Influenza Virus Infections and Cellular Kinases



Abstract
1. Introduction
2. Phosphorylation of Influenza Virus Proteins
3. Tyrosine Kinases
4. Serine/Threonine Kinases
5. Lipid Kinases
6. Linking Metabolism and Innate Immunity
7. Perspectives and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza a viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [PubMed]
- Moore, H.C.; Lehmann, D.; de Klerk, N.; Smith, D.W.; Richmond, P.C.; Keil, A.D.; Blyth, C.C. How accurate are international classification of diseases-10 diagnosis codes in detecting influenza and pertussis hospitalizations in children? J. Pediatr. Infect. Dis. Soc. 2014, 3, 255–260. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Influenza (Seasonal) Fact Sheet. Available online: http://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 12 November 2018).
- Nair, H.; Brooks, W.A.; Katz, M.; Roca, A.; Berkley, J.A.; Madhi, S.A.; Simmerman, J.M.; Gordon, A.; Sato, M.; Howie, S.; et al. Global burden of respiratory infections due to seasonal influenza in young children: A systematic review and meta-analysis. Lancet 2011, 378, 1917–1930. [Google Scholar] [CrossRef]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the united states. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef] [PubMed]
- van de Sandt, C.E.; Bodewes, R.; Rimmelzwaan, G.F.; de Vries, R.D. Influenza b viruses: Not to be discounted. Future Microbiol. 2015, 10, 1447–1465. [Google Scholar] [CrossRef] [PubMed]
- Paul Glezen, W.; Schmier, J.K.; Kuehn, C.M.; Ryan, K.J.; Oxford, J. The burden of influenza b: A structured literature review. Am. J. Public Health 2013, 103, e43–e51. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Nguyen, T.H.; Barclay, W.S.; Kedzierska, K. Knowns and unknowns of influenza b viruses. Future Microbiol. 2016, 11, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; de Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef]
- Ison, M.G. Antiviral treatments. Clin. Chest Med. 2017, 38, 139–153. [Google Scholar] [CrossRef]
- Carrat, F.; Flahault, A. Influenza vaccine: The challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza a virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N. Structural basis for substrate recognition and control in protein kinases. In Data Mining in Structural Biology; Springer: Berlin/Heidelberg, Germany, 2001; pp. 47–69. [Google Scholar]
- Jacob, T.; Van den Broeke, C.; Favoreel, H.W. Viral serine/threonine protein kinases. J. Virol. 2011, 85, 1158–1173. [Google Scholar] [CrossRef]
- Kennelly, P.J.; Krebs, E.G. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 1991, 266, 15555–15558. [Google Scholar]
- Raggiaschi, R.; Gotta, S.; Terstappen, G.C. Phosphoproteome analysis. Biosci. Rep. 2005, 25, 33–44. [Google Scholar] [CrossRef]
- Johnson, L.N.; Lewis, R.J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. Erk and p38 mapk-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Frojdo, S.; Vidal, H.; Pirola, L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochim. Biophys. Acta 2009, 1792, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Flight, M.H. Neurodegenerative diseases: New kinase targets for alzheimer’s disease. Nat. Rev. Drug Discov. 2013, 12, 739. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Hijazi, M.; Britton, D.; Cutillas, P.R. Impact of phosphoproteomics in the translation of kinase-targeted therapies. Proteomics 2017, 17, 1600235. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of fda-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Kumar, N.; Liang, Y.; Parslow, T.G.; Liang, Y. Receptor tyrosine kinase inhibitors block multiple steps of influenza a virus replication. J. Virol. 2011, 85, 2818–2827. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Sharma, N.R.; Ly, H.; Parslow, T.G.; Liang, Y. Receptor tyrosine kinase inhibitors that block replication of influenza a and other viruses. Antimicrob. Agents Chemother. 2011, 55, 5553–5559. [Google Scholar] [CrossRef]
- Kurokawa, M.; Ochiai, H.; Nakajima, K.; Niwayama, S. Inhibitory effect of protein kinase c inhibitor on the replication of influenza type a virus. J. Gen. Virol. 1990, 71, 2149–2155. [Google Scholar] [CrossRef]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the raf/mek/erk signalling cascade. Nat. Cell Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef]
- Arrese, M.; Portela, A. Serine 3 is critical for phosphorylation at the n-terminal end of the nucleoprotein of influenza virus a/victoria/3/75. J. Virol. 1996, 70, 3385–3391. [Google Scholar]
