Insights into Innate Sensing of Prototype Foamy Viruses in Myeloid Cells

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
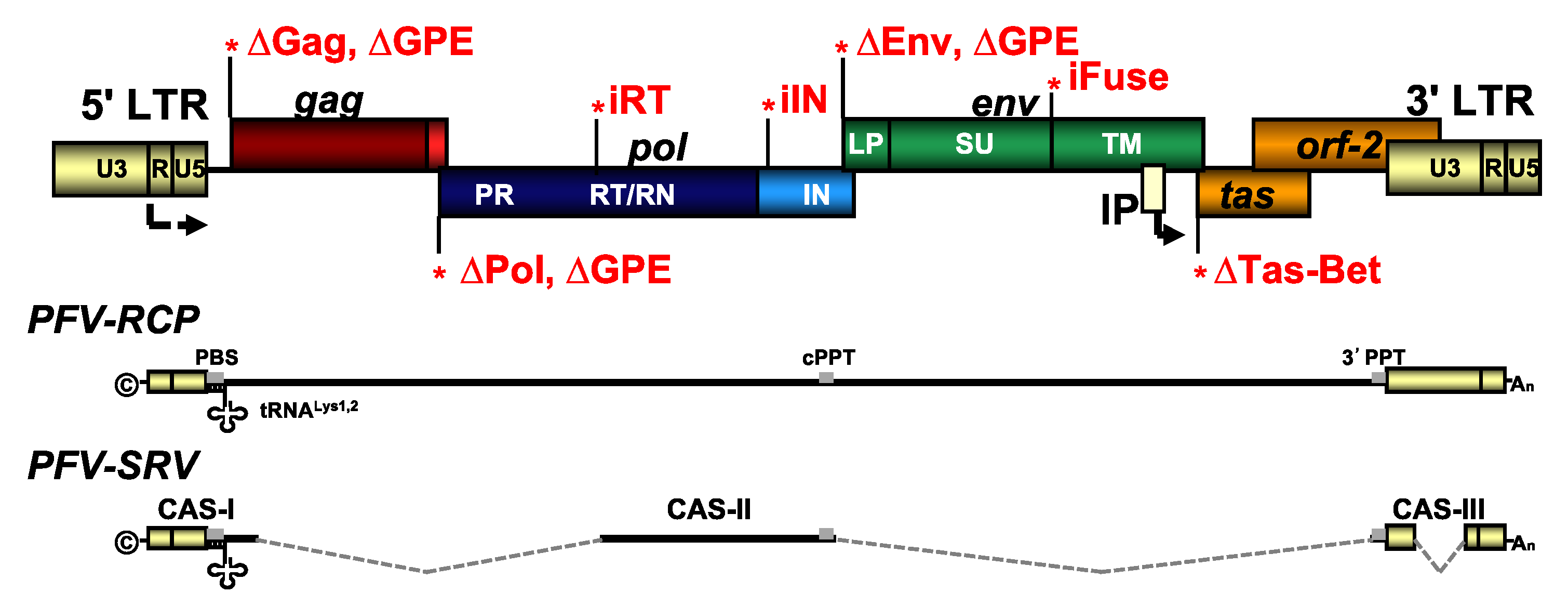
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Recombinant Plasmid DNAs
2.3. Transfection, Virus Production, and Titration
2.4. Myeloid Cell Stimulation and qPCR Analysis of ISG Induction
2.5. Quantitative PCR Analysis
2.6. Flow Cytometry Analysis
2.7. Analysis of PFV Particle Protein and Nucleic Acid Composition
2.8. Analysis of Cellular Protein Expression
2.9. Statistics
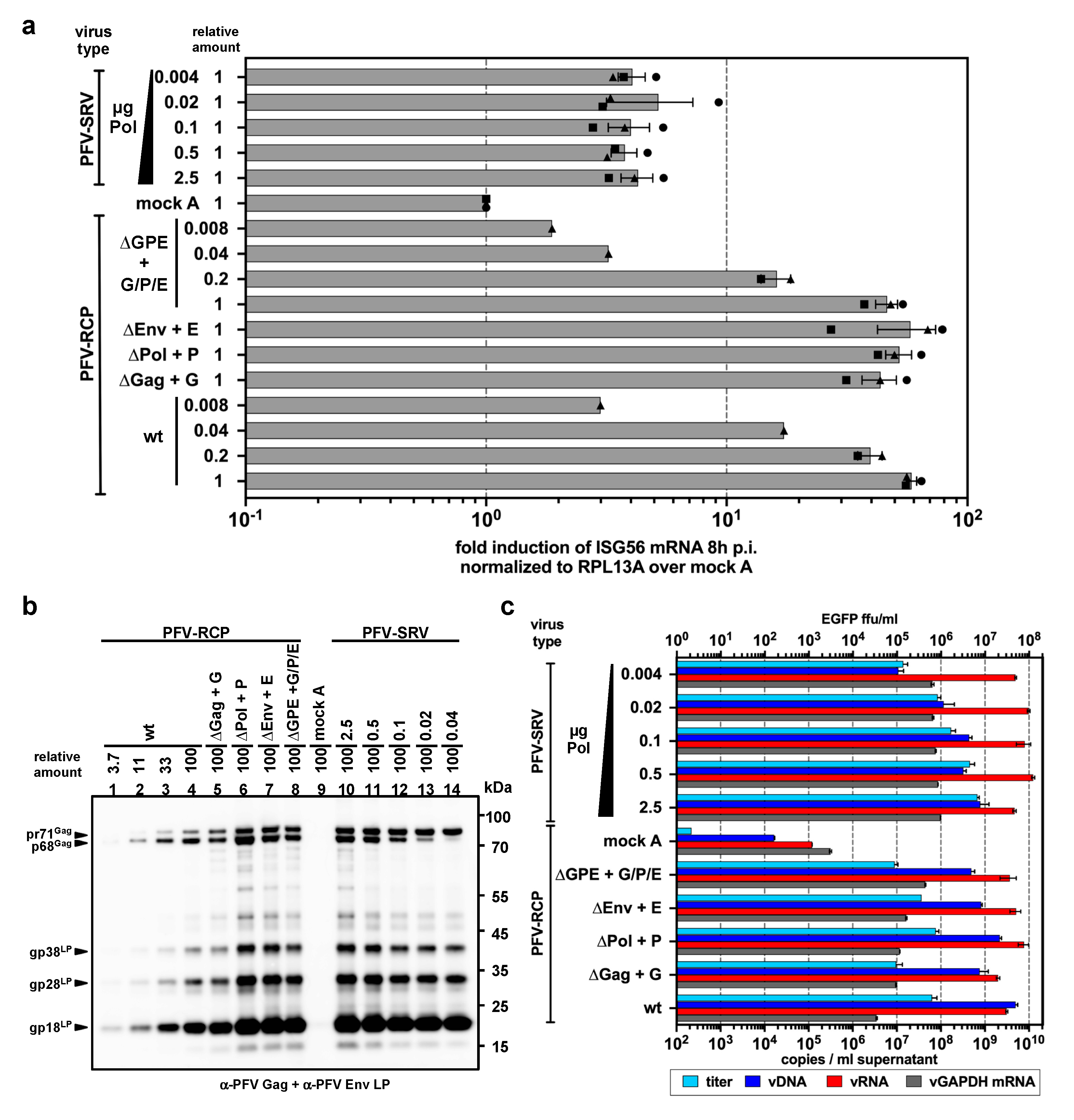
3. Results
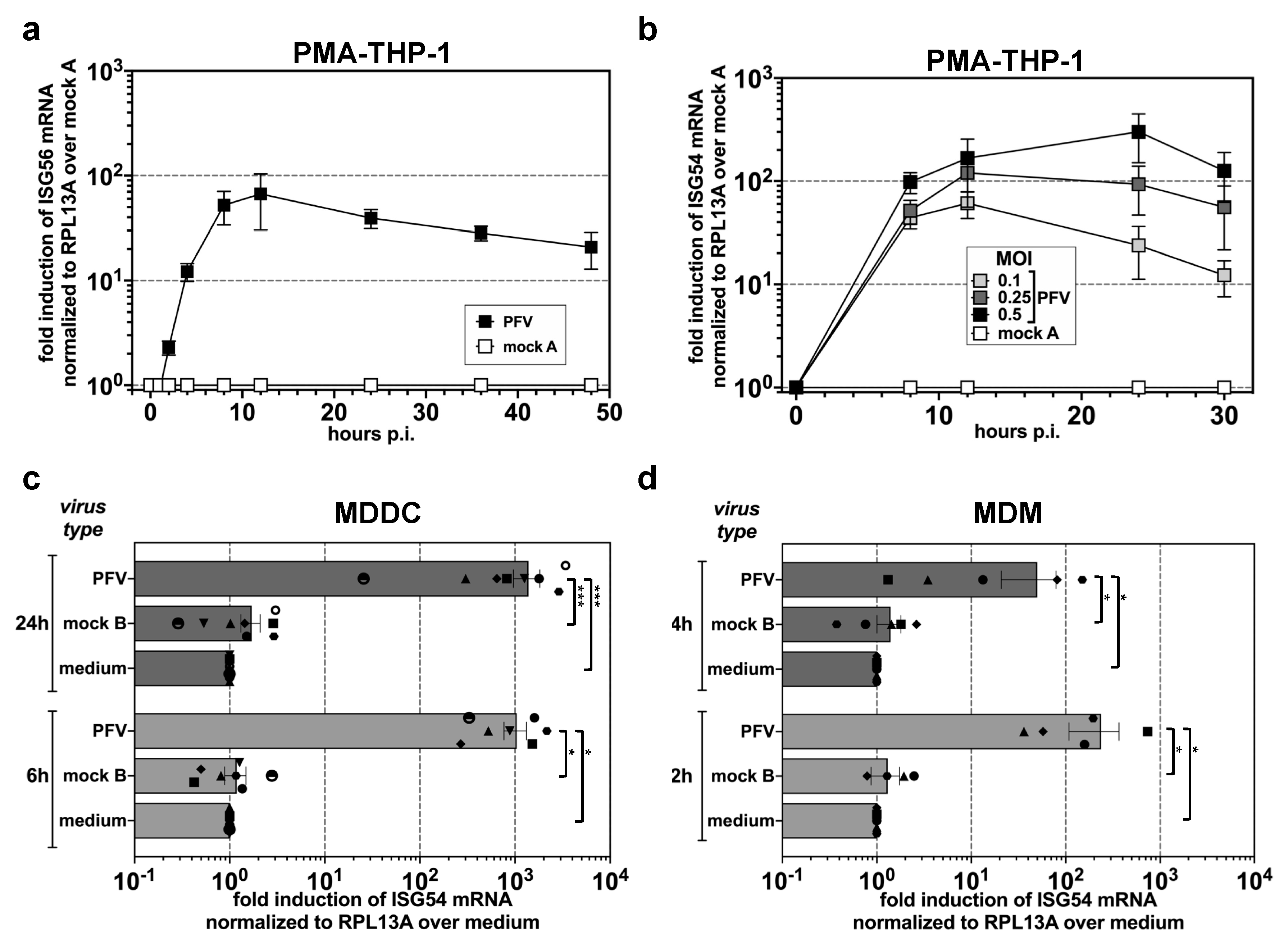
3.1. ISG Induction in Myeloid Cells upon Exposure to Replication-Competent PFV
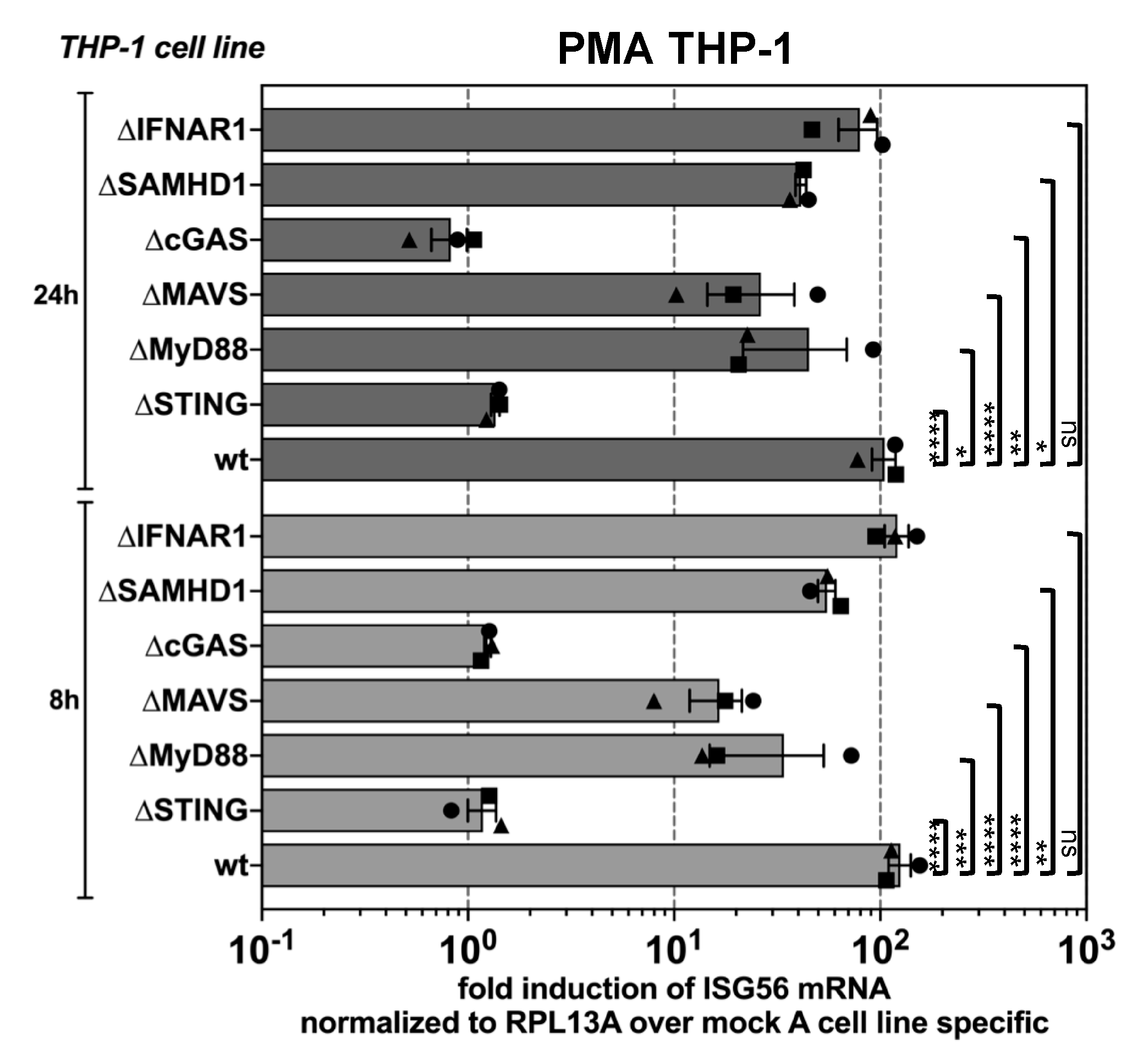
3.2. PFV is Sensed by the Cellular cGAS-STING Pathway
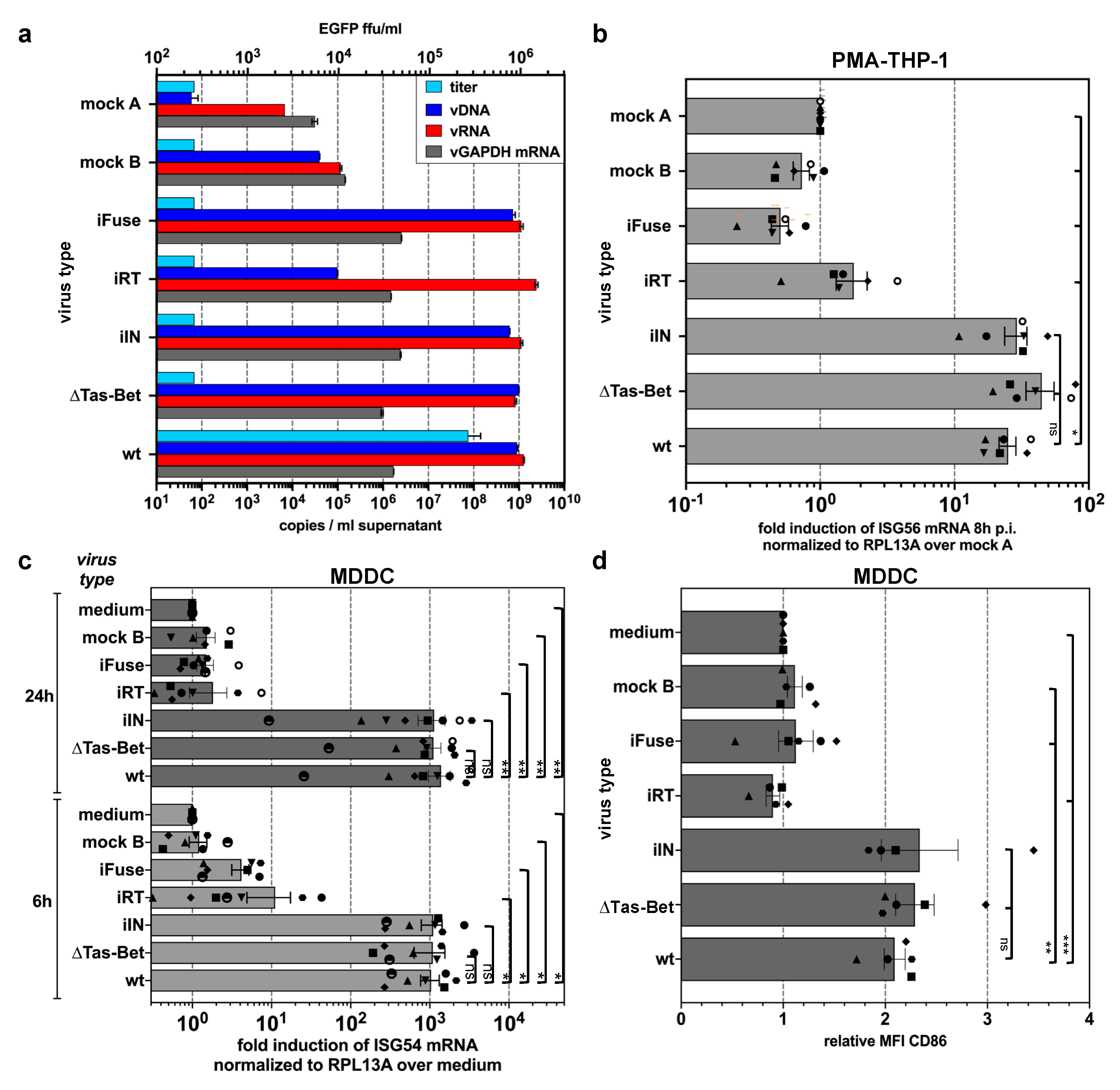
3.3. Innate Sensing of PFV Requires Cytoplasmic Access and Enzymatically Active Reverse Transcriptase
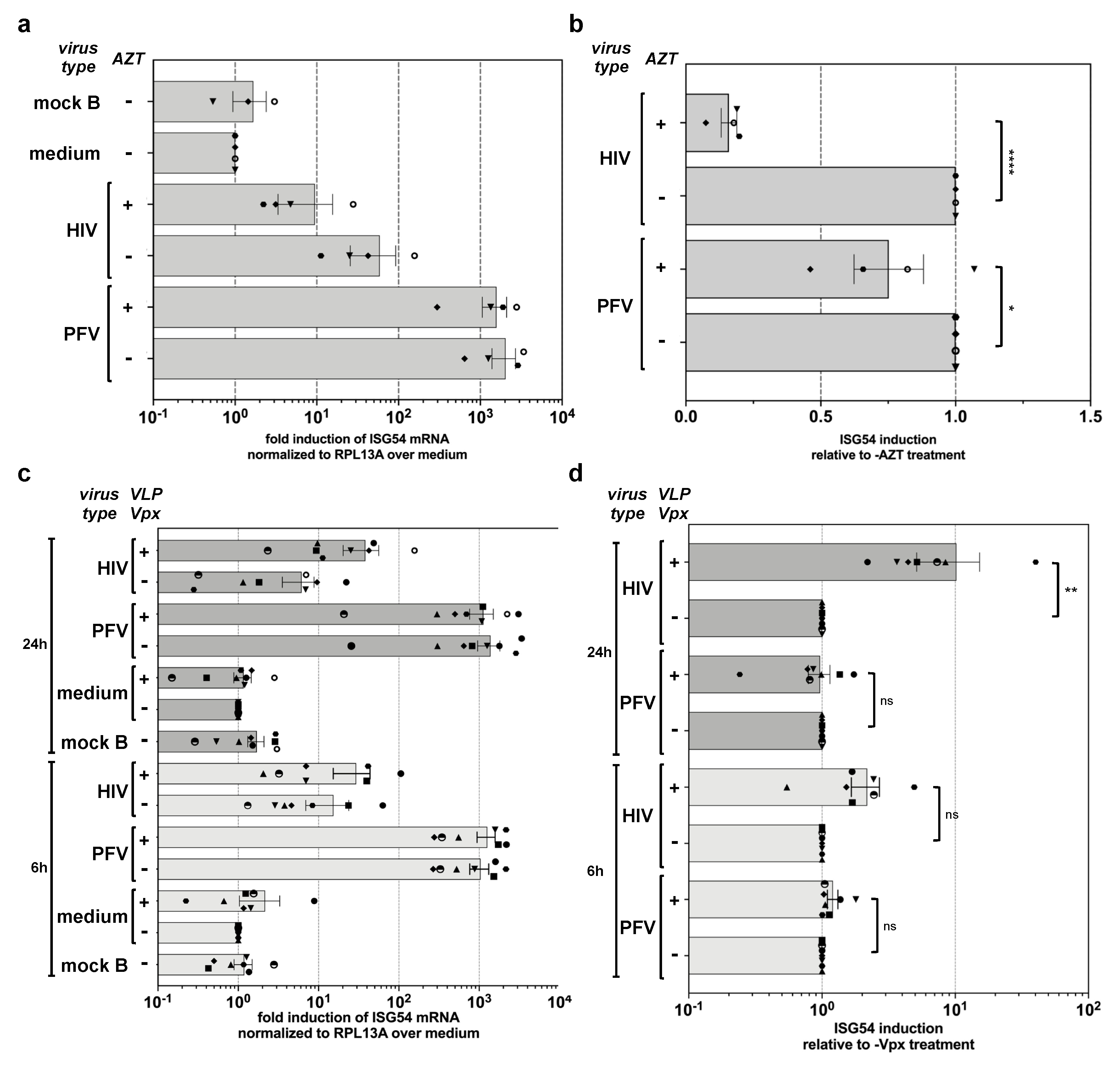
3.4. PFV ISG Induction Does Not Require Reverse Transcription upon Target Cell Entry and Is Not Suppressed by SAMHD1
3.5. PFV ISG Induction Requires Reverse Transcription of Full-Length Viral Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer/Probe | Sequence [5′-3′] | Cycle Conditions |
|---|---|---|---|
| PFV | fwd | TGCAATTCCAAAGGTGATTC | 95 °C, 8 min, 1× |
| RNA/DNA | rev | TACCTCTTTCCTTTGCCCAT | 95 °C, 30 s, 40× |
| probe | TCAAGGTGCAGCATTCACTTCTTCAA | 59 °C, 30 s, 40× | |
| 72 °C, 90 s, 40× | |||
| hGAPDH | fwd | CATCAATGGAAATCCCATCA | 95 °C, 8 min, 1× |
| rev | GACTCCACGACGTACTCAGC | 95 °C, 30 s, 40× | |
| probe | TCCAGGAGCGAGATCCCTCCA | 59 °C, 30 s, 40× | |
| 72 °C, 90 s, 40× | |||
| hISG56 | fwd | GAGATCATTCACACCTCATTAT | 95 °C, 8 min, 1× |
| rev | GGAGTGAGTCTTCTGTTCT | 95 °C, 30 s, 40× | |
| probe | TTCCCTCCCTTGCTAACTGCCATT | 58 °C, 30 s, 40× | |
| 72 °C, 90 s, 40× | |||
| hRPL13A | fwd | CCACCCTGGAGGAGAAGAGG | 95 °C, 8 min, 1× |
| rev | GAGTCCGTGGGTCTTGAGGA | 95 °C, 30 s, 40× | |
| probe | TCGGCCTGTTTCCGTAGCCTCA | 58 °C, 30 s, 40× | |
| 72 °C, 90 s, 40× |
| Target | Primer/Probe | Sequence [5′-3′] | Cycle Conditions |
|---|---|---|---|
| hISG54 | fwd | GGTGGCAGAAGAGGAAGATT | 50 °C, 30 min, 1× |
| rev | TAGGCCAGTAGGTTGCACAT | 95 °C, 15 min, 1× | |
| 94 °C, 15 s, 40× | |||
| 56 °C, 45 s, 40× | |||
| 72 °C, 45 s, 40× | |||
