Ectromelia Virus Affects Mitochondrial Network Morphology, Distribution, and Physiology in Murine Fibroblasts and Macrophage Cell Line

 , , ,
, , ,
Abstract
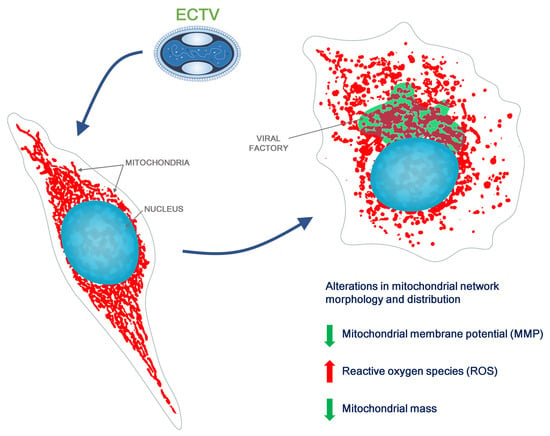
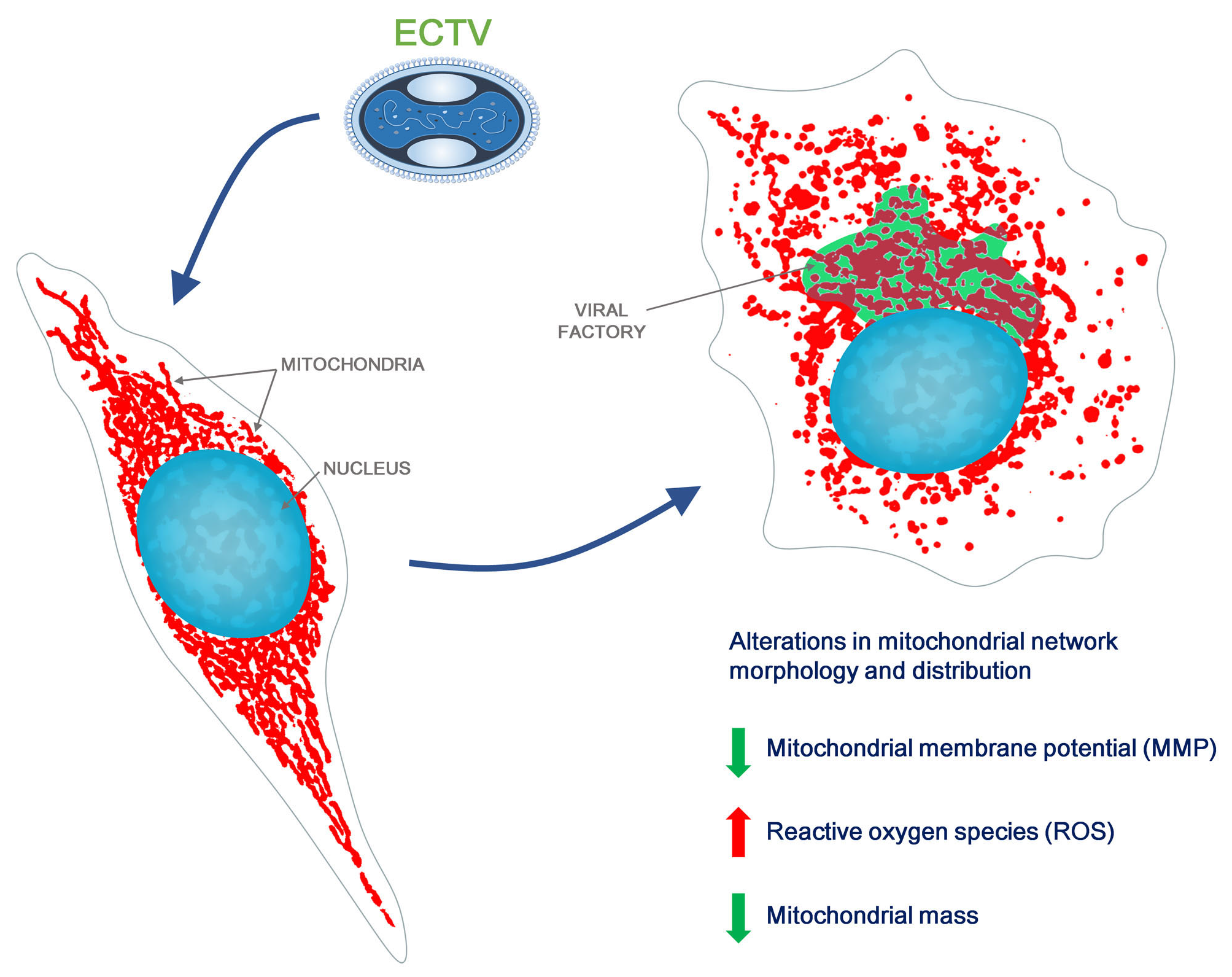{kind=link}
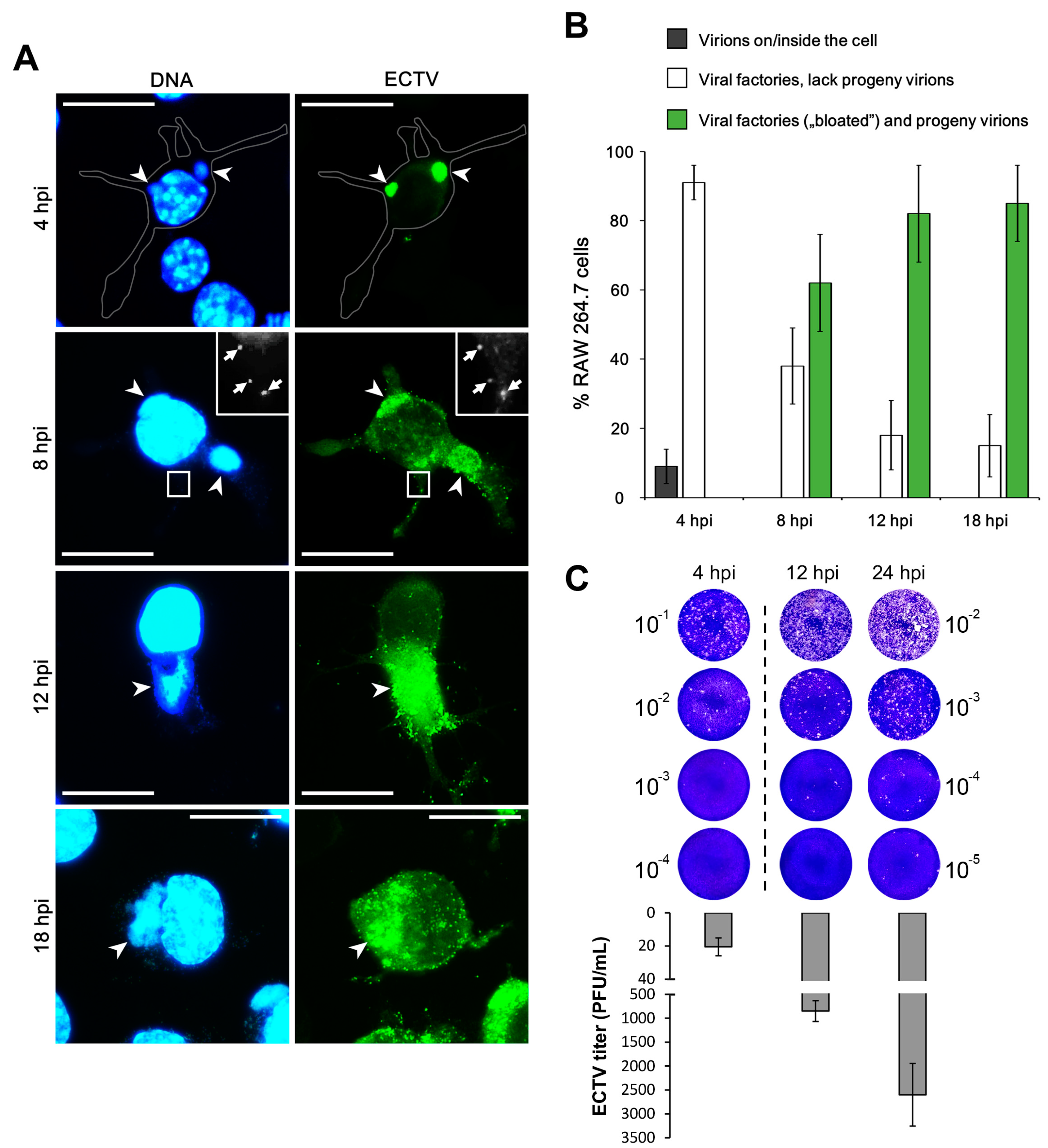{kind=link}
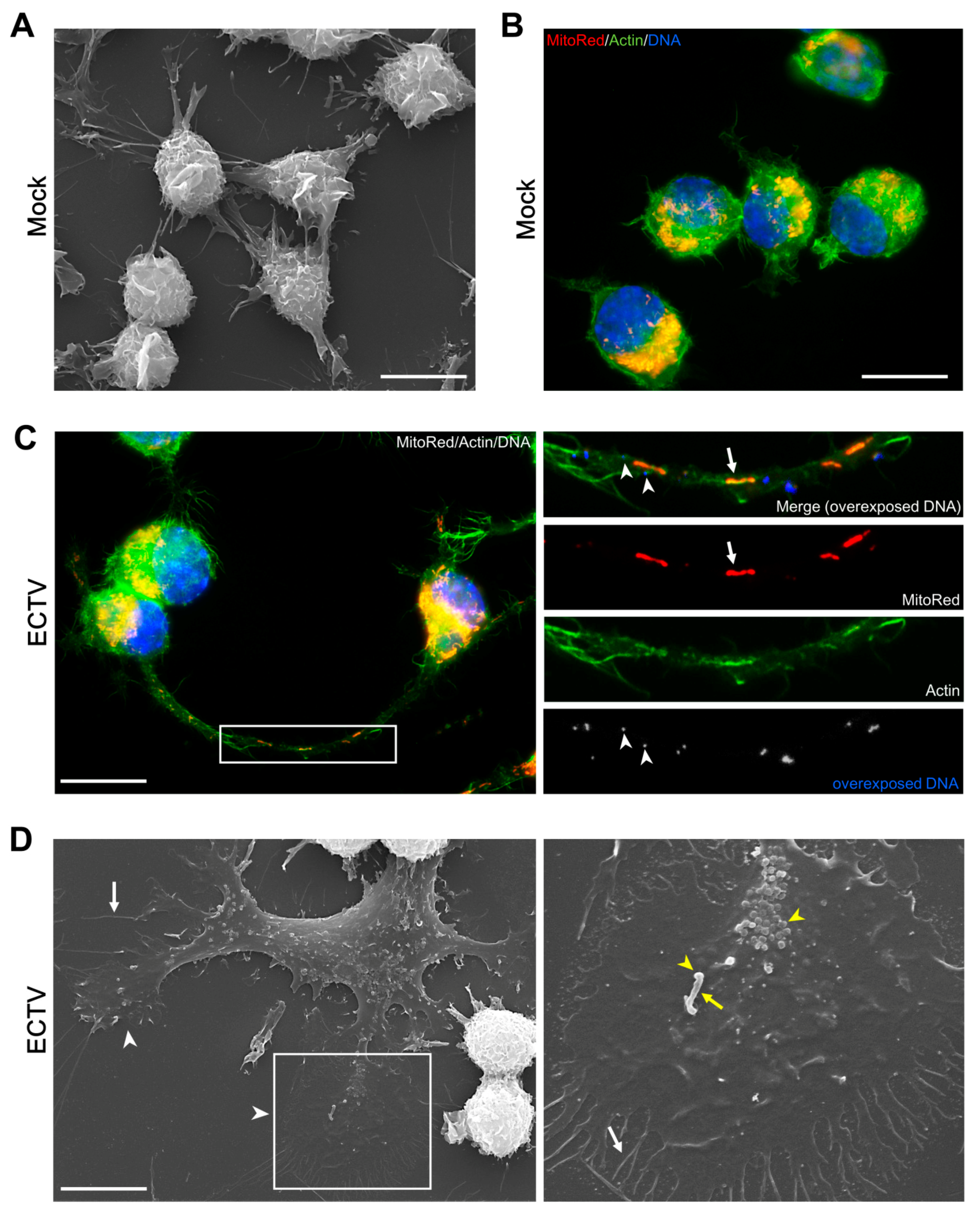{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Virus and Cell Lines
2.2. Fluorescent Probes and Antibodies
2.3. Cell Line Infection with ECTV
2.4. Plaque Assay
2.5. Immunofluorescent Staining and Microscopy Analysis
2.6. Transmission Electron Microscopy (TEM) Analysis
2.7. Scanning Electron Microscopy (SEM) Analysis
2.8. Determination of Mitochondrial Membrane Potential
2.9. Measurement of Reactive Oxygen Species Level
2.10. Measurement of Mitochondrial Mass
2.11. Determination of ATP Levels
2.12. Western Blot Analysis
2.13. Apoptosis Measurement by Flow Cytometry
2.14. Statistical Analysis
3. Results
3.1. Kinetics of ECTV Replication in Murine Cell Lines
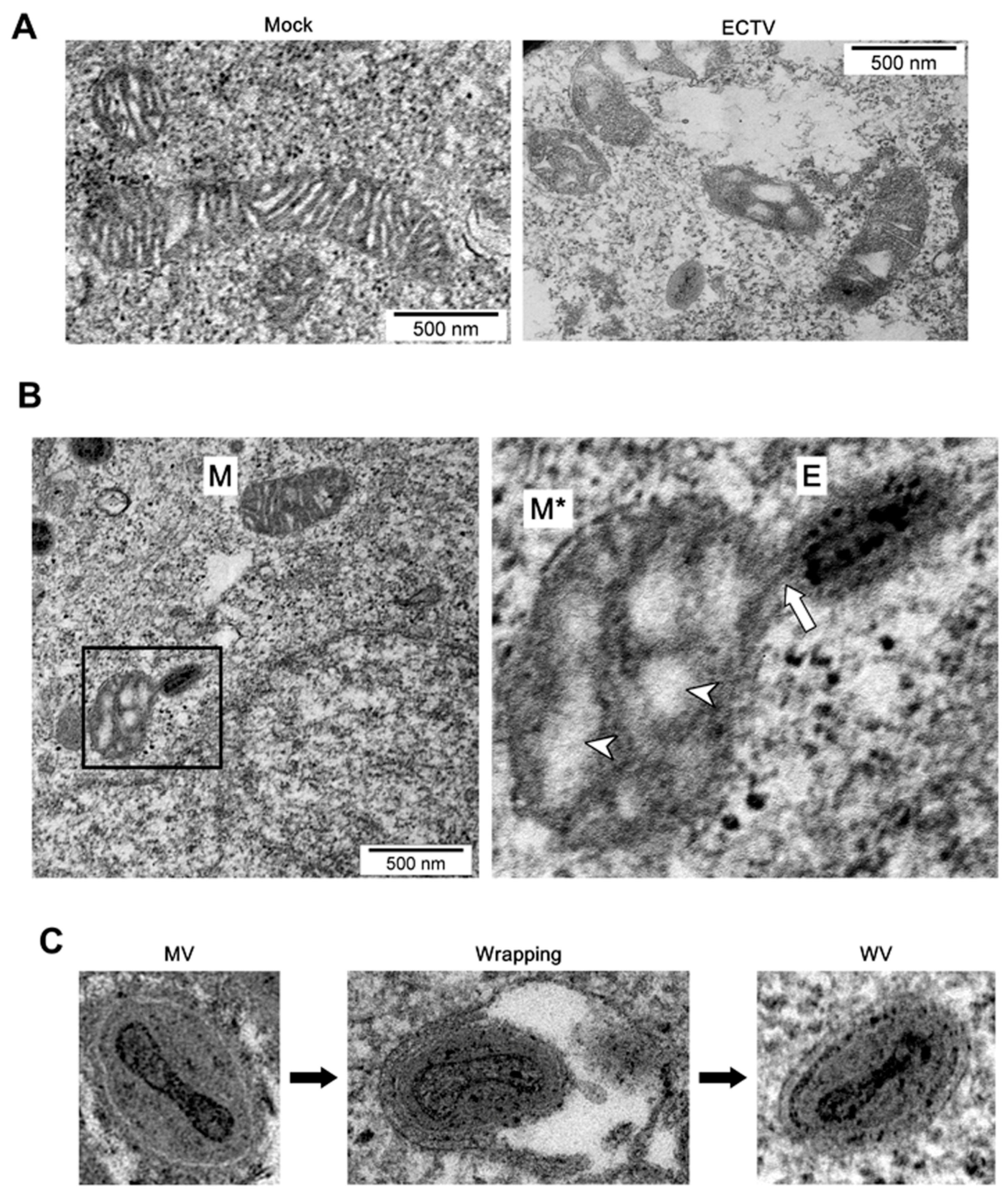
3.2. Cell Morphology during ECTV Infection
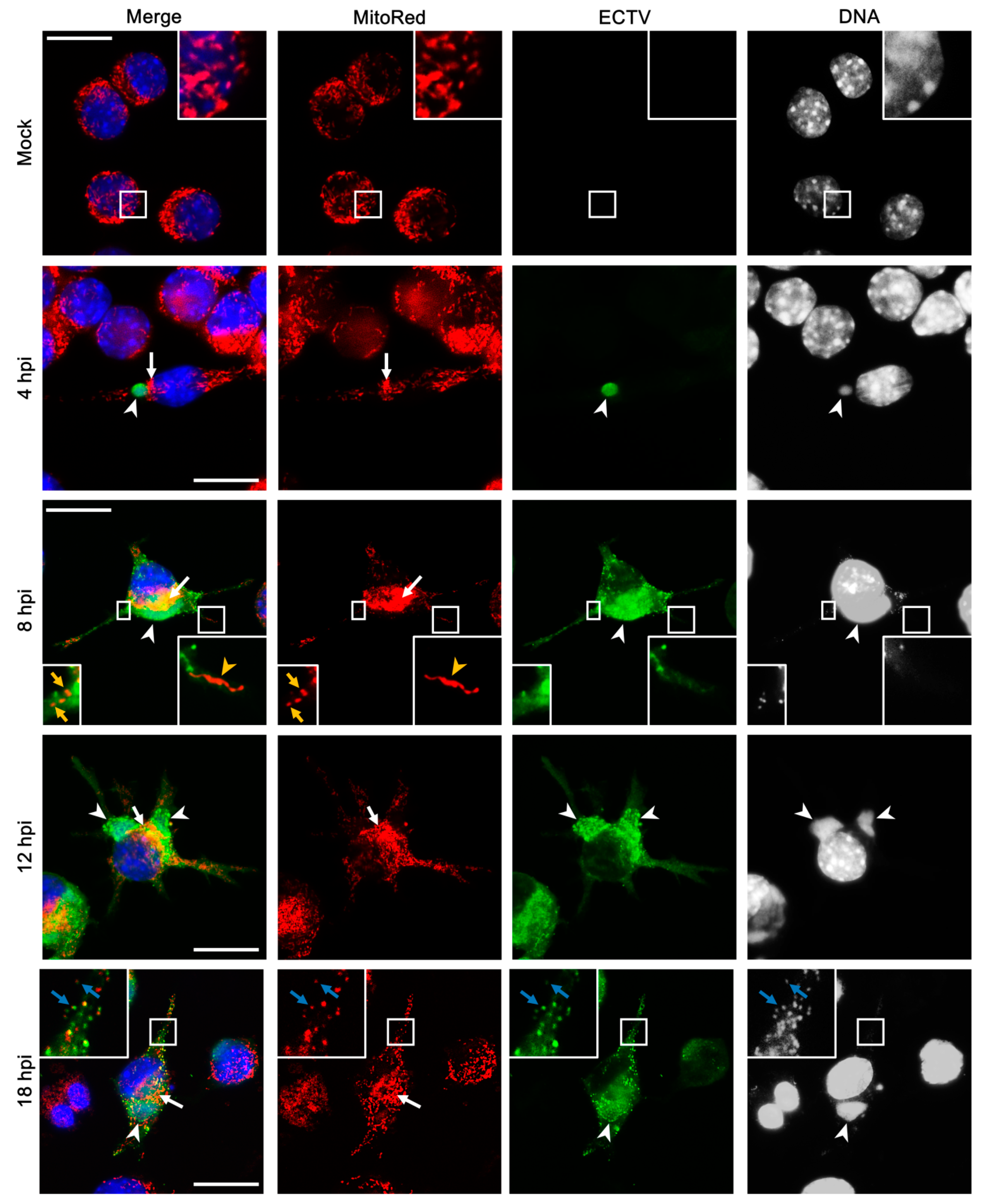
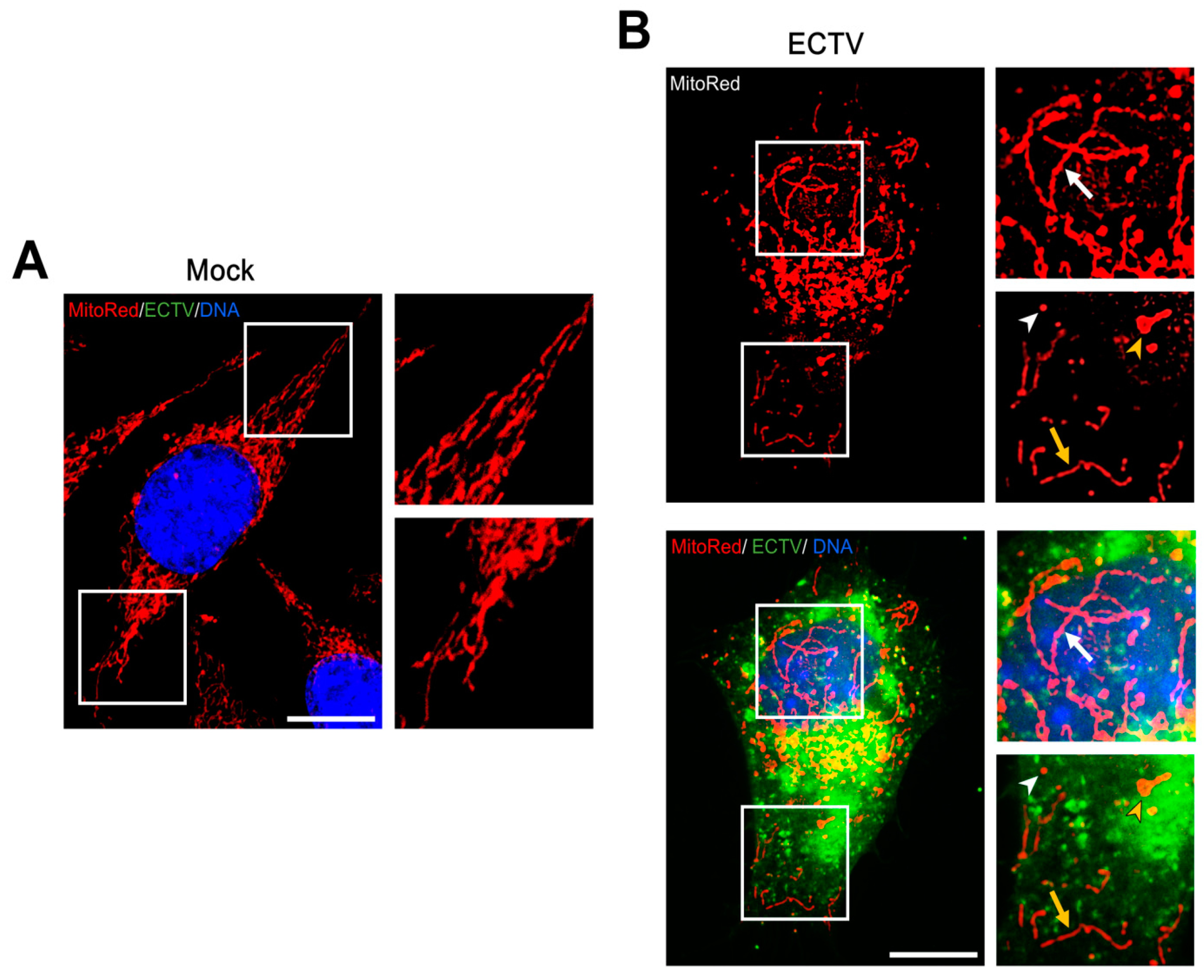
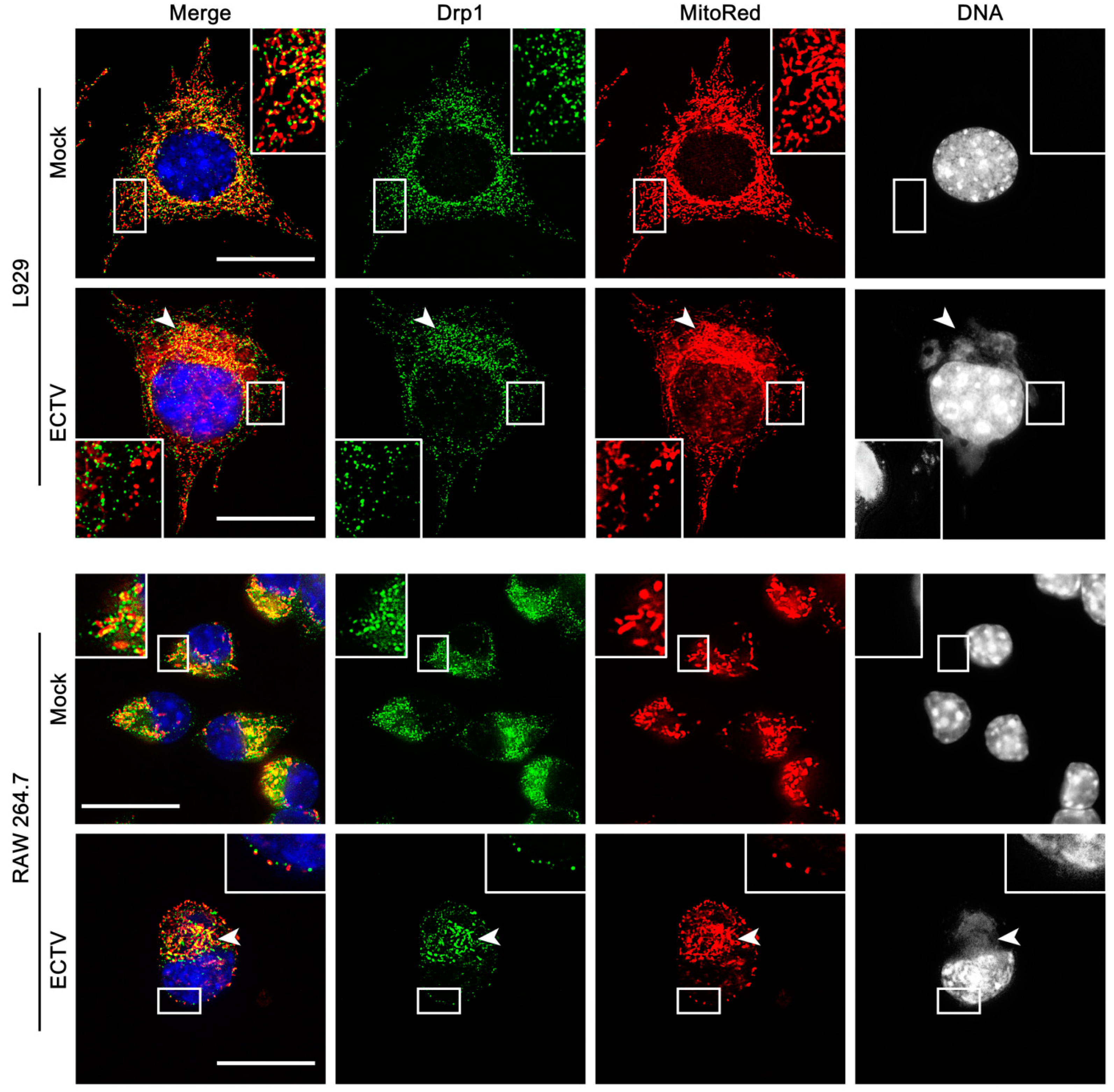
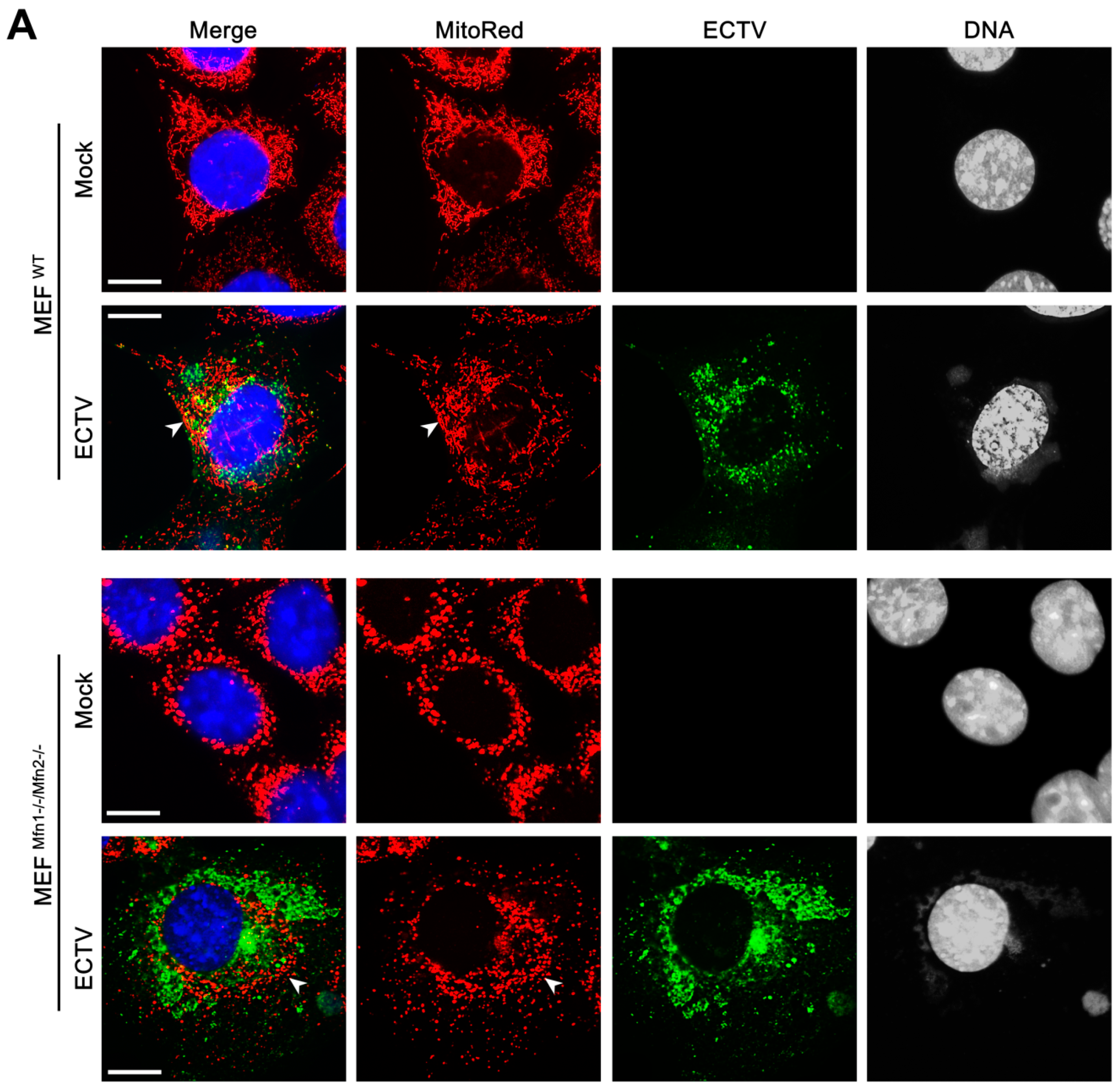
3.3. Mitochondrial Network Morphology and Distribution in Murine Cell Lines during ECTV Infection
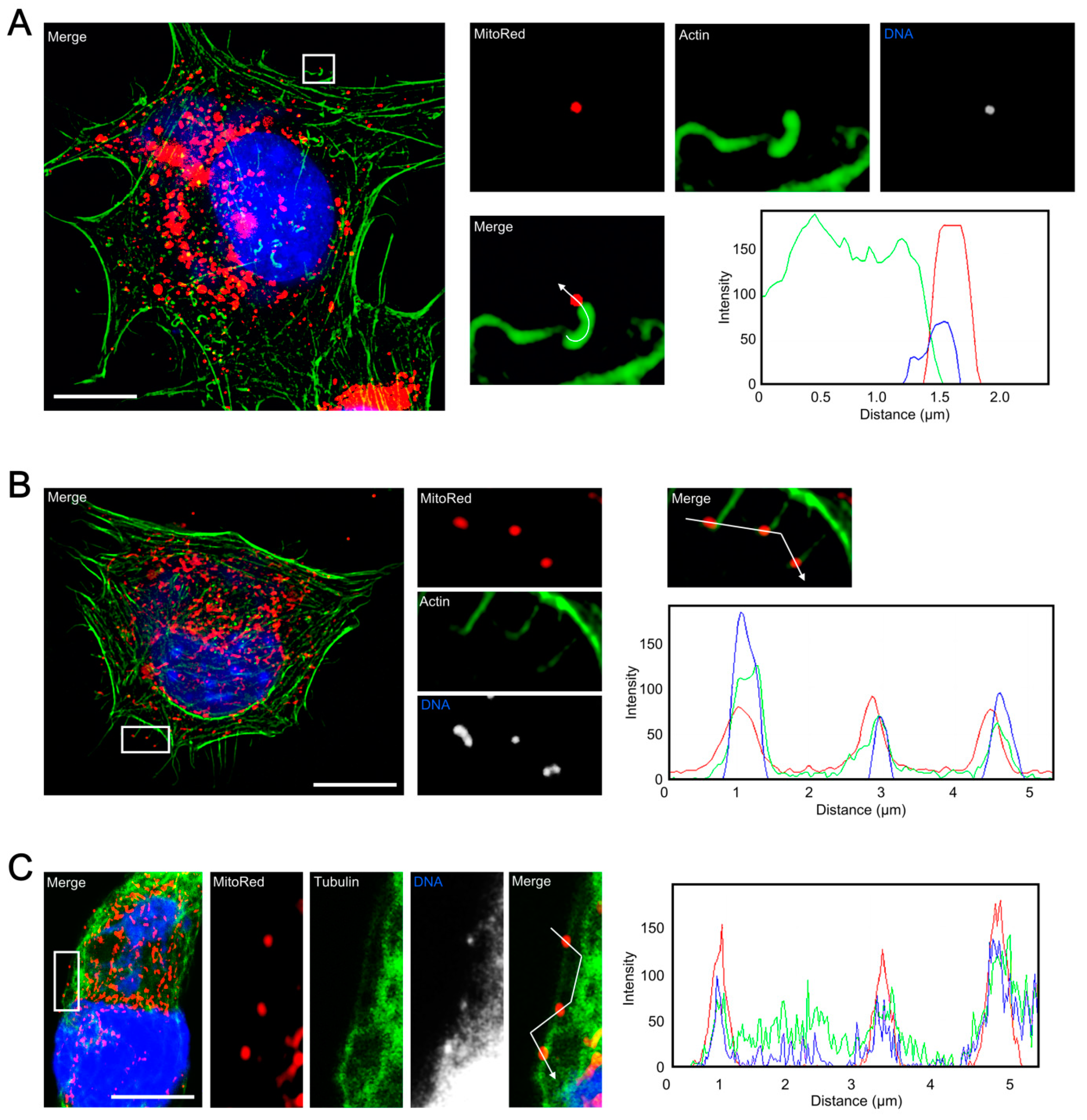
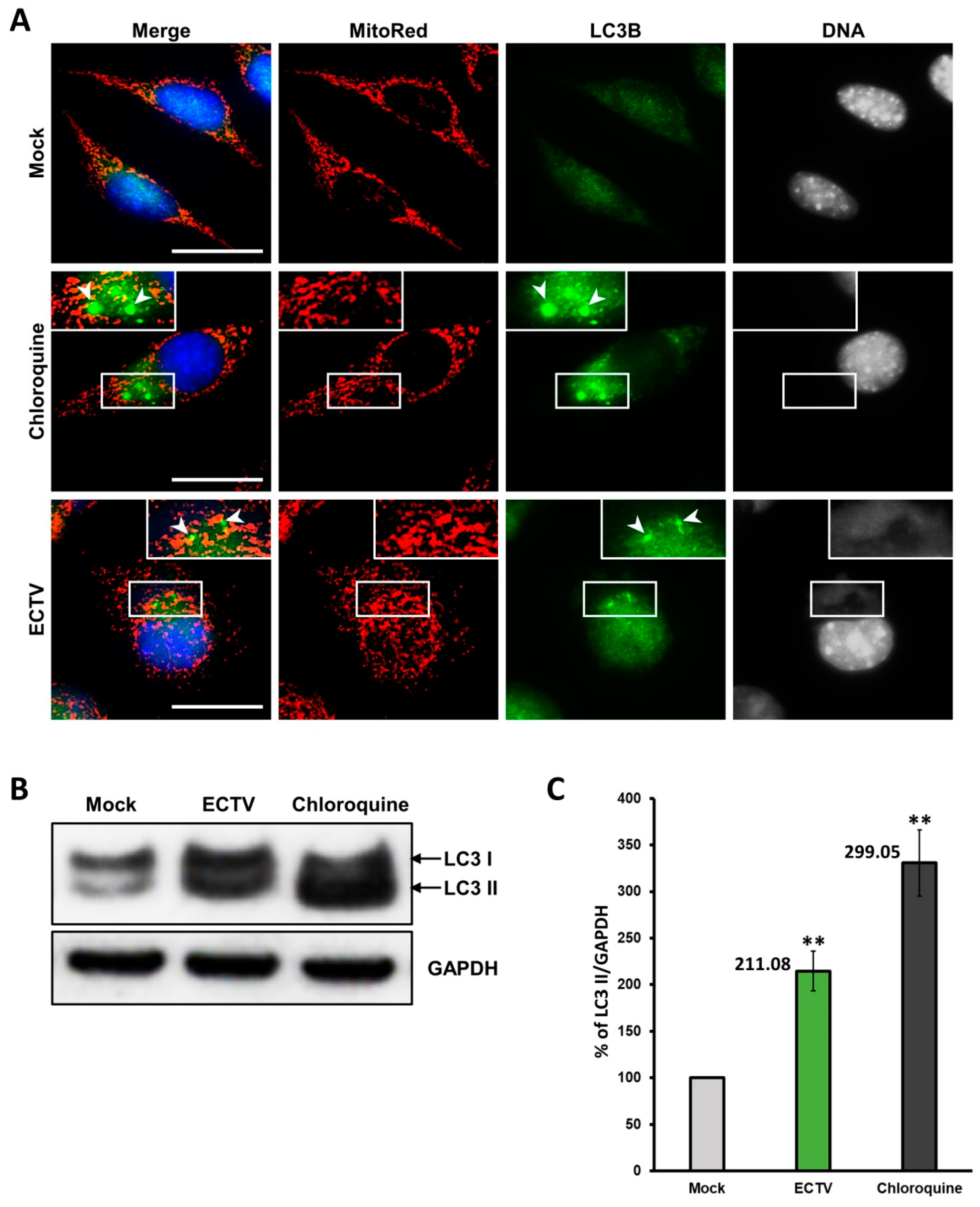
3.4. Co-Localization of Punctate Mitochondria with Progeny ECTV Virions and Their Interaction with Cytoskeleton in Murine Cell Lines
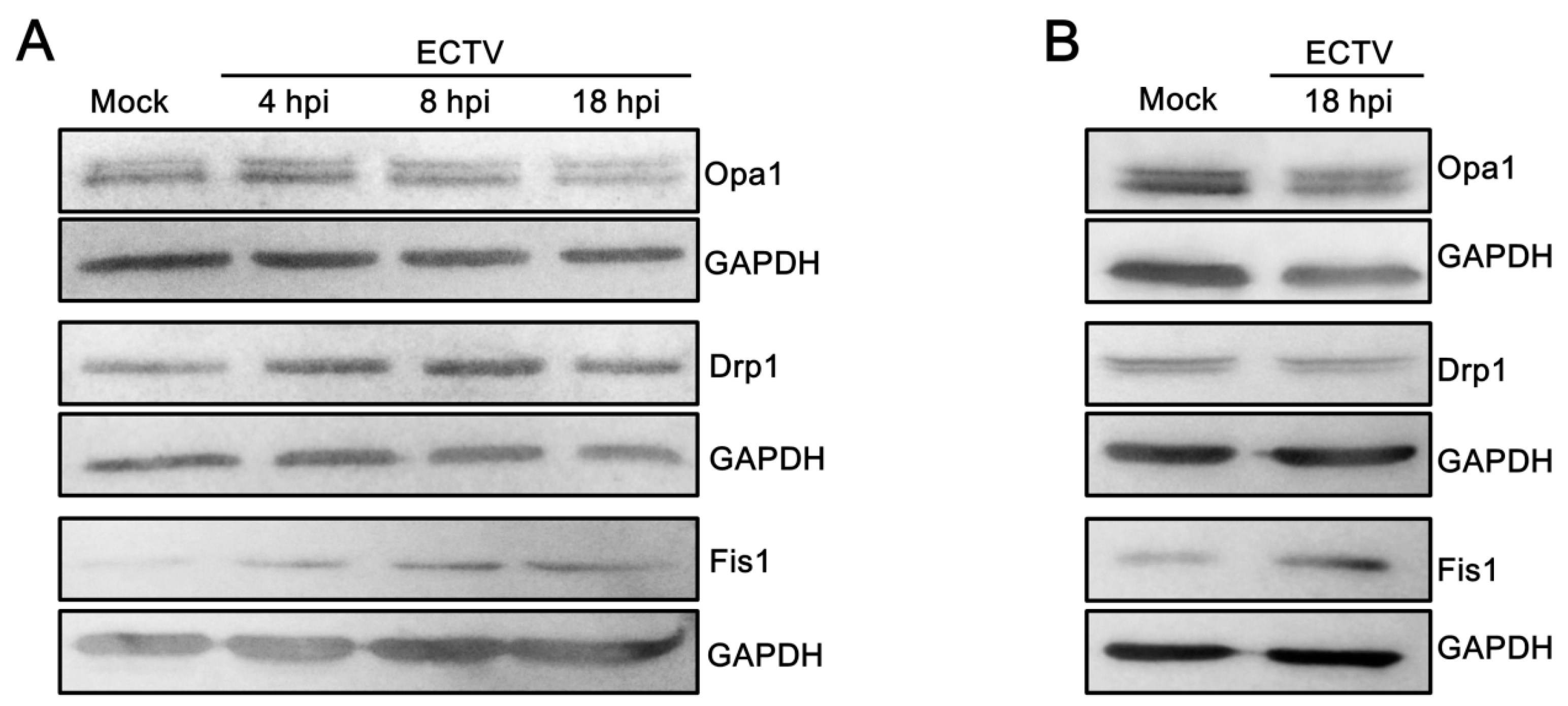
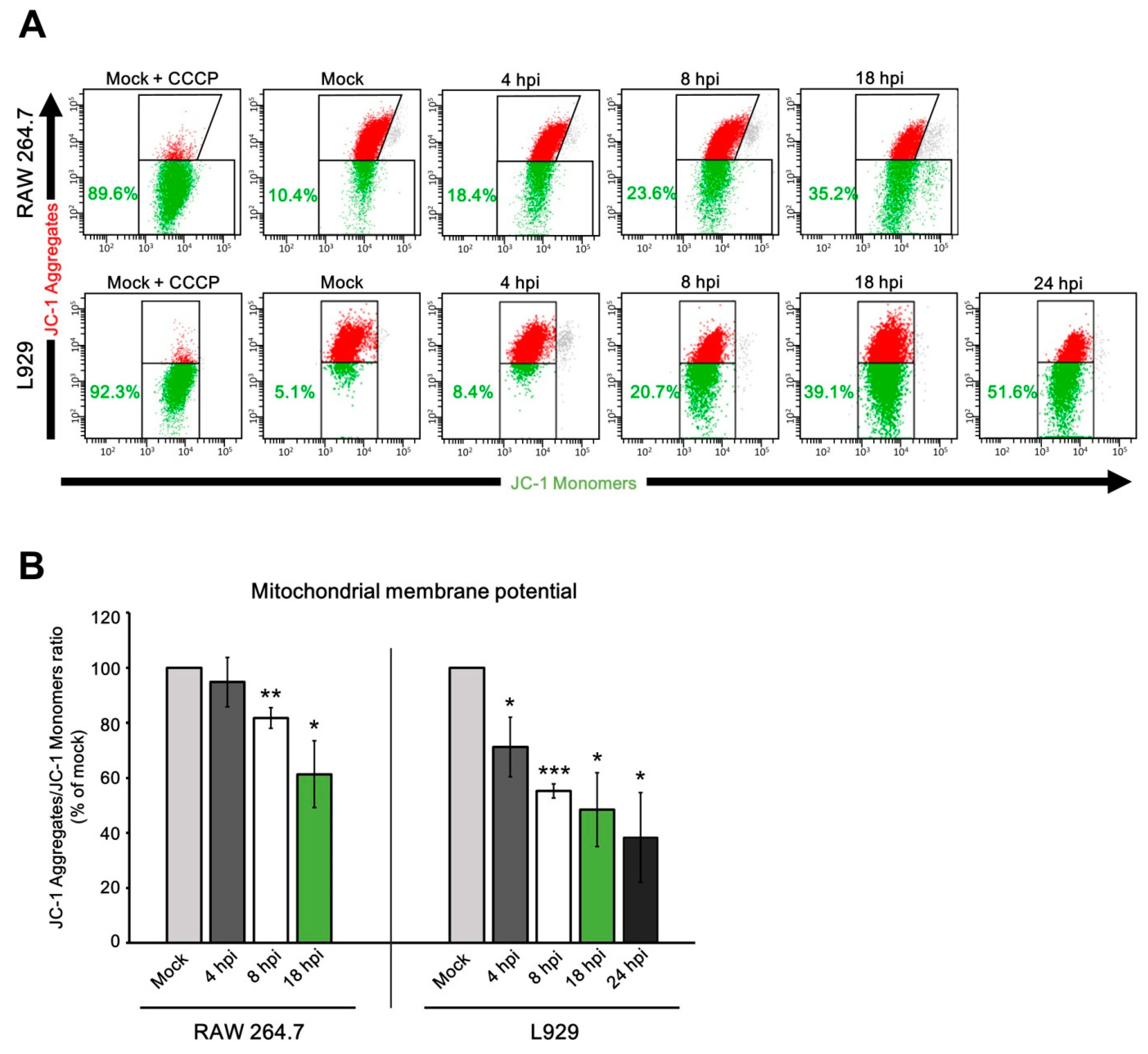
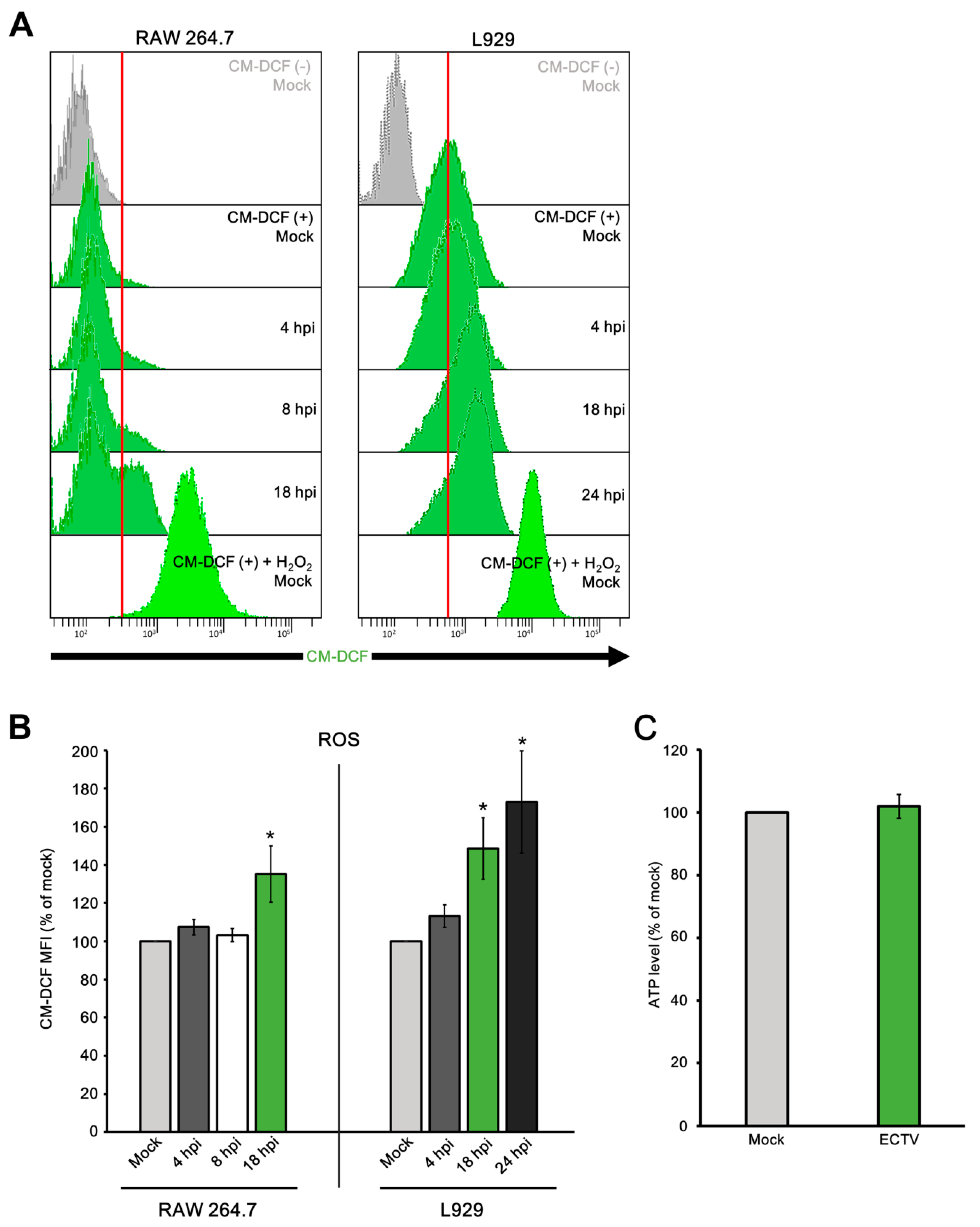
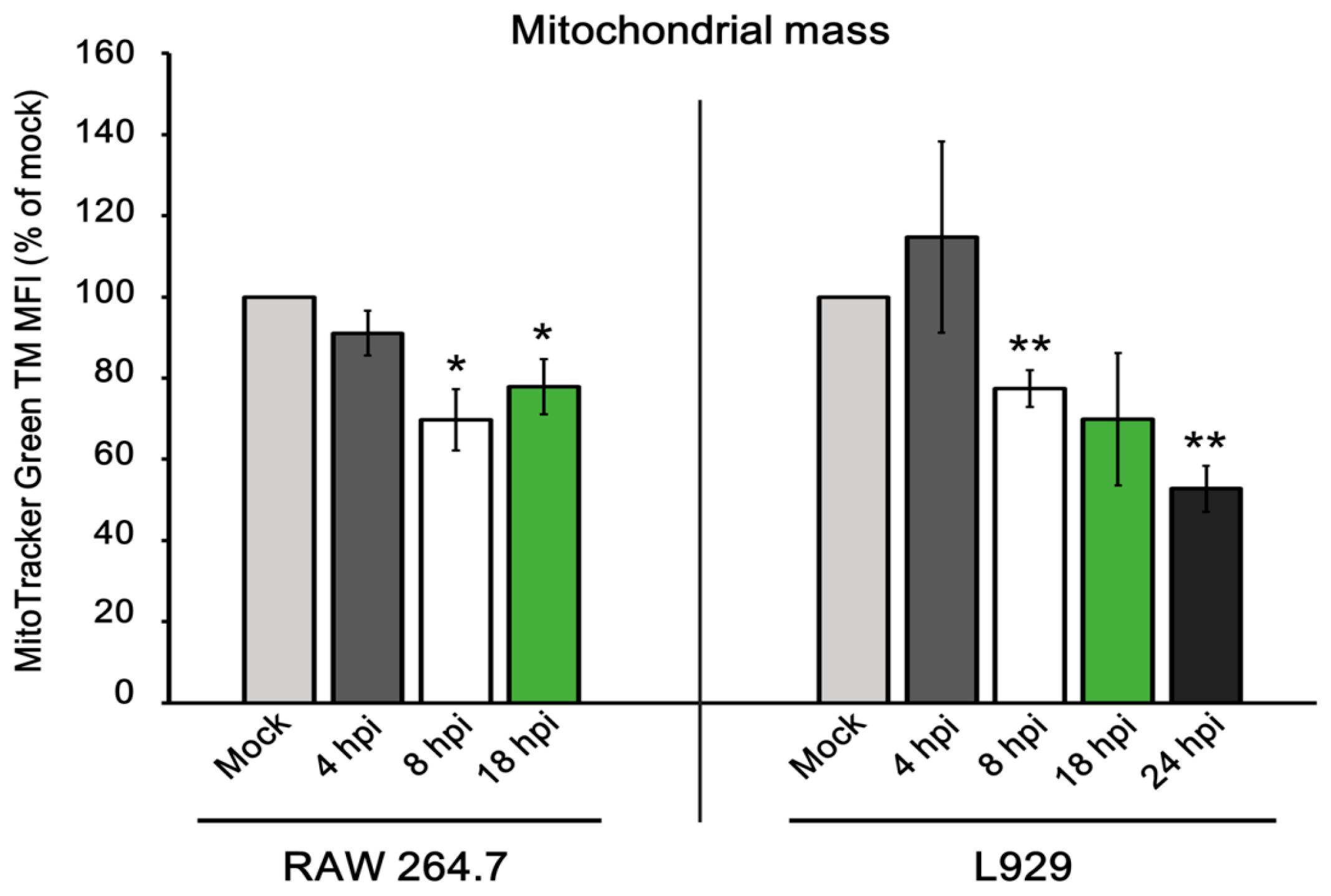
3.5. Mitochondrial Physiology in Murine Cells during ECTV Infection
