Drug Delivery Strategies for Antivirals against Hepatitis B Virus


 , ,
, ,  and
and
Abstract
1. Introduction
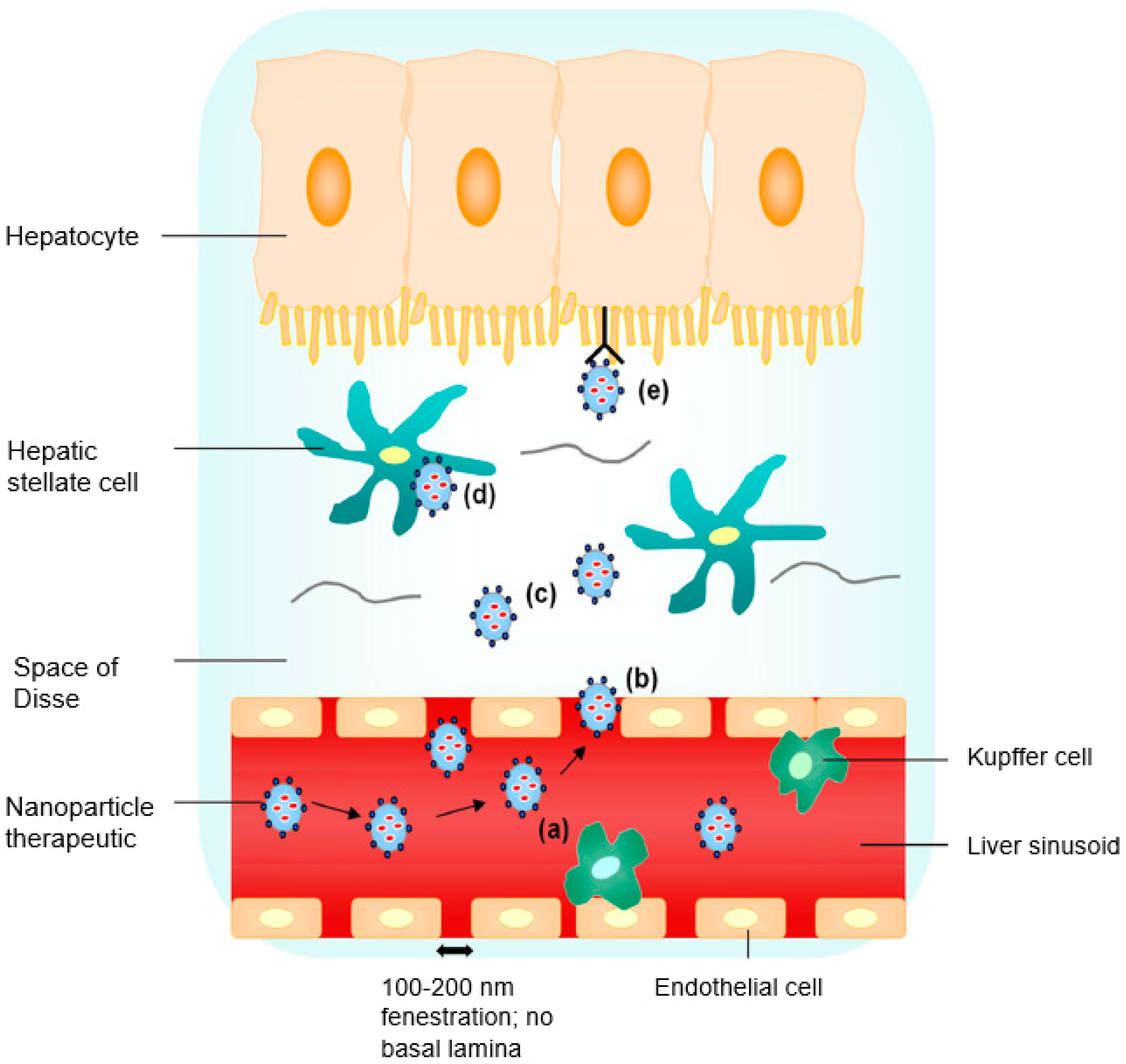
2. Site-Specific Liver Targeting
3. Conventional Anti-HBV Therapies and Drugs
3.1. Cytokines and Nucleot(s)ide Analogues
3.2. Thiazolide Anti-Infectives
3.3. Small Interfering Ribonucleic Acids
3.4. Heteroarylpyrimidines
3.5. Sulfamoyl Benzamide Capsid Assembly Modulators
4. Novel Drug Delivery Strategies for Anti-HBV Therapeutics
4.1. Nanoparticle Systems
4.1.1. Inorganic Nanoparticles
4.1.2. Polymeric Nanoparticles
4.2. Lipids
4.2.1. Ionizable Lipid Nanoparticles
4.2.2. Cationic Lipids
4.2.3. Liposomes
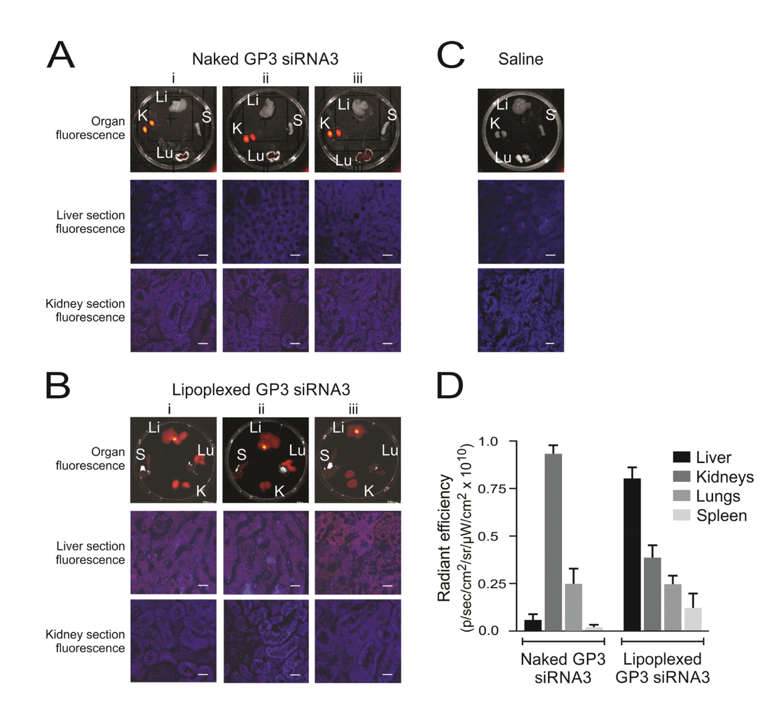
4.2.4. Lipoplexes
4.2.5. High-Density Lipoprotein
4.2.6. Solid Lipid Nanoparticles
4.3. Cell-Penetrating Peptides
4.4. Inhibitors of HBV Attachment
5. Treatment Endpoint for Chronic HBV Infection
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Ghany, M.G.; Perrillo, R.; Li, R.; Belle, S.H.; Janssen, H.L.; Terrault, N.A.; Shuhart, M.C.; Lau, D.T.; Kim, W.R.; Fried, M.W.; et al. Characteristics of adults in the hepatitis B research network in North America reflect their country of origin and hepatitis B virus genotype. Clin. Gastroenterol. Hepatol. 2015, 13, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.-M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Sancho, A.; Sheldon, J.; Soriano, V. Telbivudine: A new option for the treatment of chronic hepatitis B. Expert Opin. Biol. Ther. 2007, 7, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Leucuta, S.E. Subcellular drug targeting, pharmacokinetics and bioavailability. J. Drug Target. 2014, 22, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Cuestas, M.L.; Mathet, V.L.; Oubiña, J.R.; Sosnik, A. Drug delivery systems and liver targeting for improved pharmacotherapy of the hepatitis B virus (HBV) infection. Pharm. Res. 2010, 27, 1184–1202. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, G.; Morell, A.G. The Role of Surface Carbohydrates in the Hepatic Recognition and Transport of Circulating Glycoproteins. In Advances in Enzymology and Related Areas of Molecular Biology; Meister, A., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Stockert, R.J. The asialoglycoprotein receptor: Relationships between structure, function, and expression. Physiol. Rev. 1995, 75, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Lin, C.Y.; Wu, H.T.; Chan, H.S.; Chuang, Y.J.; Chen, C.T.; Lin, C.C. Galactose encapsulated multifunctional nanoparticle for HepG2 cell internalization. Adv. Funct. Mater. 2010, 20, 3948–3958. [Google Scholar] [CrossRef]
- Kato, Y.; Onishi, H.; Machida, Y. Biological characteristics of lactosaminated N-succinyl-chitosan as a liver-specific drug carrier in mice. J. Controll. Release 2001, 70, 295–307. [Google Scholar] [CrossRef]