| hRPL13A | fwd | CCTGGAGGAGAAGAGGAAAGAGA | 50 °C, 30 min, 1× |
| rev | TTGAGGACCTCTGTGTATTTGTCAA | 95 °C, 15 min, 1× | |
| 94 °C, 15 s, 40× | |||
| 56 °C, 45 s, 40× | |||
| 72 °C, 45 s, 40× |
| Antibody Specificity | Company | Clone | Volume per Sample in µL | Fluorophore Attached |
|---|---|---|---|---|
| CD14 | BioLegend | M5E2 | 1 | Pacific Blue |
| CD163 | BD Bioscience | GHI/61 | 2 | PE |
| CD206 | BD Bioscience | 19.2 (RUO) | 2 | APC |
| CD11c | Miltenyi | N418 | 2 | Vio Blue |
| CD1a | BioLegend | SK9 | 2 | PE |
| CD16 | BioLegend | 3G8 | 2 | APC |
| CD86 | BioLegend | IT2.2 | 1.5 | PE |
| IgG2a, κ; isotype control | BioLegend | MOPC-173 | 1 | Pacific Blue |
| IgG1, k Isotype control | Beckman Coulter | Clone GHI/61 (RUO) | 2 | PE |
| IgG1, k Isotype control | BD Bioscience | MOPC-21 | 2 | APC |
| IgG2b, isotype control | Miltenyi | IS6-11E5.11 | 2 | Vio Blue |
| IgG2b, k isotype control | BioLegend | MG2b-57 | 2 or 1.5 | PE |
References
- Khan, A.S.; Bodem, J.; Buseyne, F.; Gessain, A.; Johnson, W.; Kuhn, J.H.; Kuzmak, J.; Lindemann, D.; Linial, M.L.; Lochelt, M.; et al. Spumaretroviruses: Updated taxonomy and nomenclature. Virology 2018, 516, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Rethwilm, A.; Lindemann, D. Foamy viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins, a Wolters Kluwer business: Philadelphia, PA, USA, 2013; Volume 2, pp. 1613–1632. [Google Scholar]
- Lindemann, D.; Rethwilm, A. Foamy virus biology and its application for vector development. Viruses 2011, 3, 561–585. [Google Scholar] [CrossRef] [PubMed]
- Delelis, O.; Saib, A.; Sonigo, P. Biphasic DNA synthesis in spumaviruses. J. Virol. 2003, 77, 8141–8146. [Google Scholar] [CrossRef] [PubMed]
- Moebes, A.; Enssle, J.; Bieniasz, P.D.; Heinkelein, M.; Lindemann, D.; Bock, M.; McClure, M.O.; Rethwilm, A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 1997, 71, 7305–7311. [Google Scholar]
- Yu, S.F.; Baldwin, D.N.; Gwynn, S.R.; Yendapalli, S.; Linial, M.L. Human foamy virus replication: A pathway distinct from that of retroviruses and hepadnaviruses. Science 1996, 271, 1579–1582. [Google Scholar] [CrossRef]
- Zamborlini, A.; Renault, N.; Saib, A.; Delelis, O. Early reverse transcription is essential for productive foamy virus infection. PLoS ONE 2010, 5, e11023. [Google Scholar] [CrossRef]