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lazarou, M. Keeping the immune system in check: A role for mitophagy. Immunol. Cell Biol. 2015, 93, 3–10. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More Than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-G.; Wang, Y.-Y.; Han, K.-J.; Li, L.-Y.; Zhai, Z.; Shu, H.-B. VISA Is an Adapter Protein Required for Virus-Triggered IFN-β Signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 2009, 227, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008, 4, e1000018. [Google Scholar] [CrossRef] [PubMed]
- Irving, A.; Williams, B. Latest advances in innate antiviral defence. F1000 Biol. Rep. 2009, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Bamming, D.; Horvath, C.M. Negative regulation of cytoplasmic RNA-mediated antiviral signaling. Cytokine 2008, 43, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Basler, C.F.; Amarasinghe, G.K. Molecular mechanisms of viral inhibitors of RIG-I-like receptors. Trends Microbiol. 2012, 20, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Cherok, E.; Xu, S.; Li, S.; Das, S.; Meltzer, W.A.; Zalzman, M.; Wang, C.; Karbowski, M. Novel regulatory roles of Mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol. Biol. Cell 2017, 28, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Corrado, M.; Scorrano, L.; Campello, S. Mitochondrial Dynamics in Cancer and Neurodegenerative and Neuroinflammatory Diseases. Int. J. Cell Biol. 2012, 2012, 729290. [Google Scholar] [CrossRef] [PubMed]
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Syed, G.H.; Kim, S.-J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Esteban, D.J.; Buller, R.M.L. Ectromelia virus: The causative agent of mousepox. J. Gen. Virol. 2005, 86, 2645–2659. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Taylor, J.; Quilty, D.; Barry, M. Ectromelia virus encodes an anti-apoptotic protein that regulates cell death. Virology 2015, 475, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Pardo, J.; Gálvez, E.M.; Koskinen, A.; Simon, M.M.; Lobigs, M.; Regner, M.; Müllbacher, A. Caspase-Dependent Inhibition of Mousepox Replication by gzmB. PLoS ONE 2009, 4, e7512. [Google Scholar] [CrossRef] [PubMed]