- Yang, K.W.; Li, X.R.; Yang, Z.L.; Li, P.Z.; Wang, F.; Liu, Y. Novel polyion complex micelles for liver-targeted delivery of diammonium glycyrrhizinate: In vitro and in vivo characterization. J. Biomed. Mater. Res. A 2009, 88, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Liu, Y.; Huang, Y.; Sun, J.; Wu, Z.; Zhang, X.; Ping, Q. Glycyrrhizin surface-modified chitosan nanoparticles for hepatocyte-targeted delivery. Int. J. Pharm. 2008, 359, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Biessen, E.A.; Valentijn, A.R.; De Vreuh, R.L.; Van de Bilt, E.; Sliedregt, L.A.; Prince, P.; Bijsterbosch, M.K.; Van Boom, J.H.; Van Der Marel, G.A.; Abrahams, P.J.; et al. Novel hepatotrophic prodrugs of the antiviral nucleoside 9-(2-phosphonylmethoxyethyl) adenine with improved pharmacokinetics and antiviral activity. FASEB J. 2000, 14, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.W.; Ferry, D.R.; Anderson, D.; Hesslewood, S.; Julyan, P.J.; Poyner, R.; Doran, J.; Young, A.M.; Burtles, S.; Kerr, D.J.; et al. Hepatic drug targeting: Phase I evaluation of polymer-bound doxorubicin. J. Clin. Oncol. 2002, 20, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, H.; Ong, Z.Y.; Xu, K.; Ee, P.L.R.; Zheng, S.; Hedrick, J.L.; Yang, Y.-Y. Polymer- and lipid-based nanoparticle therapeutics for the treatment of liver diseases. Nano Today 2010, 5, 296–312. [Google Scholar] [CrossRef]
- Erion, M.D.; Reddy, K.R.; Boyer, S.H.; Matelich, M.C.; Gomez-Galeno, J.; Lemus, R.H.; Ugarkar, B.G.; Colby, T.J.; Schanzer, J.; Van Poelje, P.D. Design, synthesis, and characterization of a series of cytochrome P450 3A-activated prodrugs (hepDirect prodrugs) useful for targeting phosph(on)ate-based drugs to the liver. J. Am. Chem. Soc. 2004, 126, 5154–5163. [Google Scholar] [CrossRef] [PubMed]
- Kramer, W.; Wess, G.; Schubert, G.; Bickel, M.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Baringhaus, K.H.; Enhsen, A.; Glombik, H.; et al. Liver-specific drug targeting by coupling to bile acids. J. Biol. Chem. 1992, 267, 18598–185604. [Google Scholar] [PubMed]
- Popielarski, S.R.; Hu-Lieskovan, S.; French, S.W.; Triche, T.J.; Davis, M.E. A nanoparticle-based model delivery system to guide the rational design of gene delivery to the liver. 2. In vitro and in vivo uptake results. Bioconj. Chem. 2005, 16, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Popielarski, S.R.; Pun, S.H.; Davis, M.E. A nanoparticle-based model delivery system to guide the rational design of gene delivery to the liver. 1. Synthesis and characterization. Bioconj. Chem. 2005, 16, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Suslov, A.; Wieland, S.; Menne, S. Modulators of innate immunity as novel therapeutics for treatment of chronic hepatitis B. Curr. Opin. Virol. 2018, 30, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tassopoulos, N.C.; Koutelou, M.G.; Polychronaki, H.; Paraloglou-Ioannides, M.; Hadziyannis, S.J. Recombinant interferon-alpha therapy for acute hepatitis B: A randomized, double-blind, placebo-controlled trial. J. Viral Hepat. 