- Rua, R.; Gessain, A. Origin, evolution and innate immune control of simian foamy viruses in humans. Curr. Opin. Virol. 2015, 10, 47–55. [Google Scholar] [CrossRef]
- Kehl, T.; Tan, J.; Materniak, M. Non-simian foamy viruses: Molecular virology, tropism and prevalence and zoonotic/interspecies transmission. Viruses 2013, 5, 2169–2209. [Google Scholar] [CrossRef]
- Katzourakis, A.; Aiewsakun, P.; Jia, H.; Wolfe, N.D.; LeBreton, M.; Yoder, A.D.; Switzer, W.M. Discovery of prosimian and afrotherian foamy viruses and potential cross species transmissions amidst stable and ancient mammalian co-evolution. Retrovirology 2014, 11, 61. [Google Scholar] [CrossRef]
- Han, G.Z.; Worobey, M. Endogenous viral sequences from the cape golden mole (chrysochloris asiatica) reveal the presence of foamy viruses in all major placental mammal clades. PLoS ONE 2014, 9, e97931. [Google Scholar] [CrossRef]
- Schartl, M.; Walter, R.B.; Shen, Y.; Garcia, T.; Catchen, J.; Amores, A.; Braasch, I.; Chalopin, D.; Volff, J.N.; Lesch, K.P.; et al. The genome of the platyfish, xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat. Genet. 2013, 45, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Achong, B.G.; Mansell, P.W.A.; Epstein, M.A.; Clifford, P. An unusual virus in cultures from a human nasopharyngeal carcinoma. J. Natl. Cancer Inst. 1971, 46, 299–307. [Google Scholar] [PubMed]
- Epstein, M.A. Simian retroviral infections in human beings. Lancet 2004, 364, 138–139, author reply 139–140. [Google Scholar] [CrossRef]
- Herchenröder, O.; Turek, R.; Neumann Haefelin, D.; Rethwilm, A.; Schneider, J. Infectious proviral clones of chimpanzee foamy virus (sfvcpz) generated by long pcr reveal close functional relatedness to human foamy virus. Virology 1995, 214, 685–689. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stirnnagel, K.; Lüftenegger, D.; Stange, A.; Swiersy, A.; Müllers, E.; Reh, J.; Stanke, N.; Grosse, A.; Chiantia, S.; Keller, H.; et al. Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology 2010, 7, 45. [Google Scholar] [CrossRef]
- Mikovits, J.A.; Hoffman, P.M.; Rethwilm, A.; Ruscetti, F.W. In Vitro infection of primary and retrovirus-infected human leukocytes by human foamy virus. J. Virol. 1996, 70, 2774–2780. [Google Scholar]
- Yu, S.F.; Stone, J.; Linial, M.L. Productive persistent infection of hematopoietic cells by human foamy virus. J. Virol. 1996, 70, 1250–1254. [Google Scholar]