- Wasilenko, S.T.; Banadyga, L.; Bond, D.; Barry, M. The Vaccinia Virus F1L Protein Interacts with the Proapoptotic Protein Bak and Inhibits Bak Activation. J. Virol. 2005, 79, 14031–14043. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Speiseder, T.; Dobner, T.; Gonzalez, R.A. DNA virus replication compartments. J. Virol. 2014, 88, 1404–1420. [Google Scholar] [CrossRef] [PubMed]
- Boratynska, A.; Martyniszyn, L.; Szulc, L.; Krzyzowska, M.; Szczepanowska, J.; Niemialtowski, M.G. Contribution of rearranged actin structures to the spread of Ectromelia virus infection in vitro. Acta Virol. 2010, 54, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Gregorczyk, K.P.; Szulc-Daąbrowska, L.; Wyzewski, Z.; Struzik, J.; Niemiałtowski, M. Changes in the mitochondrial network during ectromelia virus infection of permissive L929 cells. Acta Biochim. Pol. 2014, 61, 171–177. [Google Scholar] [PubMed]
- Szulc-Dabrowska, L.; Gregorczyk, K.P.; Struzik, J.; Boratynska-Jasinska, A.; Szczepanowska, J.; Wyzewski, Z.; Toka, F.N.; Gierynska, M.; Ostrowska, A.; Niemialtowski, M.G. Remodeling of the fibroblast cytoskeletal architecture during the replication cycle of Ectromelia virus: A morphological in vitro study in a murine cell line. Cytoskeleton 2016, 73, 396–417. [Google Scholar] [CrossRef] [PubMed]
- Irwin, C.R.; Evans, D.H. Modulation of the Myxoma Virus Plaque Phenotype by Vaccinia Virus Protein F11. J. Virol. 2012, 86, 7167–7179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duteyrat, J.-L.; Gelfi, J.; Bertagnoli, S. Ultrastructural study of myxoma virus morphogenesis. Arch. Virol. 2006, 151, 2161–2180. [Google Scholar] [CrossRef] [PubMed]
- Bablanian, R.; Baxt, B.; Sonnabend, J.A.; Esteban, M. Studies on the Mechanisms of Vaccinia Virus Cytopathic Effects. II. Early Cell Rounding is Associated with Virus Polypeptide Synthesis: II. Early Cell Rounding is Associated with Virus Polypeptide Synthesis. J. Gen. Virol. 1978, 39, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.M.; Palumbo, G.J. Poxvirus pathogenesis. Microbiol. Rev. 1991, 55, 80–122. [Google Scholar] [PubMed]
