Transmissibility versus Pathogenicity of Self-Propagating Protein Aggregates


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Prion, and Prion-Like, Diseases
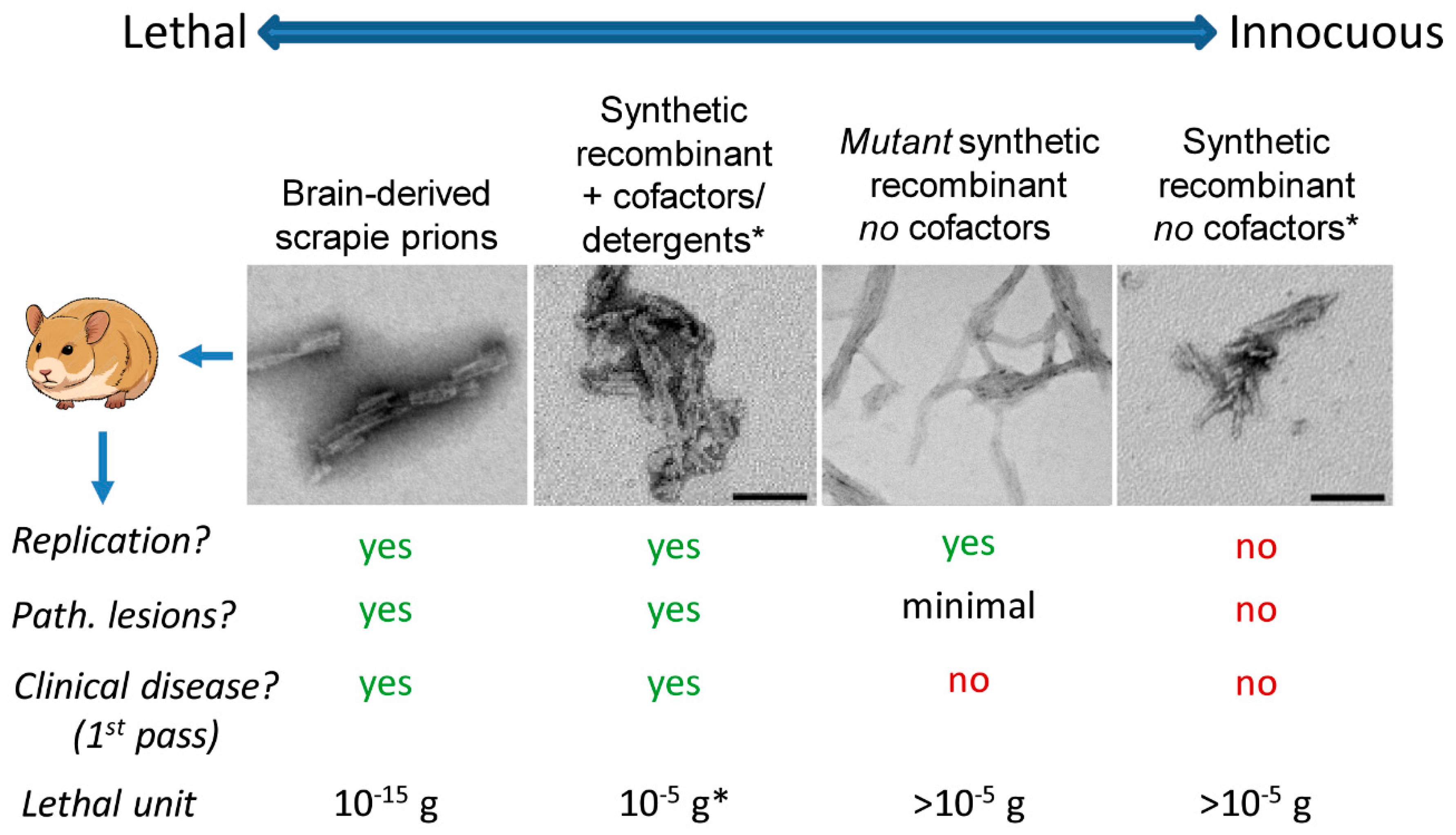
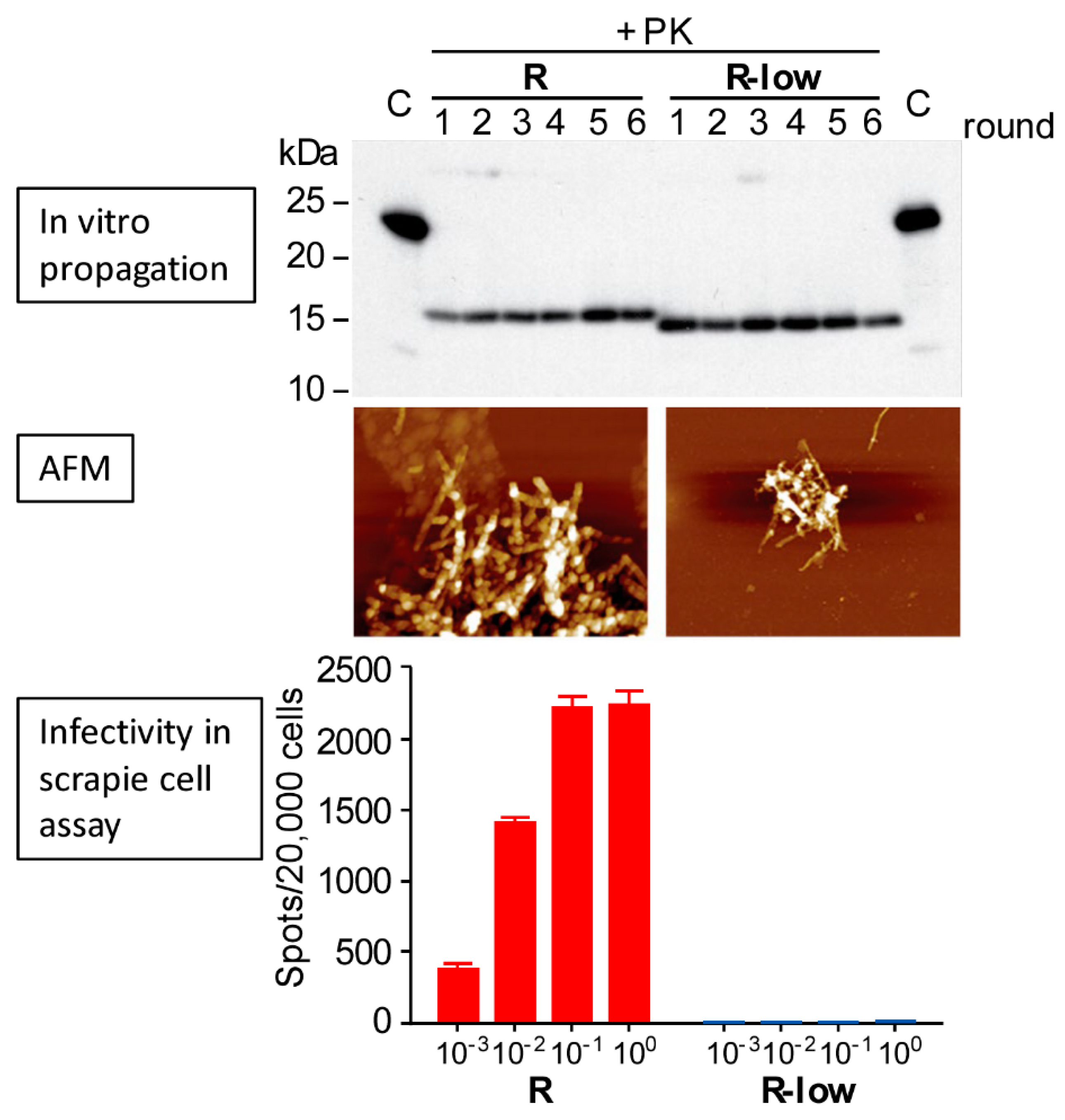
3. Transmissibility versus Pathogenicity of PrP Aggregation
4. Transmission of, and Susceptibility to, Human Prion Diseases
5. Implications for Other Types of Proteopathies
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Griffith, J.S. Self-Replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Liberski, P.P.; Gajos, A.; Sikorska, B.; Lindenbaum, S. Kuru, the first human prion disease. Viruses 2019, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Hadlow, W.J. Scrapie and kuru. Lancet 1959, 2, 289–290. [Google Scholar] [CrossRef]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Gajdusek, D.C. Unconventional virus infections as cerebral amyloidoses. In Unconventional Virus Diseases of the Central Nervous System; Court, L.A., Dormont, D., Brown, P., Eds.; Commissariat a l’Energie Atomique (CEA), Service de Documentation: Fontenay-aux-Roses, France, 1986; pp. 641–659. [Google Scholar]
- Gajdusek, D.C. Subacute spongiform encephalopathies: Transmissible cerebral amyloidoses caused by unconventional viruses. In Field’s Virology, 2nd ed.; Fields, B.N., Knipe, D.M., Eds.; Raven Press: New York, NY, USA, 1990; Volume 2, pp. 2298–2324. [Google Scholar]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Lansbury, P.T., Jr.; Caughey, B. The chemistry of scrapie infection: Implications of the ‘ice 9’ metaphor. Chem. Biol. 1995, 2, 1–5. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 109, 27–68. [Google Scholar] [CrossRef]
- Alibhai, J.; Blanco, R.A.; Barria, M.A.; Piccardo, P.; Caughey, B.; Perry, V.H.; Freeman, T.C.; Manson, J.C. Distribution of misfolded prion protein seeding activity alone does not predict regions of neurodegeneration. PLoS Biol. 2016, 14, e1002579. [Google Scholar] [CrossRef]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, microglia, and cell-association during prion disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Groveman, B.R.; Caughey, B. Prions and the potential transmissibility of protein misfolding diseases. Annu. Rev. Microbiol. 2013, 67, 543–564. [Google Scholar] [CrossRef]
- Walker, L.C.; Jucker, M. Neurodegenerative diseases: Expanding the prion concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217–226. [Google Scholar] [CrossRef]
- Watts, J.C. Calling alpha-synuclein a prion is scientifically justifiable. Acta Neuropathol. 2019. [Google Scholar] [CrossRef]