- Murray, S.M.; Picker, L.J.; Axthelm, M.K.; Hudkins, K.; Alpers, C.E.; Linial, M.L. Replication in a superficial epithelial cell niche explains the lack of pathogenicity of primate foamy virus infections. J. Virol. 2008, 82, 5981–5985. [Google Scholar] [CrossRef]
- Callahan, M.E.; Switzer, W.M.; Matthews, A.L.; Roberts, B.D.; Heneine, W.; Folks, T.M.; Sandstrom, P.A. Persistent zoonotic infection of a human with simian foamy virus in the absence of an intact orf-2 accessory gene. J. Virol. 1999, 73, 9619–9624. [Google Scholar]
- von Laer, D.; Neumann Haefelin, D.; Heeney, J.L.; Schweizer, M. Lymphocytes are the major reservoir for foamy viruses in peripheral blood. Virology 1996, 221, 240–244. [Google Scholar] [CrossRef]
- Rua, R.; Betsem, E.; Montange, T.; Buseyne, F.; Gessain, A. In vivo cellular tropism of gorilla simian foamy virus in blood of infected humans. J. Virol. 2014, 88, 13429–13435. [Google Scholar] [CrossRef] [PubMed]
- Rua, R.; Lepelley, A.; Gessain, A.; Schwartz, O. Innate sensing of foamy viruses by human hematopoietic cells. J. Virol. 2012, 86, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Bähr, A.; Singer, A.; Hain, A.; Vasudevan, A.A.; Schilling, M.; Reh, J.; Riess, M.; Panitz, S.; Serrano, V.; Schweizer, M.; et al. Interferon but not mxb inhibits foamy retroviruses. Virology 2016, 488, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Falcone, V.; Schweizer, M.; Toniolo, A.; Neumann-Haefelin, D.; Meyerhans, A. Gamma interferon is a major suppressive factor produced by activated human peripheral blood lymphocytes that is able to inhibit foamy virus-induced cytopathic effects. J. Virol. 1999, 73, 1724–1728. [Google Scholar]
- Matthes, D.; Wiktorowicz, T.; Zahn, J.; Bodem, J.; Stanke, N.; Lindemann, D.; Rethwilm, A. Basic residues in the foamy virus gag protein. J. Virol. 2011, 85, 3986–3995. [Google Scholar] [CrossRef]
- Rhodes Feuillette, A.; Lasneret, J.; Paulien, S.; Ogunkolade, W.; Peries, J.; Canivet, M. Effects of human recombinant alpha and gamma and of highly purified natural beta interferons on simian spumavirinae prototype (simian foamy virus 1) multiplication in human cells. Res. Virol. 1990, 141, 31–43. [Google Scholar] [CrossRef]
