.0 | 15.3 | 23.0-45.1 | |
| Biochemical Features | T50(°C) | 82.05 ± 3.70 | 79.48 ± 3.63 | 79.66 ± 2.30 | 65.26 ± 3.19 | 59.41 ± 6.04 | 57.11 ± 5.96 | <25 | NA |
| PK-Resistance ED50 (U/ml) | 4.14 ± 3.56 | 1.41 ± 0.89 | 0.09 ± 0.07 | 5.19 ± 2.38 | 0.13 ± 0.09 | 0.28 ± 0.21 | 0.03 ± 0.21 | 0.07 ± 0.01 | |
| Glycoform Ratio (D/U) * | 2.05 | 0.88 | 1.04 | 2.99 | 0.66 | 0.78 | 0.53 | 0° | |
| Aggregation Ratio ¥ | 8.68 ± 1.22 | 8.38 ± 1.93 | 6.43 ± 2.15 | 11.65 ± 3.35 | 10.92 ± 1.44 | 5.68 ± 0.99 | 5.47 ± 0.46 | 3.76 ± 0.28 | |
| Transmission Properties | Attack Rate (%) | 100 | 100 | 100 | 83–100 | 93 | 0 | 14 | 0 |
| Incubation Time (days) | 274 ± 4, 302 ± 9 | 288 ± 3, 329 ± 3 | 446 ± 3, 467 ± 24 | 540 ± 41, 668 ± 22 | 535 ± 32 | NA | 568 ± 0 | NA | |
| Isolated Strain | V2 | M1 | BSE | M2T | M2C | V1 | ND | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, M.; Baiardi, S.; Parchi, P. Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans. Viruses 2019, 11, 309. https://doi.org/10.3390/v11040309
Rossi M, Baiardi S, Parchi P. Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans. Viruses. 2019; 11(4):309. https://doi.org/10.3390/v11040309
Chicago/Turabian StyleRossi, Marcello, Simone Baiardi, and Piero Parchi. 2019. "Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans" Viruses 11, no. 4: 309. https://doi.org/10.3390/v11040309
APA StyleRossi, M., Baiardi, S., & Parchi, P. (2019). Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans. Viruses, 11(4), 309. https://doi.org/10.3390/v11040309