- Hsiang, T.-Y.; Zhou, L.; Krug, R.M. Roles of the phosphorylation of specific serines and threonines in the ns1 protein of human influenza a viruses. J. Virol. 2012, 86, 10370–10376. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; Denham, E.M.; Thomas, B.; Trudgian, D.C.; Hester, S.S.; Ridlova, G.; York, A.; Turrell, L.; Fodor, E. Mapping the phosphoproteome of influenza a and b viruses by mass spectrometry. PLoS Pathog. 2012, 8, e1002993. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Z.; Bi, Y.; Sun, L.; Liu, X.; Liu, W. Tyrosine 132 phosphorylation of influenza a virus m1 protein is crucial for virus replication by controlling the nuclear import of m1. J. Virol. 2013, 87, 6182–6191. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, J.; Wang, S.; Cao, S.; Jiang, J.; Chen, C.; Ding, C.; Qin, C.; Ye, X.; Gao, G.F.; et al. Phosphorylation controls the nuclear-cytoplasmic shuttling of influenza a virus nucleoprotein. J. Virol. 2015, 89, 5822–5834. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Heo, J.; Yi, C.M.; Ban, J.; Lee, N.J.; Lee, N.R.; Kim, S.W.; Kim, N.J.; Inn, K.S. A novel p38 mitogen activated protein kinase (mapk) specific inhibitor suppresses respiratory syncytial virus and influenza a virus replication by inhibiting virus-induced p38 mapk activation. Biochem. Biophys. Res. Commun. 2016, 477, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C. From virus entry to release: The diverse functions of pi3k during rna virus infections. Future Virol. 2011, 6, 1225–1239. [Google Scholar] [CrossRef]
- Elbahesh, H.; Bergmann, S.; Russell, C.J. Focal adhesion kinase (fak) regulates polymerase activity of multiple influenza a virus subtypes. Virology 2016, 499, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Elbahesh, H.; Cline, T.; Baranovich, T.; Govorkova, E.A.; Schultz-Cherry, S.; Russell, C.J. Novel roles of focal adhesion kinase in cytoplasmic entry and replication of influenza a viruses. J. Virol. 2014, 88, 6714–6728. [Google Scholar] [CrossRef]
- Marjuki, H.; Gornitzky, A.; Marathe, B.M.; Ilyushina, N.A.; Aldridge, J.R.; Desai, G.; Webby, R.J.; Webster, R.G. Influenza a virus-induced early activation of erk and pi3k mediates v-atpase-dependent intracellular ph change required for fusion. Cell. Microbiol. 2011, 13, 587–601. [Google Scholar] [CrossRef]
- Ohmichi, M.; Pang, L.; Ribon, V.; Gazit, A.; Levitzki, A.; Saltiel, A.R. The tyrosine kinase inhibitor tyrphostin blocks the cellular actions of nerve growth factor. Biochemistry 1993, 32, 4650–4658. [Google Scholar] [CrossRef]
- Planz, O. Development of cellular signaling pathway inhibitors as new antivirals against influenza. Antivir. Res. 2013, 98, 457–468. [Google Scholar] [CrossRef]
- Sieczkarski, S.B.; Brown, H.A.; Whittaker, G.R. Role of protein kinase c betaii in influenza virus entry via late endosomes. J. Virol. 2003, 77, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhang, S.; Hu, Y.; Li, D.; Cui, J.; Xue, J.; Zhang, G.; Khachigian, L.M.; Wong, J.; Sun, L.; et al. Regulatory roles of c-jun in h5n1 influenza virus replication and host inflammation. Biochim. Biophys. Acta 2014, 1842, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ruan, T.; Sheng, T.; Wang, J.; Sun, J.; Wang, J.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. Role of c-jun terminal kinase (jnk) activation in influenza a virus-induced autophagy and replication. Virology 2018, 526, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S. Targeting cell signalling pathways to fight the flu: Towards a paradigm change in anti-influenza therapy. J. Antimicrob. Chemother. 2009, 64, 1–4. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Davis, S.A.; Matthay, M.A.; Kurdowska, A.K. Inhibiting bruton’s tyrosine kinase rescues mice from lethal influenza-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L52–l58. [Google Scholar] [CrossRef] [PubMed]
- Hrincius, E.R.; Liedmann, S.; Anhlan, D.; Wolff, T.; Ludwig, S.; Ehrhardt, C. Avian influenza viruses inhibit the major cellular signalling integrator c-abl. Cell. Microbiol. 2014, 16, 1854–1874. [Google Scholar] [CrossRef]
- Hrincius, E.R.; Liedmann, S.; Finkelstein, D.; Vogel, P.; Gansebom, S.; Ehrhardt, C.; Ludwig, S.; Hains, D.S.; Webby, R.; McCullers, J.A. Nonstructural protein 1 (ns1)-mediated inhibition of c-abl results in acute lung injury and priming for bacterial co-infections: Insights into 1918 h1n1 pandemic? J. Infect. Dis. 2015, 211, 1418–1428. [Google Scholar] [CrossRef]
