Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping


Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Constructs
2.2. Infection
2.3. Plaque Assays
2.4. Fluorescent Virus Generation
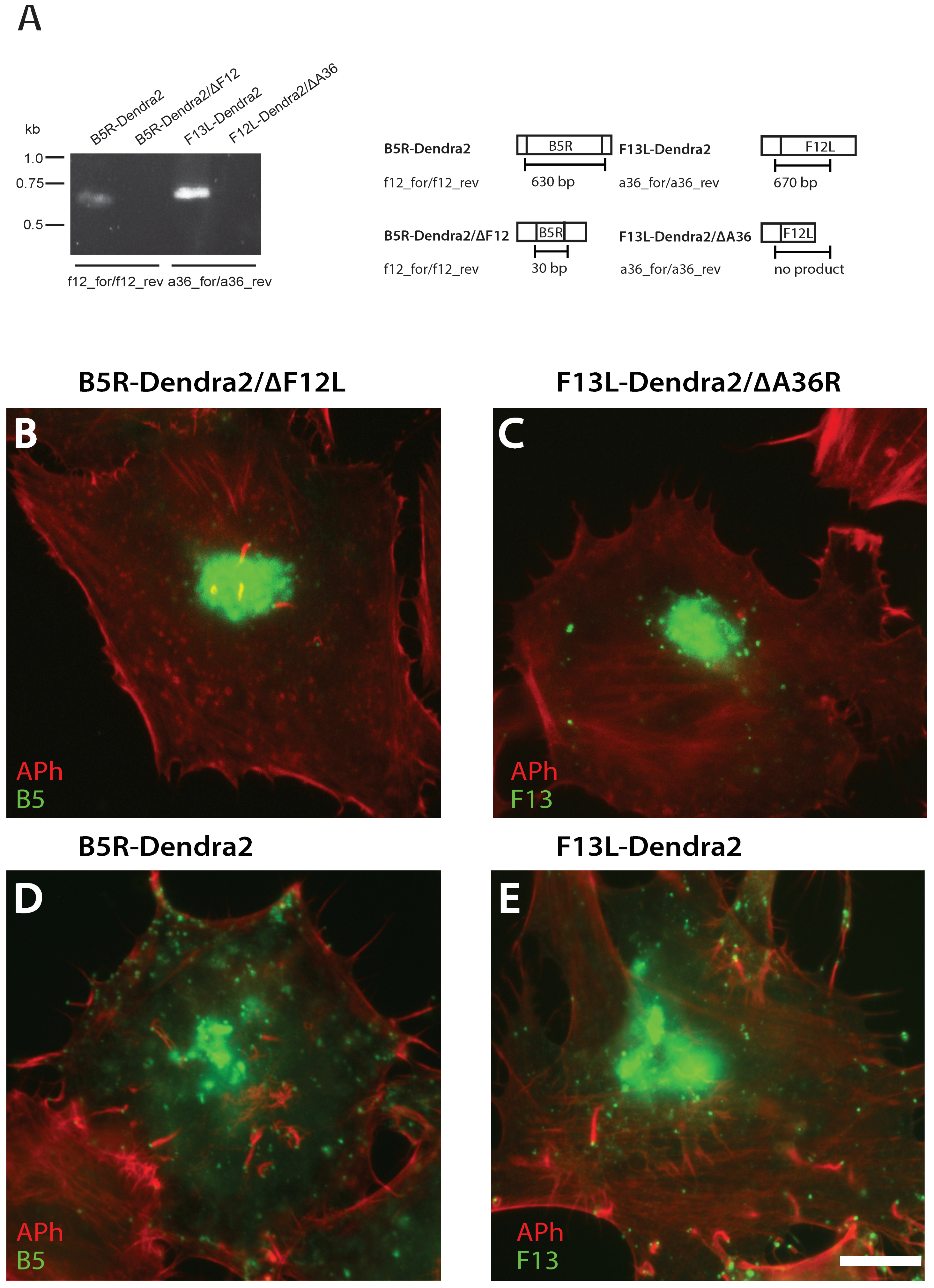
2.5. Validating B5R-Dendra2/∆F12L and F13L-Dendra2/∆A36R Null Mutants
2.6. Immunofluorescence Assays
2.7. Confocal Microscopy
3. Results
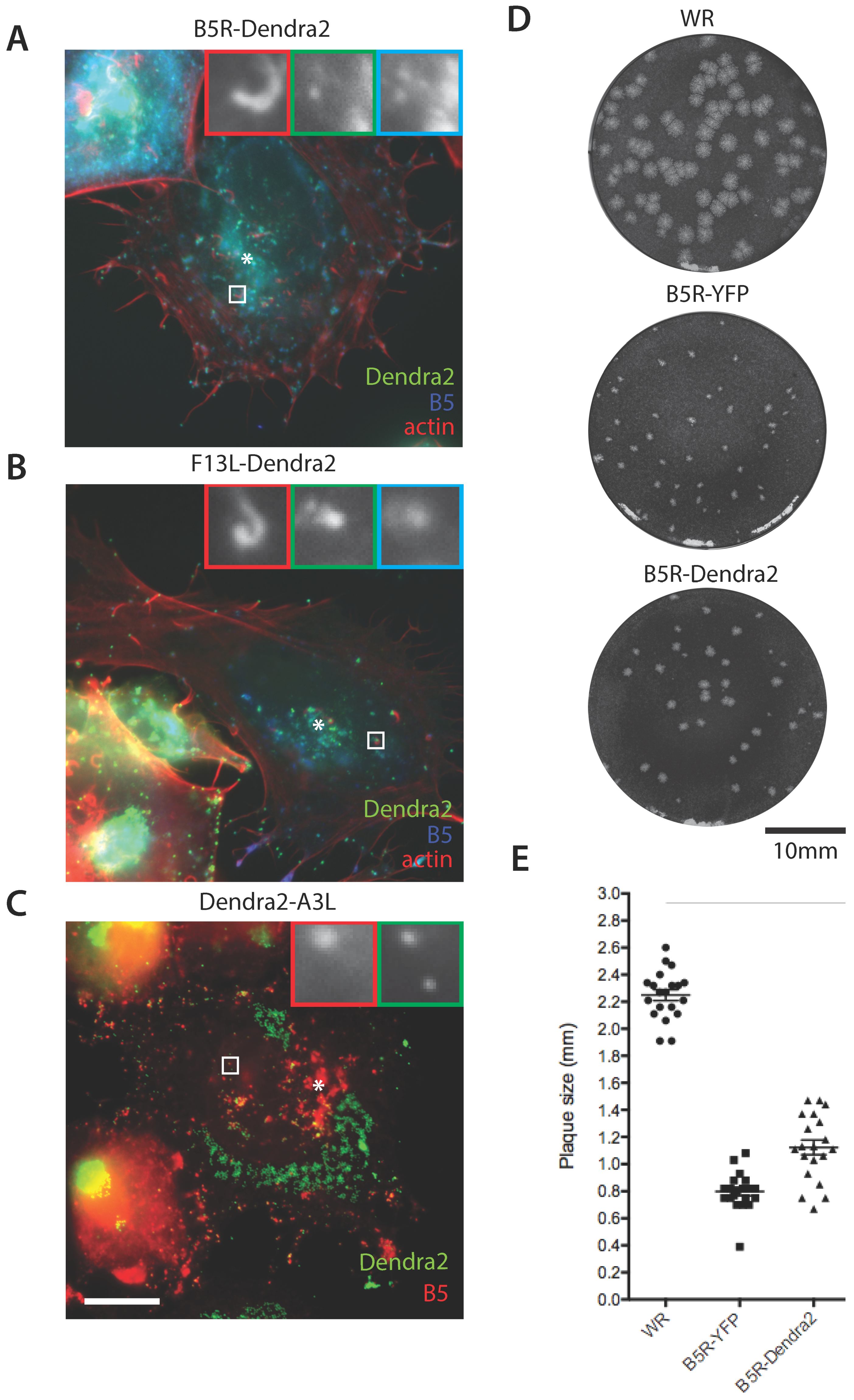
3.1. Construction of Photoconvertible VACV
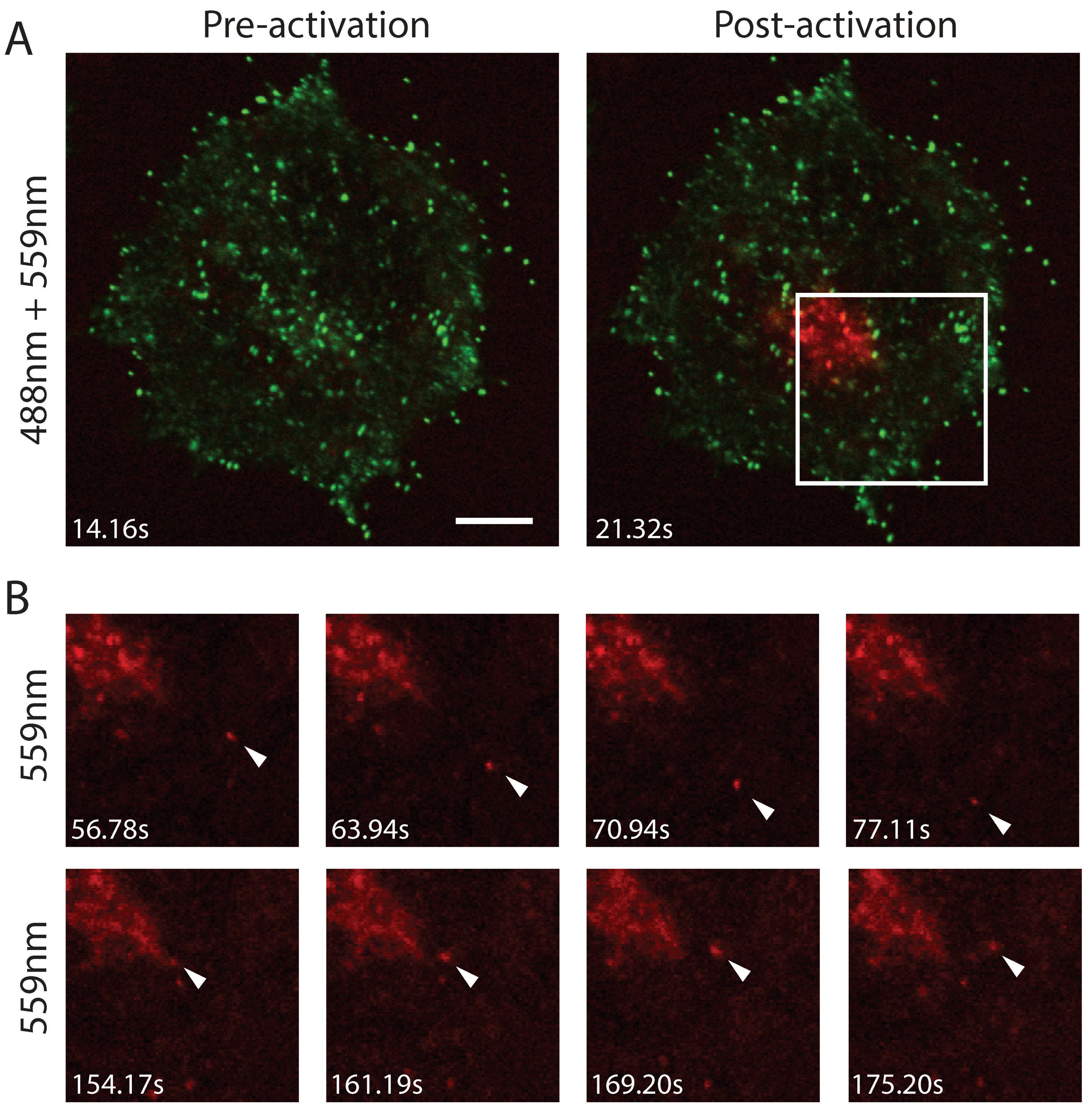
3.2. Efficient Photoconversion of Dendra2 Viruses
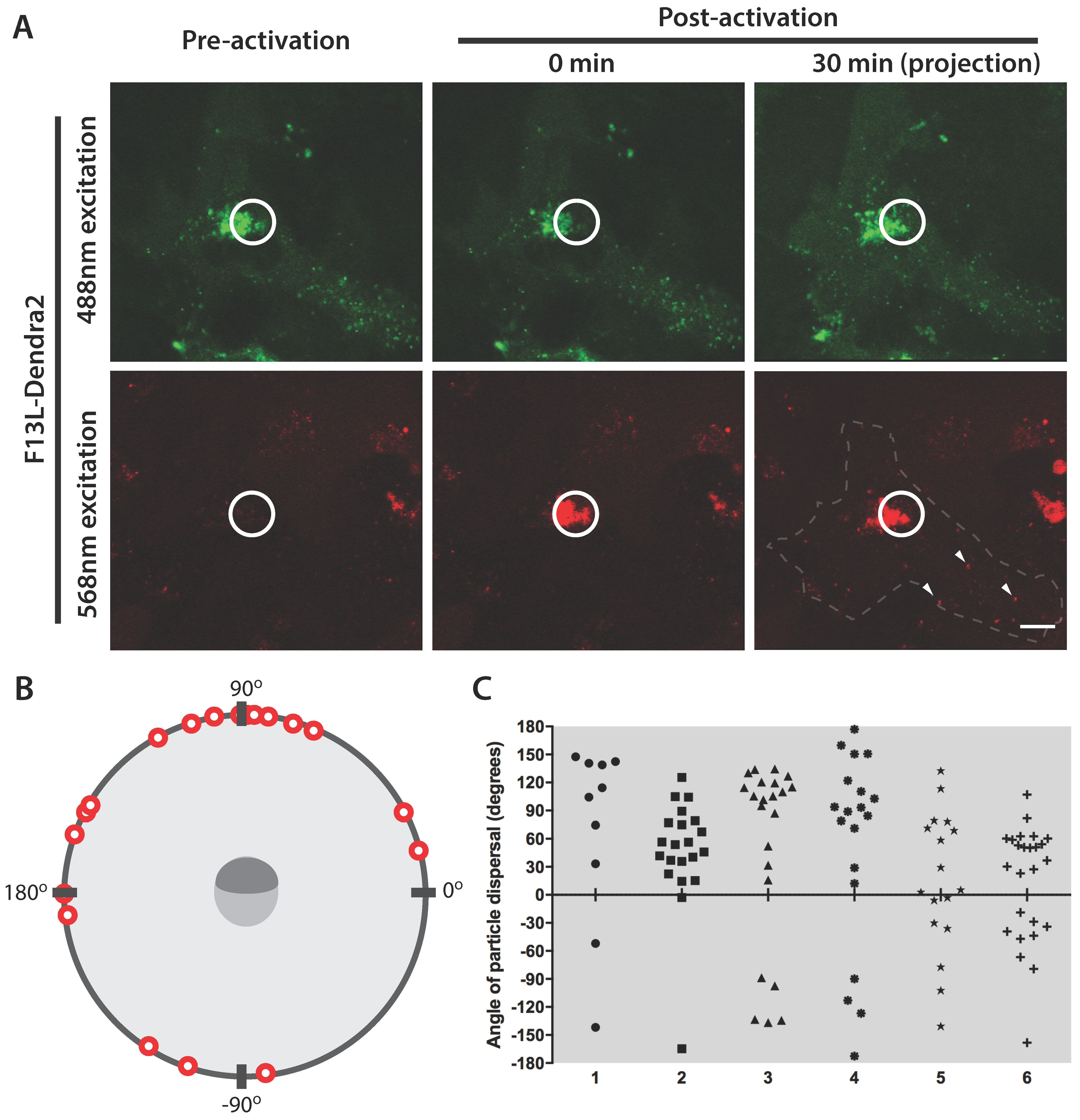
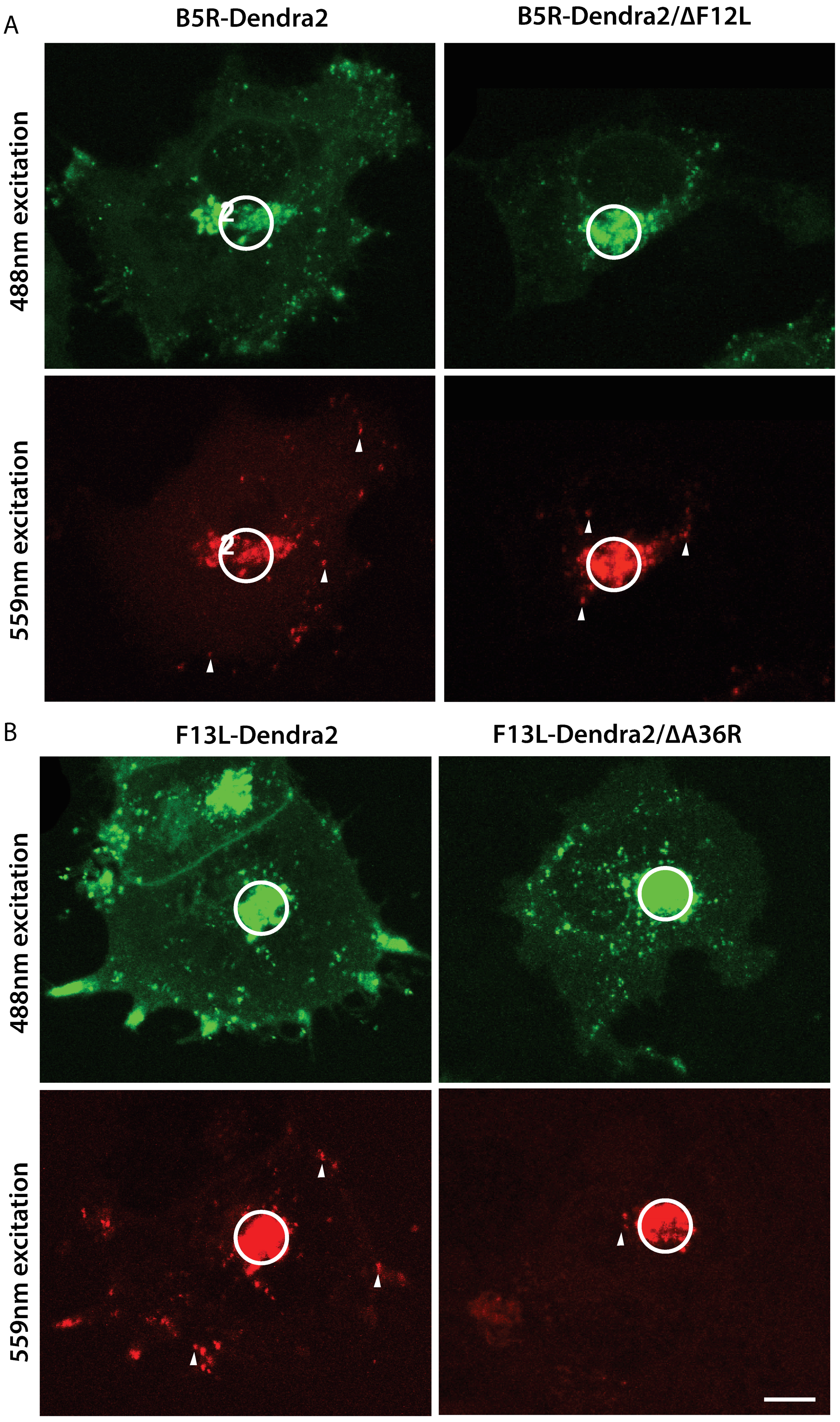
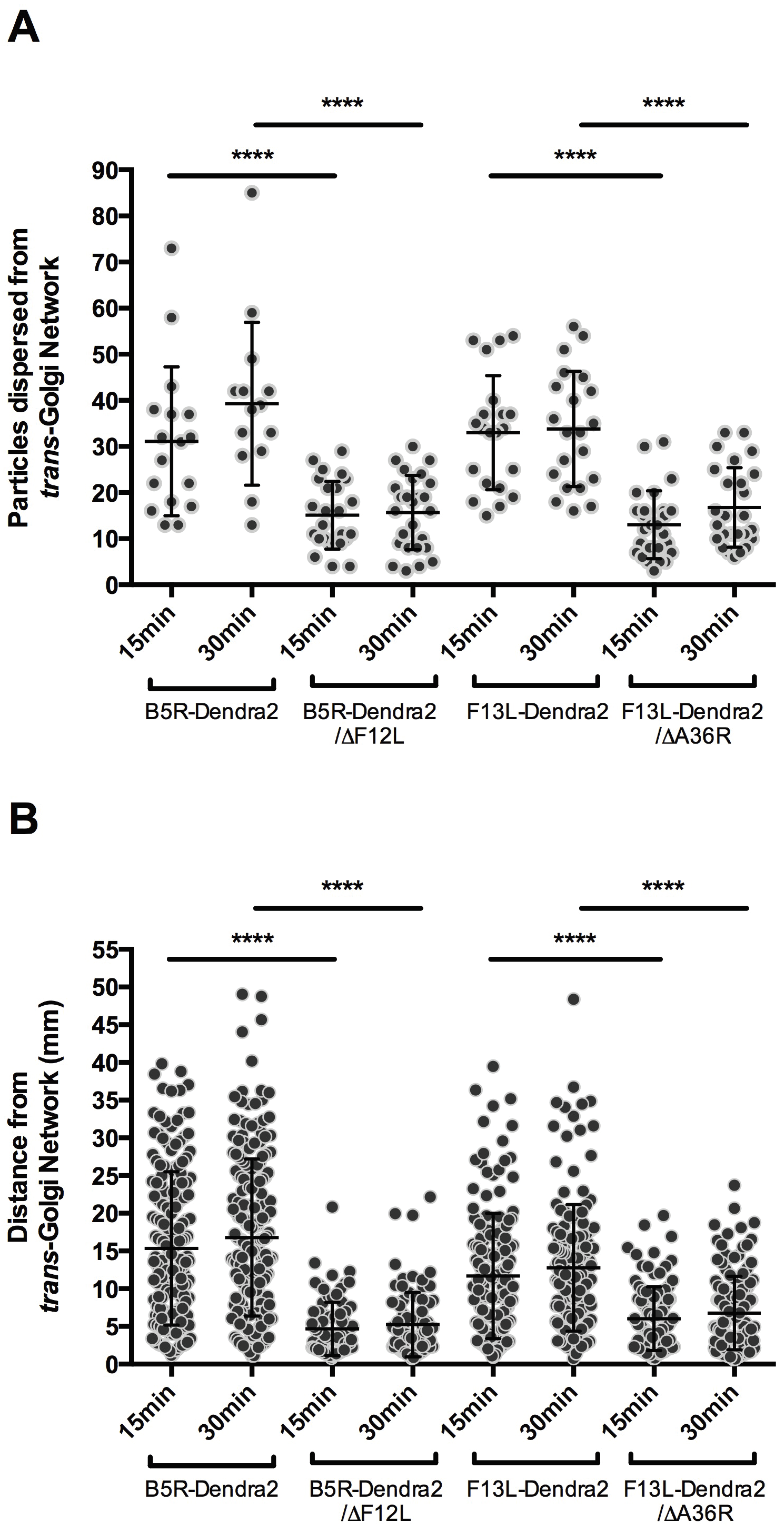
3.3. F12L and A36R Mutants Reveal a Role for Kinesin-1 in WV Exit from the TGN
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dodding, M.P.; Way, M. Coupling viruses to dynein and kinesin-1. EMBO J. 2011, 30, 3527–3539. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Lakadamyali, M.; Brandenburg, B.; Zhuang, X. Single-virus tracking in live cells. Cold Spring Harb. Protoc. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Verkhusha, V.V.; Staroverov, D.B.; Souslova, E.A.; Lukyanov, S.; Lukyanov, K.A. Photoswitchable cyan fluorescent protein for protein tracking. Nat. Biotechnol. 2004, 22, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, J.; Ivanchenko, S.; Oswald, F.; Schmitt, F.; Rocker, C.; Salih, A.; Spindler, K.D.; Nienhaus, G.U. Eosfp, a fluorescent marker protein with uv-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15905–15910. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Lukyanov, S.; Lukyanov, K.A. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat. Protoc. 2007, 2, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.H.; McCaffery, J.M.; Chan, D.C. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 2012, 50, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.; Mathur, N.; Mathur, J. Simultaneous live-imaging of peroxisomes and the er in plant cells suggests contiguity but no luminal continuity between the two organelles. Front. Physiol. 2013, 4, 196. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.; Way, M. The role of signalling and the cytoskeleton during vaccinia virus egress. Virus Res. 2015, 209, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Condit, R.C.; Moussatche, N.; Traktman, P. In a nutshell: Structure and assembly of the vaccinia virion. Adv. Virus Res. 2006, 66, 31–124. [Google Scholar] [PubMed]
- Schepis, A.; Stauber, T.; Krijnse Locker, J. Kinesin-1 plays multiple roles during the vaccinia virus life cycle. Cell. Microbiol. 2007, 9, 1960–1973. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, C.M.; Hollinshead, M.; Smith, G.L. The vaccinia virus A27L protein is needed for the microtubule-dependent transport of intracellular mature virus particles. J. Gen. Virol. 2000, 81, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Smith, G.L. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 2008, 16, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Rietdorf, J.; Ploubidou, A.; Reckmann, I.; Holmstrom, A.; Frischknecht, F.; Zettl, M.; Zimmermann, T.; Way, M. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 2001, 3, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.M.; Moss, B. Vaccinia virus A36R membrane protein provides a direct link between intracellular enveloped virions and the microtubule motor kinesin. J. Virol. 2004, 78, 2486–2493. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.P.; Mitter, R.; Humphries, A.C.; Way, M. A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 2011, 30, 4523–4538. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.N.D.; Carpentier, D.C.J.; Ewles, H.A.; Lee, S.A.; Smith, G.L. Vaccinia virus proteins a36 and f12/e2 show strong preferences for different kinesin light chain isoforms. Traffic 2017, 18, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.; Newsome, T.P.; Collinson, L.; Edwards, C.; Way, M. An E2-F12 complex is required for intracellular enveloped virus morphogenesis during vaccinia infection. Cell. Microbiol. 2009, 11, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, D.C.; Gao, W.N.; Ewles, H.; Morgan, G.W.; Smith, G.L. Vaccinia virus protein complex F12/E2 interacts with kinesin light chain isoform 2 to engage the kinesin-1 motor complex. PLoS Pathog. 