- Sabile, A.; Rhodes Feuillette, A.; Jaoui, F.Z.; Tobaly Tapiero, J.; Giron, M.L.; Lasneret, J.; Peries, J.; Canivet, M. In Vitro studies on interferon-inducing capacity and sensitivity to ifn of human foamy virus. Res. Virol. 1996, 147, 29–37. [Google Scholar] [CrossRef]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schafer, A.; McClure, M.O.; Cullen, B.R. Foamy virus bet proteins function as novel inhibitors of the apobec3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rösler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of apobec3 is inhibited by the foamy virus accessory bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef]
- Yap, M.W.; Lindemann, D.; Stanke, N.; Reh, J.; Westphal, D.; Hanenberg, H.; Ohkura, S.; Stoye, J.P. Restriction of foamy viruses by primate trim5alpha. J. Virol. 2008, 82, 5429–5439. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Hogstad, B.; Wang, Y.; Levy, D.E.; Unutmaz, D.; Littman, D.R. A cryptic sensor for hiv-1 activates antiviral innate immunity in dendritic cells. Nature 2010, 467, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Bridgeman, A.; Benlahrech, A.; Cursi, C.; Rehwinkel, J. Restriction by samhd1 limits cgas/sting-dependent innate and adaptive immune responses to hiv-1. Cell Rep. 2016, 16, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Luban, J. Innate immune sensing of hiv-1 by dendritic cells. Cell Host Microbe 2012, 12, 408–418. [Google Scholar] [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic gmp-amp synthase is an innate immune sensor of hiv and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef]
- Yoh, S.M.; Schneider, M.; Seifried, J.; Soonthornvacharin, S.; Akleh, R.E.; Olivieri, K.C.; De Jesus, P.D.; Ruan, C.; de Castro, E.; Ruiz, P.A.; et al. Pqbp1 is a proximal sensor of the cgas-dependent innate response to hiv-1. Cell 2015, 161, 1293–1305. [Google Scholar] [CrossRef]
- Leong, C.R.; Oshiumi, H.; Okamoto, M.; Azuma, M.; Takaki, H.; Matsumoto, M.; Chayama, K.; Seya, T. A mavs/ticam-1-independent interferon-inducing pathway contributes to regulation of hepatitis b virus replication in the mouse hydrodynamic injection model. J. Innate Immun. 2015, 7, 47–58. [Google Scholar] [CrossRef]
- DuBridge, R.B.; Tang, P.; Hsia, H.C.; Leong, P.M.; Miller, J.H.; Calos, M.P. Analysis of mutation in human cells by using an epstein-barr virus shuttle system. Mol. Cell. Biol 1987, 7, 379–387. [Google Scholar] [CrossRef]