- Lefkowitz, E.J.; Wang, C.; Upton, C. Poxviruses: Past, present and future. Virus Res. 2006, 117, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Vaccinia Virus Uses Macropinocytosis and Apoptotic Mimicry to Enter Host Cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Platt, N.; da Silva, R.P.; Gordon, S. Recognizing death: The phagocytosis of apoptotic cells. Trends Cell Biol. 1998, 8, 365–372. [Google Scholar] [CrossRef]
- Henson, P.M.; Bratton, D.L.; Fadok, V.A. Apoptotic cell removal. Curr. Biol. 2001, 11, R795–R805. [Google Scholar] [CrossRef]
- Albert, M.L. Death-defying immunity: Do apoptotic cells influence antigen processing and presentation? Nat. Rev. Immunol. 2004, 4, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Frederick, R.L.; Shaw, J.M. Moving Mitochondria: Establishing Distribution of an Essential Organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.M.; Smith, S.K.; Olson, V.A.; Thorne, S.H.; Bornmann, W.; Damon, I.K.; Kalman, D. Variola and monkeypox viruses utilize conserved mechanisms of virion motility and release that depend on abl and SRC family tyrosine kinases. J. Virol. 2011, 85, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Gómez, J.C.; Risco, C.; Rodríguez, D.; Cabezas, P.; Guerra, S.; Carrascosa, J.L.; Esteban, M. Differences in virus-induced cell morphology and in virus maturation between MVA and other strains (WR, Ankara, and NYCBH) of vaccinia virus in infected human cells. J. Virol. 2003, 77, 10606–10622. [Google Scholar] [CrossRef] [PubMed]