1997, 4, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Larrubia, J.R.; Benito-Martínez, S.; Miquel-Plaza, J.; Sanz-de-Villalobos, E.; González-Mateos, F.; Parra, T. Cytokines—Their pathogenic and therapeutic role in chronic viral hepatitis. Rev. Esp. Enferm. Dig. 2009, 101, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Brunetto, M.R.; Oliveri, F.; Coco, B.; Leandro, G.; Colombatto, P.; Gorin, J.M.; Bonino, F. Outcome of anti-HBe positive chronic hepatitis B in alpha-interferon treated and untreated patients: A long term cohort study. J. Hepatol. 2002, 36, 263–270. [Google Scholar] [CrossRef]
- Janssen, H.L.; Gerken, G.; Carreño, V.; Marcellin, P.; Naoumov, N.V.; Craxi, A.; Ring-Larsen, H.; Kitis, G.; van Hattum, J.; de Vries, R.A.; et al. Interferon alfa for chronic hepatitis B infection: Increased efficacy of prolonged treatment, The European Concerted Action on Viral Hepatitis (EUROHEP). Hepatology 1999, 30, 238–243. [Google Scholar] [CrossRef] [PubMed]
- van Zonneveld, M.; Honkoop, P.; Hansen, B.E.; Niesters, H.G.; Darwish Murad, S.; de Man, R.A.; Schalm, S.W.; Janssen, H.L. Long-term follow-up of alpha-interferon treatment of patients with chronic hepatitis B. Hepatology 2004, 39, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Robek, M.D.; Boyd, B.S.; Chisari, F.V. Lambda interferon inhibits hepatitis B and C virus replication. J. Virol. 2005, 79, 3851–3854. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F. Emerging drugs for hepatitis B. Expert Opin. Emerg. Drugs 2007, 12. [Google Scholar] [CrossRef] [PubMed]
- Dawood, A.; Basit, S.A.; Jayaraj, M.; Gish, R.G. Drugs in Development for Hepatitis B. Drugs 2017, 77, 1263–1280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Littler, E. Nucleoside analogs as anti-HBV agents. Curr. Top. Med. Chem. 2006, 6, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Scala, M.D.; Korolowicz, K.; Thampi, L.M.; Otano, I.; Suarez, L.; Fioravanti, J.; Aranda, F.; Ardaiz, N.; Yang, J.; et al. Liver-directed gene therapy of chronic hepadnavirus infection using interferon alpha tethered to apolipoprotein A-I. J. Hepat. 2015, 63, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T. Immunoregulation of hepatitis B virus infection--rationale and clinical application. Nagoya J. Med. Sci. 2012, 74, 217–232. [Google Scholar] [PubMed]
- De Clercq, E.; Férir, G.; Kaptein, S.; Neyts, J. Antiviral treatment of chronic hepatitis B virus (HBV) infections. Viruses 2010, 2, 1279–1305. [Google Scholar] [CrossRef] [PubMed]
- Dienstag, J.L.; Schiff, E.R.; Wright, T.L.; Perrillo, R.P.; Hann, H.W.; Goodman, Z.; Crowther, L.; Condreay, L.D.; Woessner, M.; Rubin, M.; et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N. Engl. J. Med. 1999, 341, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.; Wang, H.; Niu, J.; Chim, A.M.; Sung, J.J. Two-year lamivudine treatment for hepatitis B e antigen-negative chronic hepatitis B: A double-blind, placebo-controlled trial. Antivir. Ther. 2007, 12, 345–353. [Google Scholar] [PubMed]