2015, 11, e1004723. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.C.; Ward, B.M. The vaccinia virus protein F12 associates with intracellular enveloped virus through an interaction with A36. J. Virol. 2009, 83, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Martinez, E.; Roberts, K.L.; Hollinshead, M.; Smith, G.L. Vaccinia virus intracellular enveloped virions move to the cell periphery on microtubules in the absence of the A36R protein. J. Gen. Virol. 2005, 86, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, D.C.J.; Hollinshead, M.S.; Ewles, H.A.; Lee, S.A.; Smith, G.L. Tagging of the vaccinia virus protein F13 with mcherry causes aberrant virion morphogenesis. J. Gen. Virol. 2017, 98, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Newsome, T.P.; Marzook, N.B. Viruses that ride on the coat-tails of actin nucleation. Semin. Cell Dev. Biol. 2015, 46, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.A.; Harriman, P.D.; Wall, J.D. Escherichia coli mutants deficient in guanine-xanthine phosphoribosyltransferase. J. Bacteriol. 1976, 126, 1141–1148. [Google Scholar] [PubMed]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar] [CrossRef] [PubMed]
- Falkner, F.G.; Moss, B. Escherichia coli gpt gene provides dominant selection for vaccinia virus open reading frame expression vectors. J. Virol. 1988, 62, 1849–1854. [Google Scholar] [PubMed]