- Watts, J.C.; Prusiner, S.B. Beta-Amyloid prions and the pathobiology of Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2018, 8, a023507. [Google Scholar] [CrossRef]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef]
- Kraus, A.; Raymond, G.J.; Race, B.; Campbell, K.J.; Hughson, A.G.; Anson, K.J.; Raymond, L.D.; Caughey, B. PrP P102L and nearby lysine mutations promote spontaneous in vitro formation of transmissible prions. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Orru, C.D.; Groveman, B.R.; Surewicz, K.; Abskharon, R.; Imamura, M.; Yokoyama, T.; Kim, Y.S.; Van der Stel, K.J.; et al. Self-Propagating, protease-resistant, recombinant prion protein conformers with or without in vivo pathogenicity. PLoS Path. 2017, 13, e1006491. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.M.; Walsh, D.J.; Steele, A.D.; Agrimi, U.; Di Bari, M.A.; Watts, J.C.; Supattapone, S. Full restoration of specific infectivity and strain properties from pure mammalian prion protein. PLoS Path. 2019, 15, e1007662. [Google Scholar] [CrossRef] [PubMed]
- Wille, H.; Michelitsch, M.D.; Guenebaut, V.; Supattapone, S.; Serban, A.; Cohen, F.E.; Agard, D.A.; Prusiner, S.B. Structural studies of the scrapie prion protein by electron crystallography. Proc. Natl. Acad. Sci. USA 2002, 99, 3563–3568. [Google Scholar] [CrossRef] [Green Version]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nature 2005, 437, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Caughey, B.; Kocisko, D.A.; Raymond, G.J.; Lansbury, P.T. Aggregates of scrapie associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chem. Biol. 1995, 2, 807–817. [Google Scholar] [CrossRef]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef]
- Terry, C.; Harniman, R.L.; Sells, J.; Wenborn, A.; Joiner, S.; Saibil, H.R.; Miles, M.J.; Collinge, J.; Wadsworth, J.D.F. Structural features distinguishing infectious ex vivo mammalian prions from non-infectious fibrillar assemblies generated in vitro. Sci. Rep. 2019, 9, 376. [Google Scholar] [CrossRef]
- Atarashi, R.; Moore, R.A.; Sim, V.L.; Hughson, A.G.; Dorward, D.W.; Onwubiko, H.A.; Priola, S.A.; Caughey, B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 2007, 4, 645–650. [Google Scholar] [CrossRef]
- Kim, J.I.; Cali, I.; Surewicz, K.; Kong, Q.; Raymond, G.J.; Atarashi, R.; Race, B.; Qing, L.; Gambetti, P.; Caughey, B.; et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 2010, 285, 14083–14087. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Abskharon, R.; Ma, J. Prion infectivity is encoded exclusively within the structure of proteinase K-resistant fragments of synthetically generated recombinant PrP(Sc). Acta Neuropathol. Commun. 