- Berg, J.; Zscheppang, K.; Fatykhova, D.; Tonnies, M.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Ruckert, J.C.; Eggeling, S.; Schimek, M.; et al. Tyk2 as a target for immune regulation in human viral/bacterial pneumonia. Eur. Respir. J. 2017, 50, 1601953. [Google Scholar] [CrossRef]
- Zhang, S.; Tian, H.; Cui, J.; Xiao, J.; Wang, M.; Hu, Y. The c-jun n-terminal kinase (jnk) is involved in h5n1 influenza a virus rna and protein synthesis. Arch. Virol. 2016, 161, 345–351. [Google Scholar] [CrossRef]
- Borgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal h5n1 infection. J. Biol Chem. 2014, 289, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar] [CrossRef] [PubMed]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.; Palamara, A. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by nox4-derived ros. Cell. Microbiol. 2014, 17, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Droebner, K.; Pleschka, S.; Ludwig, S.; Planz, O. Antiviral activity of the mek-inhibitor u0126 against pandemic h1n1v and highly pathogenic avian influenza virus in vitro and in vivo. Antivir. Res. 2011, 92, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Haasbach, E.; Hartmayer, C.; Planz, O. Combination of mek inhibitors and oseltamivir leads to synergistic antiviral effects after influenza a virus infection in vitro. Antivir. Res. 2013, 98, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Haasbach, E.; Muller, C.; Ehrhardt, C.; Schreiber, A.; Pleschka, S.; Ludwig, S.; Planz, O. The mek-inhibitor ci-1040 displays a broad anti-influenza virus activity in vitro and provides a prolonged treatment window compared to standard of care in vivo. Antivir. Res. 2017, 142, 178–184. [Google Scholar] [CrossRef]
- Marjuki, H.; Alam, M.I.; Ehrhardt, C.; Wagner, R.; Planz, O.; Klenk, H.D.; Ludwig, S.; Pleschka, S. Membrane accumulation of influenza a virus hemagglutinin triggers nuclear export of the viral genome via protein kinase calpha-mediated activation of erk signaling. J. Biol. Chem. 2006, 281, 16707–16715. [Google Scholar] [CrossRef]
- Kakugawa, S.; Shimojima, M.; Goto, H.; Horimoto, T.; Oshimori, N.; Neumann, G.; Yamamoto, T.; Kawaoka, Y. Mitogen-activated protein kinase-activated kinase rsk2 plays a role in innate immune responses to influenza virus infection. J. Virol. 2009, 83, 2510–2517. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Ruckle, A.; Hrincius, E.R.; Haasbach, E.; Anhlan, D.; Ahmann, K.; Banning, C.; Reiling, S.J.; Kuhn, J.; Strobl, S.; et al. The nf-kappab inhibitor sc75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell. Microbiol. 2013, 15, 1198–1211. [Google Scholar] [CrossRef]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza a virus-encoded ns1 virulence factor protein inhibits innate immune response by targeting ikk. Cell. Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef]
- Haasbach, E.; Reiling, S.J.; Ehrhardt, C.; Droebner, K.; Ruckle, A.; Hrincius, E.R.; Leban, J.; Strobl, S.; Vitt, D.; Ludwig, S.; et al. The nf-kappab inhibitor sc75741 protects mice against highly pathogenic avian influenza a virus. Antivir. Res. 2013, 99, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Dudziak, D.; Dirmeier, U.; Hobom, G.; Riedel, A.; Schlee, M.; Staudt, L.M.; Rosenwald, A.; Behrends, U.; Bornkamm, G.W.; et al. Active nf-kappab signalling is a prerequisite for influenza virus infection. J. Gen. Virol. 2004, 85, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Ehrhardt, C.; Pleschka, S.; Berberich-Siebelt, F.; Wolff, T.; Walczak, H.; Planz, O.; Ludwig, S. Nf-kappab-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (trail) and fas/fasl is crucial for efficient influenza virus propagation. J. Biol. Chem. 2004, 279, 30931–30937. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Kohno, S.; Newstead, M.W.; Zeng, X.; Bhan, U.; Lukacs, N.W.; Kunkel, S.L.; Standiford, T.J. Critical role of il-1 receptor-associated kinase-m in regulating chemokine-dependent deleterious inflammation in murine influenza pneumonia. J. Immunol. 2010, 184, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudian, S.; Auerochs, S.; Grone, M.; Marschall, M. Influenza a virus proteins pb1 and ns1 are subject to functionally important phosphorylation by protein kinase C. J. Gen. Virol. 2009, 90, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Mitzner, D.; Dudek, S.E.; Studtrucker, N.; Anhlan, D.; Mazur, I.; Wissing, J.; Jansch, L.; Wixler, L.; Bruns, K.; Sharma, A.; et al. Phosphorylation of the influenza a virus protein pb1-f2 by pkc is crucial for apoptosis promoting functions in monocytes. Cell. Microbiol. 2009, 11, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Dawson, A.R.; Potts, G.K.; Freiberger, E.C.; Baker, S.F.; Moser, L.A.; Bernard, K.A.; Coon, J.J.; Mehle, A. Influenza virus recruits host protein kinase c to control assembly and activity of its replication machinery. Elife 2017, 6, e26910. [Google Scholar] [CrossRef] [PubMed]