- Rasheed, S.; Nelson-Rees, W.A.; Toth, E.M.; Arnstein, P.; Gardner, M.B. Characterization of a newly derived human sarcoma cell line (ht-1080). Cancer 1974, 33, 1027–1033. [Google Scholar] [CrossRef]
- Hütter, S.; Müllers, E.; Stanke, N.; Reh, J.; Lindemann, D. Prototype foamy virus protease activity is essential for intraparticle reverse transcription initiation but not absolutely required for uncoating upon host cell entry. J. Virol. 2013, 87, 3163–3176. [Google Scholar] [CrossRef]
- Wittmann, S.; Behrendt, R.; Eissmann, K.; Volkmann, B.; Thomas, D.; Ebert, T.; Cribier, A.; Benkirane, M.; Hornung, V.; Bouzas, N.F.; et al. Phosphorylation of murine samhd1 regulates its antiretroviral activity. Retrovirology 2015, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Mankan, A.K.; Schmidt, T.; Chauhan, D.; Goldeck, M.; Honing, K.; Gaidt, M.; Kubarenko, A.V.; Andreeva, L.; Hopfner, K.P.; Hornung, V. Cytosolic rna:DNA hybrids activate the cgas-sting axis. EMBO J. 2014, 33, 2937–2946. [Google Scholar] [CrossRef] [PubMed]
- Riess, M.; Fuchs, N.V.; Idica, A.; Hamdorf, M.; Flory, E.; Pedersen, I.M.; Konig, R. Interferons induce expression of samhd1 in monocytes through down-regulation of mir-181a and mir-30a. J. Biol. Chem. 2017, 292, 264–277. [Google Scholar] [CrossRef]
- Heinkelein, M.; Dressler, M.; Jarmy, G.; Rammling, M.; Imrich, H.; Thurow, J.; Lindemann, D.; Rethwilm, A. Improved primate foamy virus vectors and packaging constructs. J. Virol. 2002, 76, 3774–3783. [Google Scholar] [CrossRef] [PubMed]
- Müllers, E.; Uhlig, T.; Stirnnagel, K.; Fiebig, U.; Zentgraf, H.; Lindemann, D. Novel functions of prototype foamy virus gag glycine- arginine-rich boxes in reverse transcription and particle morphogenesis. J. Virol. 2011, 85, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Heinkelein, M.; Thurow, J.; Dressler, M.; Imrich, H.; Neumann-Haefelin, D.; McClure, M.O.; Rethwilm, A. Complex effects of deletions in the 5′ untranslated region of primate foamy virus on viral gene expression and rna packaging. J. Virol. 2000, 74, 3141–3148. [Google Scholar] [CrossRef]
- Hamann, M.V.; Müllers, E.; Reh, J.; Stanke, N.; Effantin, G.; Weissenhorn, W.; Lindemann, D. The cooperative function of arginine residues in the prototype foamy virus gag c-terminus mediates viral and cellular rna encapsidation. Retrovirology 2014, 11, 87. [Google Scholar] [CrossRef]