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Anson, K.J.; Raymond, L.D.; Martens, C.; Groveman, B.R.; Dorward, D.W.; Caughey, B. Prion protein prolines 102 and 105 and the surrounding lysine cluster impede amyloid formation. J. Biol. Chem. 2015, 290, 21510–21522. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Yuan, C.G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. USA 2012, 109, E1938–E1946. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Kovacs, G.G.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Genesis of mammalian prions: From non-infectious amyloid fibrils to a transmissible prion disease. PLoS. Pathog. 2011, 7, e1002419. [Google Scholar] [CrossRef]
- Raymond, G.J.; Race, B.; Hollister, J.R.; Offerdahl, D.K.; Moore, R.A.; Kodali, R.; Raymond, L.D.; Hughson, A.G.; Rosenke, R.; Long, D.; et al. Isolation of novel synthetic prion strains by amplification in transgenic mice coexpressing wild-type and anchorless prion proteins. J. Virol. 2012, 86, 11763–11778. [Google Scholar] [CrossRef]
- Groveman, B.R.; Raymond, G.J.; Campbell, K.J.; Race, B.; Raymond, L.D.; Hughson, A.G.; Orru, C.D.; Kraus, A.; Phillips, K.; Caughey, B. Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Path. 2017, 13, e1006623. [Google Scholar] [CrossRef]
- Diaz-Espinoza, R.; Morales, R.; Concha-Marambio, L.; Moreno-Gonzalez, I.; Moda, F.; Soto, C. Treatment with a non-toxic, self-replicating anti-prion delays or prevents prion disease in vivo. Mol. Psychiatry 2018, 23, 777–788. [Google Scholar] [CrossRef]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian evolution of prions in cell culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Baskakov, I.V. The evolution of transmissible prions: The role of deformed templating. PLoS Path. 2013, 9, e1003759. [Google Scholar] [CrossRef] [PubMed]
- Smirnovas, V.; Kim, J.I.; Lu, X.; Atarashi, R.; Caughey, B.; Surewicz, W.K. Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J. Biol. Chem. 2009, 284, 24233–24241. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, F.; Xiao, X.; Kim, C.; Bohon, J.; Kiselar, J.; Safar, J.G.; Ma, J.; Surewicz, W.K. Structural attributes of mammalian prion infectivity: Insights from studies with synthetic prions. J. Biol. Chem. 2018, 293, 18494–18503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.G.; Teplow, D.B.; Parchi, P.; Teller, J.K.; Gambetti, P.; Autilio-Gambetti, L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995, 270, 19173–19180. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Iglesias, R.; Pajares, M.A.; Ocal, C.; Espinosa, J.C.; Oesch, B.; Gasset, M. Prion protein interaction with glycosaminoglycan occurs with the formation of oligomeric complexes stabilized by Cu(II) bridges. J. Mol. Biol. 2002, 319, 527–540. [Google Scholar] [CrossRef]