- Hadziyannis, S.J.; Papatheodoridis, G.V. Adefovir dipivoxil in the treatment of chronic hepatitis B virus infection. Expert Rev. Anti-Infect. Ther. 2004, 2, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.Y.; Chuang, W.L.; Hsieh, M.Y.; Lee, L.P.; Huang, J.F.; Hou, N.J.; Lin, Z.Y.; Chen, S.C.; Hsieh, M.Y.; Wang, L.Y.; et al. Adefovir dipivoxil treatment of lamivudine-resistant chronic hepatitis B. Antivir. Res. 2007, 75, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Chang, T.T.; Lim, S.G.; Tong, M.J.; Sievert, W.; Shiffman, M.L.; Jeffers, L.; Goodman, Z.; Wulfsohn, M.S.; Xiong, S.; et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N. Engl. J. Med. 2003, 348, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Miao, J.; Li, M.; Jiang, S.; Hu, F.; Du, Y. Solid lipid nanoparticles loading adefovir dipivoxil for antiviral therapy. J. Zhejiang Univ. Sci. B 2008, 9, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Brown, R., Jr. Entecavir for treatment of chronic hepatitis B: A clinical update for the treatment of patients with decompensated cirrhosis. OJIM 2012, 2, 53–61. [Google Scholar] [CrossRef][Green Version]
- Zoutendijk, R.; Reijnders, J.G.; Brown, A.; Zoulim, F.; Mutimer, D.; Deterding, K.; Petersen, J.; Hofmann, W.P.; Buti, M.; Santantonio, T.; et al. Entecavir treatment for chronic hepatitis B: Adaptation is not needed for the majority of naïve patients with a partial virological response. Hepatology 2011, 54, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Pol, S.; Lampertico, P. First-line treatment of chronic hepatitis B with entecavir or tenofovir in ‘real-life’ settings: From clinical trials to clinical practice. J. Viral Hepat. 2012, 19, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Q.; Ding, Y.P.; Dong, Y.H. Telbivudine treatment is associated with high hepatitis B e antigen seroconversion and immune modulatory effects in chronic hepatitis B patients. J. Viral Hepat. 2013, 20, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Wursthorn, K.; Jung, M.; Riva, A.; Goodman, Z.D.; Lopez, P.; Bao, W.; Manns, M.P.; Wedemeyer, H.; Naoumov, N.V. Kinetics of hepatitis B surface antigen decline during 3 years of telbivudine treatment in hepatitis B e antigen-positive patients. Hepatology 2010, 52, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Korba, B.E.; Elazar, M.; Lui, P.; Rossignol, J.F.; Glenn, J.S. Potential for hepatitis C virus resistance to nitazoxanide or tizoxanide. Antimicrob. Agents Chemother. 2008, 52, 4069–4071. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; Keeffe, E.B. Thiazolides: A new class of drugs for the treatment of chronic hepatitis B and C. Future Microbiol. 2008, 3, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Keeffe, E.B.; Rossignol, J.F. Treatment of chronic viral hepatitis with nitazoxanide and second generation thiazolides. World J. Gastroenterol. 2009, 15, 1805–1808. [Google Scholar] [CrossRef] [PubMed]