- Negre, D.; Mangeot, P.E.; Duisit, G.; Blanchard, S.; Vidalain, P.O.; Leissner, P.; Winter, A.J.; Rabourdin-Combe, C.; Mehtali, M.; Moullier, P.; et al. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (sivmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 2000, 7, 1613–1623. [Google Scholar] [CrossRef]
- Schindler, M.; Munch, J.; Kirchhoff, F. Human immunodeficiency virus type 1 inhibits DNA damage-triggered apoptosis by a nef-independent mechanism. J. Virol. 2005, 79, 5489–5498. [Google Scholar] [CrossRef]
- Ho, Y.P.; Schnabel, V.; Swiersy, A.; Stirnnagel, K.; Lindemann, D. A small-molecule-controlled system for efficient pseudotyping of prototype foamy virus vectors. Mol. Ther. 2012, 20, 1167–1176. [Google Scholar] [CrossRef]
- Schott, K.; Fuchs, N.V.; Derua, R.; Mahboubi, B.; Schnellbacher, E.; Seifried, J.; Tondera, C.; Schmitz, H.; Shepard, C.; Brandariz-Nunez, A.; et al. Dephosphorylation of the hiv-1 restriction factor samhd1 is mediated by pp2a-b55alpha holoenzymes during mitotic exit. Nat. Commun. 2018, 9, 2227. [Google Scholar] [CrossRef] [PubMed]
- Mannigel, I.; Stange, A.; Zentgraf, H.; Lindemann, D. Correct capsid assembly mediated by a conserved yxxlgl motif in prototype foamy virus gag is essential for infectivity and reverse transcription of the viral genome. J. Virol. 2007, 81, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Arfi, V.; Pertel, T.; Luban, J.; Lienard, J.; Rigal, D.; Darlix, J.L.; Cimarelli, A. Characterization of simian immunodeficiency virus sivsm/human immunodeficiency virus type 2 vpx function in human myeloid cells. J. Virol. 2008, 82, 12335–12345. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Sato, M.; Hata, N.; Asagiri, M.; Suemori, H.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Gene induction pathways mediated by distinct irfs during viral infection. Biochem. Biophys. Res. Commun. 2001, 283, 1150–1156. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Servant, M.J.; Sen, G.C.; Balachandran, S.; Barber, G.N.; Lin, R.; Hiscott, J. Transcriptional profiling of interferon regulatory factor 3 target genes: Direct involvement in the regulation of interferon-stimulated genes. J. Virol. 2002, 76, 5532–5539. [Google Scholar] [CrossRef]
- McCauley, S.M.; Kim, K.; Nowosielska, A.; Dauphin, A.; Yurkovetskiy, L.; Diehl, W.E.; Luban, J. Intron-containing rna from the hiv-1 provirus activates type i interferon and inflammatory cytokines. Nat. Commun. 2018, 9, 5305. [Google Scholar] [CrossRef]