- Wang, F.; Yin, S.; Wang, X.; Zha, L.; Sy, M.S.; Ma, J. Role of the highly conserved middle region of prion protein (PrP) in PrP-lipid interaction. Biochemistry 2010, 49, 8169–8176. [Google Scholar] [CrossRef]
- Brown, P.; Gibbs, C.J., Jr.; Rodgers-Johnson, P.; Asher, D.M.; Sulima, M.P.; Bacote, A.; Goldfarb, L.G.; Gajdusek, D.C. Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann. Neurol. 1994, 35, 513–529. [Google Scholar] [CrossRef]
- Race, B.; Williams, K.; Hughson, A.G.; Jansen, C.; Parchi, P.; Rozemuller, A.J.M.; Chesebro, B. Familial human prion diseases associated with prion protein mutations Y226X and G131V are transmissible to transgenic mice expressing human prion protein. Acta Neuropathol. Commun. 2018, 6, 13. [Google Scholar] [CrossRef]
- Hsiao, K.K.; Groth, D.; Scott, M.; Serban, H.; Rapp, D.; Foster, D.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc. Natl. Acad. Sci. USA 1994, 91, 9126–9130. [Google Scholar] [CrossRef]
- Chiesa, R.; Drisaldi, B.; Quaglio, E.; Migheli, A.; Piccardo, P.; Ghetti, B.; Harris, D.A. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 5574–5579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, I.; Mikhail, F.; Qin, K.; Gregory, C.; Solanki, A.; Martinez, M.C.; Zhao, L.; Appleby, B.; Gambetti, P.; Norstrom, E.; et al. Impaired transmissibility of atypical prions from genetic CJD(G114V). Neurol. Genet. 2018, 4, e253. [Google Scholar] [CrossRef] [PubMed]
- Nazor, K.E.; Kuhn, F.; Seward, T.; Green, M.; Zwald, D.; Purro, M.; Schmid, J.; Biffiger, K.; Power, A.M.; Oesch, B.; et al. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J. 2005, 24, 2472–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.C.; Prusiner, S.B. Experimental models of inherited PrP prion diseases. Cold Spring Harb. Perspect. Med. 2017, 7, a027151. [Google Scholar] [CrossRef]
- Wadsworth, J.D.; Asante, E.A.; Desbruslais, M.; Linehan, J.M.; Joiner, S.; Gowland, I.; Welch, J.; Stone, L.; Lloyd, S.E.; Hill, A.F.; et al. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science 2004, 306, 1793–1796. [Google Scholar] [CrossRef]
- Kaski, D.; Mead, S.; Hyare, H.; Cooper, S.; Jampana, R.; Overell, J.; Knight, R.; Collinge, J.; Rudge, P. Variant CJD in an individual heterozygous for PRNP codon 129. Lancet 2009, 374, 2128. [Google Scholar] [CrossRef]