- Frischknecht, F.; Cudmore, S.; Moreau, V.; Reckmann, I.; Rottger, S.; Way, M. Tyrosine phosphorylation is required for actin-based motility of vaccinia but not listeria or shigella. Curr. Biol. 1999, 9, 89–92. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Sisler, J.R.; Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques 1997, 23, 1094–1097. [Google Scholar] [PubMed]
- Marzook, N.B.; Procter, D.J.; Lynn, H.; Yamamoto, Y.; Horsington, J.; Newsome, T.P. Methodology for the efficient generation of fluorescently tagged vaccinia virus proteins. J. Vis. Exp. 2014, 83, e51151. [Google Scholar] [CrossRef] [PubMed]
- Engelstad, M.; Smith, G.L. The vaccinia virus 42-kda envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 1993, 194, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Wolffe, E.J.; Isaacs, S.N.; Moss, B. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J. Virol. 1993, 67, 4732–4741. [Google Scholar] [PubMed]
- Zhang, W.H.; Wilcock, D.; Smith, G.L. Vaccinia virus F12L protein is required for actin tail formation, normal plaque size, and virulence. J. Virol. 2000, 74, 11654–11662. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J.E.; Smith, G.L. Vaccinia virus gene A36R encodes a Mr 43-50 K protein on the surface of extracellular enveloped virus. Virology 1994, 204, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, S.N. Vaccinia virus and Poxvirology: Methods and Protocols; Humana Press Inc.: Totowa, NJ, USA, 2004; Volume 269. [Google Scholar]