- Rietdorf, J.; Ploubidou, A.; Reckmann, I.; Holmström, A.; Frischknecht, F.; Zettl, M.; Zimmermann, T.; Way, M. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 2001, 3, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; McCrae, M.A. The molecular biology of rotaviruses X: Intercellular dissemination of rotavirus NSP4 requires glycosylation and is mediated by direct cell-cell contact through cytoplasmic extrusions. Arch. Virol. 2012, 157, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.D.; Tzeng, W.-P.; Frey, T.K. Analysis of the function of cytoplasmic fibers formed by the rubella virus nonstructural replicase proteins. Virology 2010, 406, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Ploubidou, A.; Moreau, V.; Ashman, K.; Reckmann, I.; González, C.; Way, M. Vaccinia virus infection disrupts microtubule organization and centrosome function. EMBO J. 2000, 19, 3932. [Google Scholar] [CrossRef] [PubMed]
- Rojo, G.; Chamorro, M.; Salas, M.L.; Viñuela, E.; Cuezva, J.M.; Salas, J. Migration of mitochondria to viral assembly sites in African swine fever virus-infected cells. J. Virol. 1998, 72, 7583–7588. [Google Scholar] [PubMed]
- Cobbold, C.; Brookes, S.M.; Wileman, T. Biochemical requirements of virus wrapping by the endoplasmic reticulum: Involvement of ATP and endoplasmic reticulum calcium store during envelopment of African swine fever virus. J. Virol. 2000, 74, 2151–2160. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.L.; Wileman, T.E. African swine fever virus organelle rearrangements. Virus Res. 2013, 173, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.; Li, H.-C.; Hsu, C.-F.; Chang, C.-Y.; Lo, S.-Y. Increased ATP generation in the host cell is required for efficient vaccinia virus production. J. Biomed. Sci. 2009, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Greseth, M.D.; Traktman, P. De novo Fatty Acid Biosynthesis Contributes Significantly to Establishment of a Bioenergetically Favorable Environment for Vaccinia Virus Infection. PLoS Pathog. 2014, 10, e1004021. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.L.; Smith, V.L.; Chow, D.; Mocarski, E.S. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 2003, 77, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.D.; Basak, N.P.; Banerjee, A.S.; Banerjee, S. Epstein–Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis 2014, 35, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Syed, G.H.; Khan, M.; Chiu, W.-W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Enquist, L.W. Alphaherpesvirus Infection Disrupts Mitochondrial Transport in Neurons. Cell Host Microbe 2012, 11, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Wasilenko, S.T.; Stewart, T.L.; Meyers, A.F.A.; Barry, M. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 14345–14350. [Google Scholar] [CrossRef] [PubMed]
- Everett, H.; Barry, M.; Lee, S.F.; Sun, X.; Graham, K.; Stone, J.; Bleackley, R.C.; McFadden, G. M11L: A novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J. Exp. Med. 2000, 191, 1487–1498. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.; Horsington, J.; Eldi, P.; Al Rumaih, Z.; Karupiah, G.; Newsome, T. Loss of Actin-Based Motility Impairs Ectromelia Virus Release In Vitro but Is Not Critical to Spread In Vivo. Viruses 2018, 10, 111. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Szulc-Dąbrowska, L.; Struzik, J.; Ostrowska, A.; Guzera, M.; Toka, F.N.; Bossowska-Nowicka, M.; Gieryńska, M.M.; Winnicka, A.; Nowak, Z.; Niemiałtowski, M.G. Functional paralysis of GM-CSF–derived bone marrow cells productively infected with ectromelia virus. PLoS ONE 2017, 12, e0179166. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.M.; Moss, B. Vaccinia Virus Intracellular Movement Is Associated with Microtubules and Independent of Actin Tails. J. Virol. 2001, 75, 11651–11663. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.P.; Way, M. Nck- and N-WASP-Dependent Actin-Based Motility Is Conserved in Divergent Vertebrate Poxviruses. Cell Host Microbe 2009, 6, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Rottner, K.; Stradal, T.E.B. Poxviruses Taking a Ride on Actin: New Users of Known Hardware. Cell Host Microbe 2009, 6, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Varadi, A.; Johnson-Cadwell, L.I.; Cirulli, V.; Yoon, Y.; Allan, V.J.; Rutter, G.A. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J. Cell Sci. 2004, 117, 4389–4400. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Choi, S.; Gibson, G.A.; Watkins, S.C.; Bakkenist, C.J.; Van Houten, B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 2012, 125, 5745–5757. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Fadeel, B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxvirus cell entry: How many proteins does it take? Viruses 2012, 4, 688–707. [Google Scholar] [CrossRef] [PubMed]
- Bidgood, S.R.; Mercer, J. Cloak and Dagger: Alternative Immune Evasion and Modulation Strategies of Poxviruses. Viruses 2015, 7, 4800–4825. [Google Scholar] [CrossRef] [PubMed]
- Hawes, P.C.; Netherton, C.L.; Wileman, T.E.; Monaghan, P. The envelope of intracellular African swine fever virus is composed of a single lipid bilayer. J. Virol. 2008, 82, 7905–7912. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Melo-Silva, C.R.; Tscharke, D.C.; Lobigs, M.; Koskinen, A.; Wong, Y.C.; Buller, R.M.; Mullbacher, A.; Regner, M. The Ectromelia Virus SPI-2 Protein Causes Lethal Mousepox by Preventing NK Cell Responses. J. Virol. 2011, 85, 11170–11182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.-H.; Lai, C.-K.; Jeng, K.-S.; Sung, V.M.-H.; Lai, M.M.C. Hepatitis C Virus Triggers Mitochondrial Permeability Transition with Production of Reactive Oxygen Species, Leading to DNA Damage and STAT3 Activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Tolkovsky, A.M. Mitophagy. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Figge, M.T.; Reichert, A.S.; Meyer-Hermann, M.; Osiewacz, H.D. Deceleration of fusion-fission cycles improves mitochondrial quality control during aging. PLoS Comput. Biol. 2012, 8, e1002576. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Shirihai, O.S. The Interplay Between Mitochondrial Dynamics and Mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- El-Bacha, T.; Da Poian, A.T. Virus-induced changes in mitochondrial bioenergetics as potential targets for therapy. Int. J. Biochem. Cell Biol. 2013, 45, 41–46. [Google Scholar] [CrossRef] [PubMed]


















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregorczyk, K.P.; Wyżewski, Z.; Szczepanowska, J.; Toka, F.N.; Mielcarska, M.B.; Bossowska-Nowicka, M.; Gieryńska, M.; Boratyńska-Jasińska, A.; Struzik, J.; Niemiałtowski, M.G.; et al. Ectromelia Virus Affects Mitochondrial Network Morphology, Distribution, and Physiology in Murine Fibroblasts and Macrophage Cell Line. Viruses 2018, 10, 266. https://doi.org/10.3390/v10050266
Gregorczyk KP, Wyżewski Z, Szczepanowska J, Toka FN, Mielcarska MB, Bossowska-Nowicka M, Gieryńska M, Boratyńska-Jasińska A, Struzik J, Niemiałtowski MG, et al. Ectromelia Virus Affects Mitochondrial Network Morphology, Distribution, and Physiology in Murine Fibroblasts and Macrophage Cell Line. Viruses. 2018; 10(5):266. https://doi.org/10.3390/v10050266
Chicago/Turabian StyleGregorczyk, Karolina P., Zbigniew Wyżewski, Joanna Szczepanowska, Felix N. Toka, Matylda B. Mielcarska, Magdalena Bossowska-Nowicka, Małgorzata Gieryńska, Anna Boratyńska-Jasińska, Justyna Struzik, Marek G. Niemiałtowski, and et al. 2018. "Ectromelia Virus Affects Mitochondrial Network Morphology, Distribution, and Physiology in Murine Fibroblasts and Macrophage Cell Line" Viruses 10, no. 5: 266. https://doi.org/10.3390/v10050266
APA StyleGregorczyk, K. P., Wyżewski, Z., Szczepanowska, J., Toka, F. N., Mielcarska, M. B., Bossowska-Nowicka, M., Gieryńska, M., Boratyńska-Jasińska, A., Struzik, J., Niemiałtowski, M. G., & Szulc-Dąbrowska, L. (2018). Ectromelia Virus Affects Mitochondrial Network Morphology, Distribution, and Physiology in Murine Fibroblasts and Macrophage Cell Line. Viruses, 10(5), 266. https://doi.org/10.3390/v10050266