- Mead, S.; Whitfield, J.; Poulter, M.; Shah, P.; Uphill, J.; Campbell, T.; Al-Dujaily, H.; Hummerich, H.; Beck, J.; Mein, C.A.; et al. A novel protective prion protein variant that colocalizes with kuru exposure. N. Engl. J. Med. 2009, 361, 2056–2065. [Google Scholar] [CrossRef]
- Collinge, J.; Palmer, M.S.; Dryden, A.J. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 1991, 337, 1441–1442. [Google Scholar] [CrossRef]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991, 352, 340–342. [Google Scholar] [CrossRef]
- Kobayashi, A.; Teruya, K.; Matsuura, Y.; Shirai, T.; Nakamura, Y.; Yamada, M.; Mizusawa, H.; Mohri, S.; Kitamoto, T. The influence of PRNP polymorphisms on human prion disease susceptibility: An update. Acta Neuropathol. 2015, 130, 159–170. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimhan, S.; Guo, J.L.; Changolkar, L.; Stieber, A.; McBride, J.D.; Silva, L.V.; He, Z.; Zhang, B.; Gathagan, R.J.; Trojanowski, J.Q.; et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 2017, 37, 11406–11423. [Google Scholar] [CrossRef] [PubMed]
- Vaquer-Alicea, J.; Diamond, M.I. Propagation of protein aggregation in neurodegenerative diseases. Annu. Rev. Biochem. 2019, 88, 785–810. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morales-Scheihing, D.; Salvadores, N.; Moreno-Gonzalez, I.; Gonzalez, C.; Taylor-Presse, K.; Mendez, N.; Shahnawaz, M.; Gaber, A.O.; Sabek, O.M.; et al. Induction of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J. Exp. Med. 2017, 214, 2591–2610. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Jicha, G.A.; Liu, H.; Schmitt, F.A. Lewy body pathology in normal elderly subjects. J. Neuropathol. Exp. Neurol. 2009, 68, 816–822. [Google Scholar] [CrossRef]
- Lowe, V.J.; Wiste, H.J.; Senjem, M.L.; Weigand, S.D.; Therneau, T.M.; Boeve, B.F.; Josephs, K.A.; Fang, P.; Pandey, M.K.; Murray, M.E.; et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 2018, 141, 271–287. [Google Scholar] [CrossRef]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Braak, H.; Brion, J.P.; Buee, L.; Del Tredici, K.; Goedert, M.; Halliday, G.; Neumann, M.; Spillantini, M.G.; Tolnay, M.; et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015, 129, 749–756. [Google Scholar] [CrossRef]
- Josephs, K.A.; Murray, M.E.; Tosakulwong, N.; Whitwell, J.L.; Knopman, D.S.; Machulda, M.M.; Weigand, S.D.; Boeve, B.F.; Kantarci, K.; Petrucelli, L.; et al. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: A clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol. 2017, 133, 705–715. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Alafuzoff, I.; Attems, J.; Beach, T.G.; Cairns, N.J.; Crary, J.F.; Dickson, D.W.; Hof, P.R.; Hyman, B.T.; Jack, C.R., Jr.; et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 2015, 129, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.K.; Del Tredici, K.; Thomas, T.L.; Braak, H.; Diamond, M.I. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol. 2018, 136, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Saijo, E.; Metrick, M.A.I.; Newell, K.; Sigurdson, C.; Zanusso, G.; Ghetti, B.; Caughey, B. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. 2019, 137, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef]
- Clavaguera, F.; Lavenir, I.; Falcon, B.; Frank, S.; Goedert, M.; Tolnay, M. “Prion-Like” templated misfolding in tauopathies. Brain Pathol. 2013, 23, 342–349. [Google Scholar] [CrossRef]
- Peelaerts, W.; Baekelandt, V. Alpha-Synuclein folds: The cards are on the table. Nat. Struct. Mol. Biol. 2016, 23, 359–360. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. Alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef]
- Lavenir, I.; Passarella, D.; Masuda-Suzukake, M.; Curry, A.; Holton, J.L.; Ghetti, B.; Goedert, M. Silver staining (Campbell-Switzer) of neuronal alpha-synuclein assemblies induced by multiple system atrophy and Parkinson’s disease brain extracts in transgenic mice. Acta Neuropathol. Commun. 2019, 7, 148. [Google Scholar] [CrossRef]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Zhang, K.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration, a four-repeat tauopathy. bioRxiv 2019. [Google Scholar] [CrossRef]
- Brown, P.; Brandel, J.P.; Sato, T.; Nakamura, Y.; MacKenzie, J.; Will, R.G.; Ladogana, A.; Pocchiari, M.; Leschek, E.W.; Schonberger, L.B. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg. Infect. Dis. 2012, 18, 901–907. [Google Scholar] [CrossRef]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of amyloid-beta protein pathology from cadaveric pituitary growth hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef]
- Irwin, D.J.; Abrams, J.Y.; Schonberger, L.B.; Leschek, E.W.; Mills, J.L.; Lee, V.M.; Trojanowski, J.Q. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 2013, 70, 462–468. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caughey, B.; Kraus, A. Transmissibility versus Pathogenicity of Self-Propagating Protein Aggregates. Viruses 2019, 11, 1044. https://doi.org/10.3390/v11111044
Caughey B, Kraus A. Transmissibility versus Pathogenicity of Self-Propagating Protein Aggregates. Viruses. 2019; 11(11):1044. https://doi.org/10.3390/v11111044
Chicago/Turabian StyleCaughey, Byron, and Allison Kraus. 2019. "Transmissibility versus Pathogenicity of Self-Propagating Protein Aggregates" Viruses 11, no. 11: 1044. https://doi.org/10.3390/v11111044