- Yanguez, E.; Hunziker, A.; Dobay, M.P.; Yildiz, S.; Schading, S.; Elshina, E.; Karakus, U.; Gehrig, P.; Grossmann, J.; Dijkman, R.; et al. Phosphoproteomic-based kinase profiling early in influenza virus infection identifies grk2 as antiviral drug target. Nat. Commun. 2018, 9, 3679. [Google Scholar] [CrossRef]
- Moseley, C.E.; Webster, R.G.; Aldridge, J.R. Peroxisome proliferator-activated receptor and amp-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respir. Viruses 2010, 4, 307–311. [Google Scholar] [CrossRef]
- Pohl, M.O.; von Recum-Knepper, J.; Rodriguez-Frandsen, A.; Lanz, C.; Yanguez, E.; Soonthornvacharin, S.; Wolff, T.; Chanda, S.K.; Stertz, S. Identification of polo-like kinases as potential novel drug targets for influenza a virus. Sci. Rep. 2017, 7, 8629. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Ludwig, S. A new player in a deadly game: Influenza viruses and the pi3k/akt signalling pathway. Cell. Microbiol. 2009, 11, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Marjuki, H.; Wolff, T.; Nurnberg, B.; Planz, O.; Pleschka, S.; Ludwig, S. Bivalent role of the phosphatidylinositol-3-kinase (pi3k) during influenza virus infection and host cell defence. Cell. Microbiol. 2006, 8, 1336–1348. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza a virus ns1 protein activates the pi3k/akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Effect of the phosphatidylinositol 3-kinase/akt pathway on influenza a virus propagation. J. Gen. Virol. 2007, 88, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Pritzl, C.J.; Vijayan, M.; Bomb, K.; McClain, M.E.; Alexander, S.; Hahm, B. Sphingosine kinase 1 serves as a pro-viral factor by regulating viral rna synthesis and nuclear export of viral ribonucleoprotein complex upon influenza virus infection. PLoS ONE 2013, 8, e75005. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Seo, Y.J.; Studstill, C.J.; Vijayan, M.; Wolf, J.J.; Hahm, B. Transient inhibition of sphingosine kinases confers protection to influenza a virus infected mice. Antivir. Res. 2018, 158, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Root, C.N.; Wills, E.G.; McNair, L.L.; Whittaker, G.R. Entry of influenza viruses into cells is inhibited by a highly specific protein kinase c inhibitor. J. Gen. Virol. 2000, 81, 2697–2705. [Google Scholar] [CrossRef]
- Ayllon, J.; Garcia-Sastre, A. The ns1 protein: A multitasking virulence factor. Curr. Top Microbiol. Immunol. 2015, 386, 73–107. [Google Scholar]
- Krug, R.M. Functions of the influenza a virus ns1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional ns1 protein of influenza a viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Hale, B.G.; Knebel, A.; Botting, C.H.; Galloway, C.S.; Precious, B.L.; Jackson, D.; Elliott, R.M.; Randall, R.E. Cdk/erk-mediated phosphorylation of the human influenza a virus ns1 protein at threonine-215. Virology 2009, 383, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Hirata, N.; Suizu, F.; Matsuda-Lennikov, M.; Edamura, T.; Bala, J.; Noguchi, M. Inhibition of akt kinase activity suppresses entry and replication of influenza virus. Biochem. Biophys. Res. Commun. 2014, 450, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Chenavas, S.; Estrozi, L.F.; Slama-Schwok, A.; Delmas, B.; Di Primo, C.; Baudin, F.; Li, X.; Crepin, T.; Ruigrok, R.W. Monomeric nucleoprotein of influenza a virus. PLoS Pathog. 2013, 9, e1003275. [Google Scholar] [CrossRef] [PubMed]
- Cheshenko, N.; Liu, W.; Satlin, L.M.; Herold, B.C. Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J. Biol. Chem. 2005, 280, 31116–31125. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, P.M.; Keiser, N.W.; Yan, Z.; Lei-Butters, D.C.; Engelhardt, J.F. Directing integrin-linked endocytosis of recombinant aav enhances productive fak-dependent transduction. Mol. Ther. 2012, 20, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Sharma-Walia, N.; Streblow, D.N.; Naranatt, P.P.; Chandran, B. Focal adhesion kinase is critical for entry of kaposi’s sarcoma-associated herpesvirus into target cells. J. Virol. 2006, 80, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Wang, L.; Schneider, R.J. Activation of focal adhesion kinase by hepatitis b virus hbx protein: Multiple functions in viral replication. J. Virol. 