- Akiyama, H.; Miller, C.M.; Ettinger, C.R.; Belkina, A.C.; Snyder-Cappione, J.E.; Gummuluru, S. Hiv-1 intron-containing rna expression induces innate immune activation and t cell dysfunction. Nat. Commun. 2018, 9, 3450. [Google Scholar] [CrossRef]
- Gramberg, T.; Kahle, T.; Bloch, N.; Wittmann, S.; Müllers, E.; Daddacha, W.; Hofmann, H.; Kim, B.; Lindemann, D.; Landau, N.R. Restriction of Diverse Retroviruses by Samhd1. Retrovirology 2013, 10, 26. [Google Scholar] [CrossRef]
- Herzner, A.M.; Hagmann, C.A.; Goldeck, M.; Wolter, S.; Kubler, K.; Wittmann, S.; Gramberg, T.; Andreeva, L.; Hopfner, K.P.; Mertens, C.; et al. Sequence-specific activation of the DNA sensor cgas by y-form DNA structures as found in primary hiv-1 cdna. Nat. Immunol. 2015, 16, 1025–1033. [Google Scholar] [CrossRef]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lassig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. Cgas senses long and hmgb/tfam-bound u-turn DNA by forming protein-DNA ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Tengchuan, J.; Laustsen, A.; Hansen, K.; Ostergaard, L.; Fitzgerald, K.A.; et al. Ifi16 senses DNA forms of the lentiviral replication cycle and controls hiv-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergez, M.; Weber, J.; Riess, M.; Erdbeer, A.; Seifried, J.; Stanke, N.; Munz, C.; Hornung, V.; König, R.; Lindemann, D. Insights into Innate Sensing of Prototype Foamy Viruses in Myeloid Cells. Viruses 2019, 11, 1095. https://doi.org/10.3390/v11121095
Bergez M, Weber J, Riess M, Erdbeer A, Seifried J, Stanke N, Munz C, Hornung V, König R, Lindemann D. Insights into Innate Sensing of Prototype Foamy Viruses in Myeloid Cells. Viruses. 2019; 11(12):1095. https://doi.org/10.3390/v11121095
Chicago/Turabian StyleBergez, Maïwenn, Jakob Weber, Maximilian Riess, Alexander Erdbeer, Janna Seifried, Nicole Stanke, Clara Munz, Veit Hornung, Renate König, and Dirk Lindemann. 2019. "Insights into Innate Sensing of Prototype Foamy Viruses in Myeloid Cells" Viruses 11, no. 12: 1095. https://doi.org/10.3390/v11121095
APA StyleBergez, M., Weber, J., Riess, M., Erdbeer, A., Seifried, J., Stanke, N., Munz, C., Hornung, V., König, R., & Lindemann, D. (2019). Insights into Innate Sensing of Prototype Foamy Viruses in Myeloid Cells. Viruses, 11(12), 1095. https://doi.org/10.3390/v11121095