- Geada, M.M.; Galindo, I.; Lorenzo, M.M.; Perdiguero, B.; Blasco, R. Movements of vaccinia virus intracellular enveloped virions with GFP tagged to the F13L envelope protein. J. Gen. Virol. 2001, 82, 2747–2760. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, Y.; Cordeiro, J.V.; Schleich, S.; Newsome, T.P.; Way, M. The release of vaccinia from infected cells requires rhoa-mdia modulation of cortical actin. Cell Host Microbe 2007, 1, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Koizumi, K.; Macrae-Crerar, A.; Gallagher, K.L. Assessing the utility of photoswitchable fluorescent proteins for tracking intercellular protein movement in the arabidopsis root. PLoS ONE 2011, 6, e27536. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.Y.; Hope, T.J. Mobility of human immunodeficiency virus type 1 pr55gag in living cells. J. Virol. 2006, 80, 8796–8806. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Gretton, S.N.; McLauchlan, J.; Targett-Adams, P. Mobility analysis of an NS5A-GFP fusion protein in cells actively replicating hepatitis C virus subgenomic RNA. J. Gen. Virol. 2007, 88, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Sardo, L.; Hatch, S.C.; Chen, J.; Nikolaitchik, O.; Burdick, R.C.; Chen, D.; Westlake, C.J.; Lockett, S.; Pathak, V.K.; Hu, W.S. Dynamics of HIV-1 RNA near the plasma membrane during virus assembly. J. Virol. 2015, 89, 10832–10840. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, G.; Ried, M.U.; Endress, T.; Buning, H.; Hallek, M.; Brauchle, C. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science 2001, 294, 1929–1932. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Pomeranz, L.; Gross, S.P.; Enquist, L.W. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. USA 2004, 101, 16034–16039. [Google Scholar] [CrossRef] [PubMed]
- Willard, M. Rapid directional translocations in virus replication. J. Virol. 2002, 76, 5220–5232. [Google Scholar] [CrossRef] [PubMed]
- Dohner, K.; Wolfstein, A.; Prank, U.; Echeverri, C.; Dujardin, D.; Vallee, R.; Sodeik, B. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 2002, 13, 2795–2809. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Bidirectional transport along microtubules. Curr. Biol. 2004, 14, R525–R537. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Beerli, C.; Pereira, P.M.; Scherer, K.M.; Samolej, J.; Bleck, C.K.; Mercer, J.; Henriques, R. Virusmapper: Open-source nanoscale mapping of viral architecture through super-resolution microscopy. Sci. Rep. 2016, 6, 29132. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| a3_1 | AAGTCGACCGTTGACGCCGAGCAATGC |
| a3_2 | TTGGATCCCGAGAATGAATAAGTACTAAAGG |
| a3_3 | AAGGCGGCCGCGAAGCCGTGGTCAATAGCG |
| a3_4 | TTAAGCTTCGAGAATGAATAAGTACTAAAGG |
| a3_5 | AAAGATCTGTCGACCGTTGACGCCGAGCAATGC |
| a3_6 | GTTAATTCCCGGGGTGTTCATTATTTATATTCGTAGTTTTTAC |
| a3_7 | GTAAAAACTACGAATATAAATAATGAACACCCCGGGAATTAAC |