2006, 80, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Fouquet, B.; Nikolic, J.; Larrous, F.; Bourhy, H.; Wirblich, C.; Lagaudriere-Gesbert, C.; Blondel, D. Focal adhesion kinase is involved in rabies virus infection through its interaction with viral phosphoprotein p. J. Virol. 2015, 89, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Ni, B.; Wen, L.B.; Wang, R.; Hao, H.P.; Huan, C.C.; Wang, X.; Huang, L.; Miao, J.F.; Fan, H.J.; Mao, X. The involvement of fak-pi3k-akt-rac1 pathway in porcine reproductive and respiratory syndrome virus entry. Biochem. Biophys. Res. Commun. 2015, 458, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Connolly, S.F.; Reinl, E.L.; Houtman, J.C. Focal adhesion kinase negatively regulates lck function downstream of the t cell antigen receptor. J. Immunol. 2013, 191, 6208–6221. [Google Scholar] [CrossRef]
- St-Pierre, J.; Ostergaard, H.L. A role for the protein tyrosine phosphatase cd45 in macrophage adhesion through the regulation of paxillin degradation. PLoS ONE 2013, 8, e71531. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Wolfram, P.; Canty, K.; Harley, B.; Nombela-Arrieta, C.; Pivarnik, G.; Manis, J.; Beggs, H.E.; Silberstein, L.E. Focal adhesion kinase regulates the localization and retention of pro-b cells in bone marrow microenvironments. J. Immunol. 2013, 190, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Bergman, S.; Elbahesh, H. Targeting the proviral host kinase, FAK, limits in vivo and in vitro influenza A virus pathogenesis and NFkB-regulated pro-inflammatory responses. Virology 2019. under review. [Google Scholar]
- Hrincius, E.R.; Wixler, V.; Wolff, T.; Wagner, R.; Ludwig, S.; Ehrhardt, C. Crk adaptor protein expression is required for efficient replication of avian influenza a viruses and controls jnk-mediated apoptotic responses. Cell. Microbiol. 2010, 12, 831–843. [Google Scholar] [CrossRef]
- Short, K.R.; Kroeze, E.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Krupa, A.; Fudala, R.; Florence, J.M.; Tucker, T.; Allen, T.C.; Standiford, T.J.; Luchowski, R.; Fol, M.; Rahman, M.; Gryczynski, Z.; et al. Bruton’s tyrosine kinase mediates fcgammariia/toll-like receptor-4 receptor crosstalk in human neutrophils. Am. J. Respir. Cell. Mol. Biol. 2013, 48, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Henkel, T.J.A. Function and activation of nf-kappab in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Graves, J.; Campbell, J.; Krebs, E.J.A. Protein serine/threonine kinases of the mapk cascade. Ann. N. Y. Acad. Sci. 1995, 766, 320–343. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase c: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef]
- Nencioni, L.; De Chiara, G.; Sgarbanti, R.; Amatore, D.; Aquilano, K.; Marcocci, M.E.; Serafino, A.; Torcia, M.; Cozzolino, F.; Ciriolo, M.R.; et al. Bcl-2 expression and p38mapk activity in cells infected with influenza a virus: Impact on virally induced apoptosis and viral replication. J. Biol. Chem. 2009, 284, 16004–16015. [Google Scholar] [CrossRef]
- Lucia, N.; Rossella, S.; Donatella, A.; Paola, C.; Ignacio, C.; Dolores, L.; Simona, A.; Anna Teresa, P.; Enrico, G. Intracellular redox signaling as therapeutic target for novel antiviral strategy. Curr. Pharm. Des. 2011, 17, 3898–3904. [Google Scholar]
- Jaulmes, A.; Sansilvestri-Morel, P.; Rolland-Valognes, G.; Bernhardt, F.; Gaertner, R.; Lockhart, B.P.; Cordi, A.; Wierzbicki, M.; Rupin, A.; Verbeuren, T.J. Nox4 mediates the expression of plasminogen activator inhibitor-1 via p38 mapk pathway in cultured human endothelial cells. Thromb. Res. 2009, 124, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-derived h2o2 mediates endoplasmic reticulum signaling through local ras activation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef] [PubMed]
- Celestino, I.; Checconi, P.; Amatore, D.; De Angelis, M.; Coluccio, P.; Dattilo, R.; Alunni Fegatelli, D.; Clemente, A.M.; Matarrese, P.; Torcia, M.G.; et al. Differential redox state contributes to sex disparities in the response to influenza virus infection in male and female mice. Front. Immunol. 2018, 9, 1747. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M.; Ostertag, D.; Li, Z.W.; Chang, L.; Chen, Y.; Hu, Y.; Williams, B.; Perrault, J.; Karin, M. Jnk2 and ikkbeta are required for activating the innate response to viral infection. Immunity 1999, 11, 721–731. [Google Scholar] [CrossRef]
- Ludwig, S.; Wolff, T.; Ehrhardt, C.; Wurzer, W.J.; Reinhardt, J.; Planz, O.; Pleschka, S. Mek inhibition impairs influenza b virus propagation without emergence of resistant variants. FEBS Lett. 2004, 561, 37–43. [Google Scholar] [CrossRef]
- Carriere, A.; Ray, H.; Blenis, J.; Roux, P.P. The rsk factors of activating the ras/mapk signaling cascade. Front. Biosci. 2008, 13, 4258–4275. [Google Scholar] [CrossRef]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza a virus ns1 protein to pkr mediates the inhibition of its activation by either pact or double-stranded rna. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef]
- Vaid, R.; Sharma, N.; Chauhan, S.; Deshta, A.; Dev, K.; Sourirajan, A. Functions of polo-like kinases: A journey from yeast to humans. Protein Pept. Lett. 2016, 23, 185–197. [Google Scholar] [CrossRef]
- Sun, D.; Luthra, P.; Li, Z.; He, B. Plk1 down-regulates parainfluenza virus 5 gene expression. PLoS Pathog. 2009, 5, e1000525. [Google Scholar] [CrossRef]
- Chen, Y.C.; Su, W.C.; Huang, J.Y.; Chao, T.C.; Jeng, K.S.; Machida, K.; Lai, M.M. Polo-like kinase 1 is involved in hepatitis c virus replication by hyperphosphorylating ns5a. J. Virol. 2010, 84, 7983–7993. [Google Scholar] [CrossRef] [PubMed]
- Bosco, R.; Melloni, E.; Celeghini, C.; Rimondi, E.; Vaccarezza, M.; Zauli, G.J.M. Fine tuning of protein kinase c (pkc) isoforms in cancer: Shortening the distance from the laboratory to the bedside. Mini Rev. Med. Chem. 2011, 11, 185–199. [Google Scholar] [CrossRef]
- Hrincius, E.R.; Dierkes, R.; Anhlan, D.; Wixler, V.; Ludwig, S.; Ehrhardt, C. Phosphatidylinositol-3-kinase (pi3k) is activated by influenza virus vrna via the pathogen pattern receptor rig-i to promote efficient type i interferon production. Cell. Microbiol. 2011, 13, 1907–1919. [Google Scholar] [CrossRef] [PubMed]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic ras/mapk and survival pi3k/akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The ras-erk and pi3k-mtor pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Cantrell, D.A. Phosphoinositide 3-kinase signalling pathways. J. Cell Sci. 2001, 114, 1439–1445. [Google Scholar] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Ali, K.; Bilancio, A.; Geering, B.; Foukas, L.C. Signalling by pi3k isoforms: Insights from gene-targeted mice. Trends Biochem. Sci. 2005, 30, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Garcia-Sastre, A.; Hale, B.G. Influenza a viruses and pi3k: Are there time, place and manner restrictions? Virulence 2012, 3, 411–414. [Google Scholar] [CrossRef]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza a virus ns1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Wolff, T.; Ludwig, S. Activation of phosphatidylinositol 3-kinase signaling by the nonstructural ns1 protein is not conserved among type a and b influenza viruses. J. Virol. 2007, 81, 12097–12100. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem. Sci. 2011, 36, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, H.S.; Duan, S.; Morfouace, M.; Rezinciuc, S.; Shulkin, B.L.; Shelat, A.; Zink, E.E.; Milasta, S.; Bajracharya, R.; Oluwaseum, A.J.; et al. Targeting metabolic reprogramming by influenza infection for therapeutic intervention. Cell Rep. 2017, 19, 1640–1653. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.L.; Cronin, J.G.; Noleto, P.G.; Sheldon, I.M. Glucose availability and amp-activated protein kinase link energy metabolism and innate immunity in the bovine endometrium. PLoS ONE 2016, 11, e0151416. [Google Scholar] [CrossRef] [PubMed]
- Towler, M.C.; Hardie, D.G. Amp-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Winder, W.W.; Hardie, D.G. Amp-activated protein kinase, a metabolic master switch: Possible roles in type 2 diabetes. Am. J. Physiol.-Endocrinol. Metab. 1999, 277, E1–E10. [Google Scholar] [CrossRef]
- Tao, R.; Gong, J.; Luo, X.; Zang, M.; Guo, W.; Wen, R.; Luo, Z. Ampk exerts dual regulatory effects on the pi3k pathway. J. Mol. Signal. 2010, 5, 1. [Google Scholar] [CrossRef]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic dinucleotides trigger ulk1 (atg1) phosphorylation of sting to prevent sustained innate immune signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef]
- Prantner, D.; Perkins, D.; Vogel, S. Ampk regulates innate immune signaling and viral control through ulk-1 dependent decrease of sting expression (inm7p.430). J. Immunol. 2014, 192, 123.8. [Google Scholar]