| den_1 | TTGCGGCCGCCCCACACCTGGCTGGGCAGGGG |
| den_2 | AAAGATCTACCATGAACACCCCGGGAATTAAC |
| den_3 | AAGCGGCCGCCAACACCCCGGGAATTAACCTG |
| den_4 | TTGGATCCTTACCACACCTGGCTGGGCAG |
| b5_1 | GTTCCATAAATTGCTACCG |
| b5_2 | AAAGGATCCTATACCATTAAGTGTATCCATCACC |
| f13_1 | AAAGATCTACCATGTGGCCATTTGCATCG |
| f13_2 | TTGCGGCCGCCAATTTTTAACGATTTACTGTG |
| Name | Sequence |
|---|---|
| f12_for | GAATATCCTGCTCTGATAGCAG |
| f12_rev | GCTAGGGTTATTTGGATGGATGCG |
| a36_for | ATGATGCTGGTACCTATCACG |
| a36_rev | CACGAACAGGGAGATATAGCAC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lynn, H.; Howell, L.M.; Diefenbach, R.J.; Newsome, T.P. Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping. Viruses 2018, 10, 390. https://doi.org/10.3390/v10080390
Lynn H, Howell LM, Diefenbach RJ, Newsome TP. Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping. Viruses. 2018; 10(8):390. https://doi.org/10.3390/v10080390
Chicago/Turabian StyleLynn, Helena, Liam M. Howell, Russell J. Diefenbach, and Timothy P. Newsome. 2018. "Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping" Viruses 10, no. 8: 390. https://doi.org/10.3390/v10080390
APA StyleLynn, H., Howell, L. M., Diefenbach, R. J., & Newsome, T. P. (2018). Phototracking Vaccinia Virus Transport Reveals Dynamics of Cytoplasmic Dispersal and a Requirement for A36R and F12L for Exit from the Site of Wrapping. Viruses, 10(8), 390. https://doi.org/10.3390/v10080390