- Prantner, D.; Perkins, D.J.; Vogel, S.N. Amp-activated kinase (ampk) promotes innate immunity and antiviral defense through modulation of stimulator of interferon genes (sting) signaling. J. Biol. Chem. 2017, 292, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Dumit, V.I.; Dengjel, J. Autophagosomal protein dynamics and influenza virus infection. Front. Immunol. 2012, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Vallerie, S.N.; Hotamisligil, G.S. The role of jnk proteins in metabolism. Sci. Transl. Med. 2010, 2, 60rv65. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Jain, K.; Basu, A. Regulation of autophagy by kinases. Cancers 2011, 3, 2630–2654. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-stranded rna-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Lee, Y.J. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem. J. 2003, 373, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. Mapk signalling in cellular metabolism: Stress or wellness? EMBO Rep. 2010, 11, 834–840. [Google Scholar] [CrossRef]
- Maines, M.D. Biliverdin reductase: Pkc interaction at the cross-talk of mapk and pi3k signaling pathways. Antioxid. Redox Signal. 2007, 9, 2187–2196. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in nf-κb signaling pathways. Nat. Immunol. 2011, 12, 695. [Google Scholar] [CrossRef]
- Zubair, H.; Azim, S.; Srivastava, S.K.; Ahmad, A.; Bhardwaj, A.; Khan, M.A.; Patel, G.K.; Arora, S.; Carter, J.E.; Singh, S.; et al. Glucose metabolism reprogrammed by overexpression of ikk-epsilon promotes pancreatic tumor growth. Cancer Res. 2016, 76, 7254–7264. [Google Scholar] [CrossRef]
- Tornatore, L.; Thotakura, A.K.; Bennett, J.; Moretti, M.; Franzoso, G. The nuclear factor kappa b signaling pathway: Integrating metabolism with inflammation. Trends Cell Biol. 2012, 22, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Invest. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C. The tail wagging the dog—Regulation of lipid metabolism by protein kinase C. FEBS J. 2013, 280, 5371–5383. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: The role of pi3k/akt/mtor signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef]
- Liu, D.D.; Han, C.C.; Wan, H.F.; He, F.; Xu, H.Y.; Wei, S.H.; Du, X.H.; Xu, F. Effects of inhibiting pi3k-akt-mtor pathway on lipid metabolism homeostasis in goose primary hepatocytes. Animal 2016, 10, 1319–1327. [Google Scholar] [CrossRef]
- Li, C.J.; Jiang, C.; Liu, Y.; Bell, T.; Ma, W.; Ye, Y.; Huang, S.; Guo, H.; Zhang, H.; Wang, L.; et al. Pleiotropic action of novel bruton’s tyrosine kinase inhibitor bgb-3111 in mantle cell lymphoma. Mol. Cancer Ther. 2018, 18, 267–277. [Google Scholar] [CrossRef]
- Cicchini, C.; Laudadio, I.; Citarella, F.; Corazzari, M.; Steindler, C.; Conigliaro, A.; Fantoni, A.; Amicone, L.; Tripodi, M. Tgfbeta-induced emt requires focal adhesion kinase (fak) signaling. Exp. Cell Res. 2008, 314, 143–152. [Google Scholar] [CrossRef]
- Lim, S.T.; Miller, N.L.; Chen, X.L.; Tancioni, I.; Walsh, C.T.; Lawson, C.; Uryu, S.; Weis, S.M.; Cheresh, D.A.; Schlaepfer, D.D. Nuclear-localized focal adhesion kinase regulates inflammatory vcam-1 expression. J. Cell Biol. 2012, 197, 907–919. [Google Scholar] [CrossRef]
- Schaller, M.D. Cellular functions of fak kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef]
- Calalb, M.B.; Polte, T.R.; Hanks, S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for src family kinases. Mol. Cell. Biol. 1995, 15, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Genna, A.; Gil-Henn, H. Fak family kinases: The yin and yang of cancer cell invasiveness. Mol. Cell. Oncol. 2018, 5, e1449584. [Google Scholar] [CrossRef] [PubMed]
- Bozym, R.A.; Delorme-Axford, E.; Harris, K.; Morosky, S.; Ikizler, M.; Dermody, T.S.; Sarkar, S.N.; Coyne, C.B. Focal adhesion kinase is a component of antiviral rig-i-like receptor signaling. Cell Host Microbe 2012, 11, 153–166. [Google Scholar] [CrossRef]
- Dwyer, S.F.; Gao, L.; Gelman, I.H. Identification of novel focal adhesion kinase substrates: Role for fak in nfkappab signaling. Int. J. Biol. Sci. 2015, 11, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M. Targeting focal adhesion kinase in cancer-part i. Anticancer Agents Med. Chem. 2010, 10, 713. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Focal adhesion kinase as a cancer therapy target. Anticancer Agents Med. Chem. 2010, 10, 735–741. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Editorial: Focal adhesion kinase signaling in cancer--part ii. Anticancer Agents Med. Chem. 2011, 11, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Dehart, J.P.; Murphy, J.M.; Lim, S.T. Understanding the roles of fak in cancer: Inhibitors, genetic models, and new insights. J. Histochem. Cytochem. 2015, 63, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Figel, S.; Ho, B.T.; Johnson, C.P.; Yemma, M.; Huang, G.; Zheng, M.; Nyberg, C.; Magis, A.; Ostrov, D.A.; et al. A small molecule focal adhesion kinase (fak) inhibitor, targeting y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3.1.1(3,7)]decane; bromide effectively inhibits fak autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis 2012, 33, 1004–1013. [Google Scholar]
- Golubovskaya, V.M.; Nyberg, C.; Zheng, M.; Kweh, F.; Magis, A.; Ostrov, D.; Cance, W.G. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J. Med. Chem. 2008, 51, 7405–7416. [Google Scholar] [CrossRef]
- O’Brien, S.; Golubovskaya, V.M.; Conroy, J.; Liu, S.; Wang, D.; Liu, B.; Cance, W.G. Fak inhibition with small molecule inhibitor y15 decreases viability, clonogenicity, and cell attachment in thyroid cancer cell lines and synergizes with targeted therapeutics. Oncotarget 2014, 5, 7945–7959. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Curtin, L.; Groman, A.; Sexton, S.; Cance, W.G. In vivo toxicity, metabolism and pharmacokinetic properties of fak inhibitor 14 or y15 (1, 2, 4, 5-benzenetetramine tetrahydrochloride). Arch. Toxicol. 2015, 89, 1095–1101. [Google Scholar] [CrossRef] [PubMed]


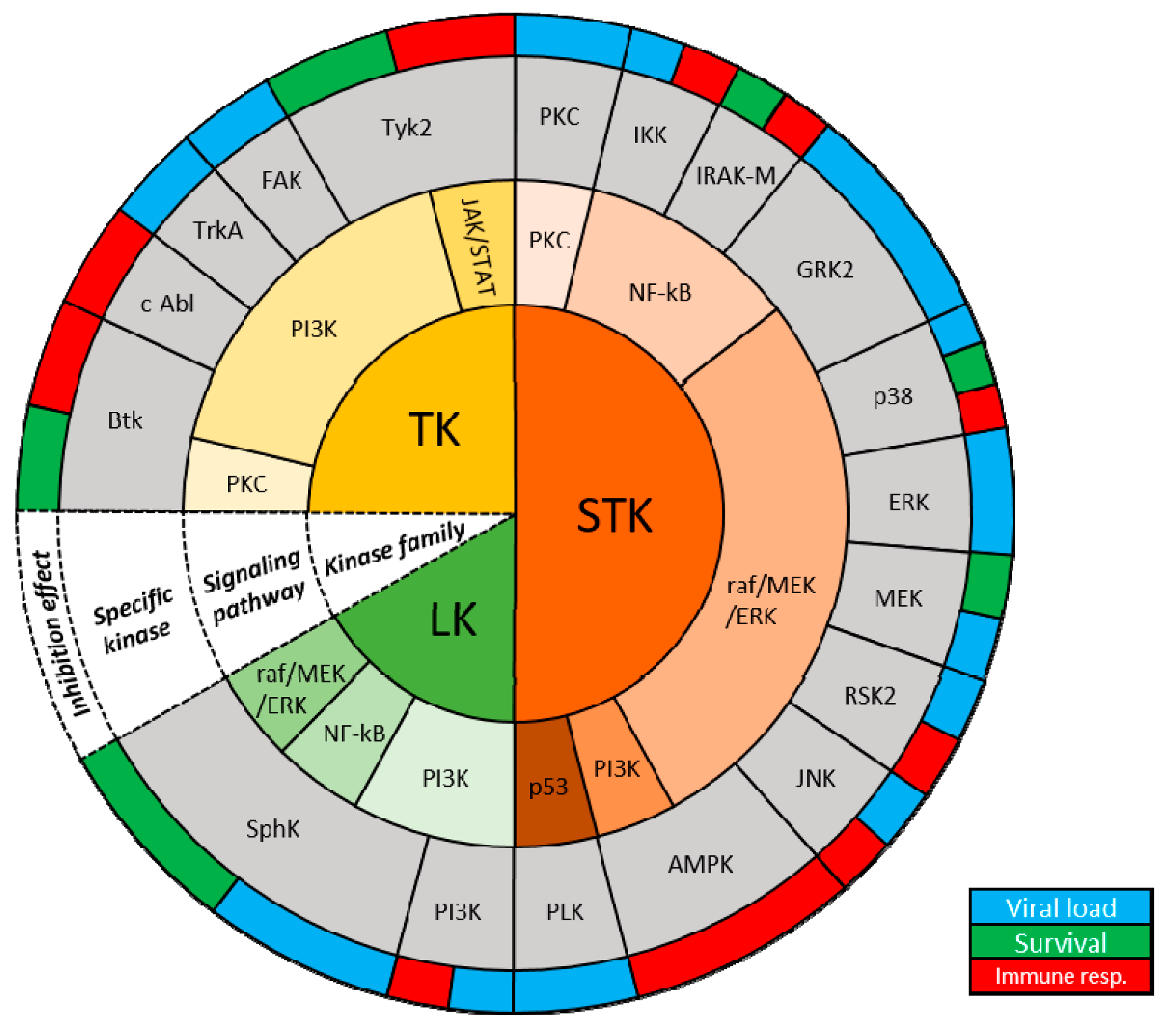{kind=link}
| Name | IAV Effect | In Vitro, In Vivo or Ex Vivo | Inh. vs. KO | Reference | |
|---|---|---|---|---|---|
| Tyrosine | FAK | -Virus entry -Polymerase activity | In vitro | Inhibition | Elbahesh et al., 2014, 2016 [39,40] |
| TrkA | -vRNA synthesis -RNP export -Budding | In vitro | Inhibition | Kumar et al., 2011a, 2011b [28,29] | |
| Btk | -Neutrophil regulation | In vivo | Inhibition | Florence et al., 2018 [49] | |
| c-Abl | -Pathogenicity mediator | In vivo | Inhibition | Hrincius et al., 2014, 2015 [50,51] | |
| Tyk2 | -Cytokine regulation | Ex vivo | Inhibition | Berg et al., 2017 [52] | |
| Serine/Threonine | JNK1 / JNK2 | -vRNA synthesis -Autophagy -Cytokine regulation | In vivo | Inhibition | Zhang et al., 2016, 2018; Xie et al., 2014 [45,46,53] |
| P38 MAPK | -vRNA synthesis -RNP export -Prevents apoptosis -Cytokine regulation -Virus entry | In vivo | Inhibition | Borgeling et al., 2014; Choi et al., 2016; Marchant et al., 2010; Amatore et al., 2014 [37,54,55,56] | |
| MEK | -RNP export | In vivo | Inhibition | Haasbach et al., 2017, 2013; Droebner et al., 2011 [57,58,59] | |
| ERK | -RNP import -RNP export | In vivo | Inhibition | Pleschka et al., 2001, Marjuki et al., 2006 [31,60] | |
| RSK2 | -Polymerase activity | In vitro | Knockdown | Kakugawa et al., 2009 [61] | |
| IKK | -Cytokine regulation -Caspase regulation -RNP export -Antiviral response modification | In vitro | Inhibition | Erhardt et al., 2013; Haasbach et al., 2013; Gao et al., 2012; Nimmerjahn et al., 2004; Wurzer et al., 2004 [62,63,64,65,66] | |
| IRAK-M | -Neutrophil interaction -Cytokine reg. | In vivo | KO | Seki et al., 2010 [67] | |
| PKC | -Endosomal entry -RNP assembly -Polymerase activity -Prevents apoptosis | In vivo | Inhibition | Mondal et al., 2017; Mitzner et al., 2009; Mahmoudian et al., 2009; Sieczkarski et al., 2003; Kurokawa et al., 1990 [30,44,68,69,70] | |
| GRK2 | -viral uncoating | In vivo | Inhibition | Yanguez et al., 2018 [71] | |
| AMPK | -antiviral response | In vivo | Activation | Moseley et al., 2010 [72] | |
| PLK1/3/4 | -unknown | ex vivo | KO | Pohl et al., 2017 [73] | |
| Lipid | PI3K | -Virus entry -Prevents apoptosis -vRNA synthesis -RNP export -antiviral response modification | In vitro | Inhibition | Erhardt et al., 2006,2007; Shin et al., 2007; Erhardt and Ludwig, 2009; Ehrhardt, 2011; Marjuki et al., 2011 [38,41,74,75,76,77] |
| SphK1 / SphK2 | -vRNA synthesis -RNP export | In vivo | Inhibition | Xia et al., 2018; Seo et al., 2013 [78,79] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meineke, R.; Rimmelzwaan, G.F.; Elbahesh, H. Influenza Virus Infections and Cellular Kinases. Viruses 2019, 11, 171. https://doi.org/10.3390/v11020171
Meineke R, Rimmelzwaan GF, Elbahesh H. Influenza Virus Infections and Cellular Kinases. Viruses. 2019; 11(2):171. https://doi.org/10.3390/v11020171
Chicago/Turabian StyleMeineke, Robert, Guus F. Rimmelzwaan, and Husni Elbahesh. 2019. "Influenza Virus Infections and Cellular Kinases" Viruses 11, no. 2: 171. https://doi.org/10.3390/v11020171
APA StyleMeineke, R., Rimmelzwaan, G. F., & Elbahesh, H. (2019). Influenza Virus Infections and Cellular Kinases. Viruses, 11(2), 171. https://doi.org/10.3390/v11020171