- Korba, B.E.; Montero, A.B.; Farrar, K.; Gaye, K.; Mukerjee, S.; Ayers, M.S.; Rossignol, J.F. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antivir. Res. 2008, 77, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Y.; Chen, H.-S. Emerging antivirals for the treatment of hepatitis B. World J. Gastroenterol. 2014, 20, 7707–7717. [Google Scholar] [CrossRef] [PubMed]
- Stachulski, A.V.; Pidathala, C.; Row, E.C.; Sharma, R.; Berry, N.G.; Iqbal, M.; Bentley, J.; Allman, S.A.; Edwards, G.; Helm, A.; et al. Thiazolides as novel antiviral agents. 1. Inhibition of hepatitis B virus replication. J. Med. Chem. 2011, 54, 4119–4132. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F. Thiazolides: A new class of antiviral drugs. Expert Opin. Drug Metab. Toxicol. 2009, 5, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Rozema, D.B.; Lewis, D.L.; Wakefield, D.H.; Wong, S.C.; Klein, J.J.; Roesch, P.L.; Bertin, S.L.; Reppen, T.W.; Chu, Q.; Blokhin, A.V.; et al. Dynamic polyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 12982–12987. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Jorgensen, M.R.; Kolli, S.; Crowther, C.; Salazar, F.H.; Marion, P.L.; Fujino, M.; Natori, Y.; Thanou, M.; Arbuthnot, P.; et al. Controlling HBV replication in vivo by intravenous administration of triggered PEGylated siRNA-nanoparticles. Mol. Pharm. 2008, 6, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Feng, S.; Wang, S.; Chen, Z. Evaluation of cationic nanoparticles of biodegradable copolymers as siRNA delivery system for hepatitis B treatment. Int. J. Pharm. 2010, 400, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Tang, Z.; Gao, X.; Zhao, F.; Zhong, H.; Wen, M.R.; Sun, X.; Song, H.F.; Qian, X.H. Optimal design and validation of antiviral siRNA for targeting hepatitis B virus. Acta Pharmacol. Sin. 2008, 29, 1522–1528. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.; Cheng, G.; Mahato, R.I. RNAi for treating hepatitis B viral infection. Pharm. Res. 2009, 25, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Giladi, H.; Ketzinel-Gilad, M.; Rivkin, L.; Felig, Y.; Nussbaum, O.; Galun, E. Small interfering RNA inhibits hepatitis B virus replication in mice. Mol. Ther. 2003, 8, 769–776. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, Z.; Ye, J.; Yao, H.; Zheng, S.; Ding, J. Combination of small interfering RNAs mediates greater inhibition of human hepatitis B virus replication and antigen expression. J. Zhejiang Univ. Sci. B 2005, 6, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, D.V.; Lee, P.A.; Johnson, D.A.; Overly, S.L.; McSwiggen, J.A.; Beigelman, L.; Mokler, V.R.; Maloney, L.; Vargeese, C.; Bowman, K.; et al. Characterization of nuclease-resistant ribozymes directed against hepatitis B virus RNA. J. Viral Hepat. 2002, 9, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Shin, D.; Choi, T.H.; Lee, J.C.; Cheon, G.J.; Kim, K.Y.; Park, M.; Kim, M. Systemic and specific delivery of small interfering RNAs to the liver mediated by apolipoprotein A-I’. Mol. Ther. 2007, 15, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, J.O.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Uprichard, S.L.; Boyd, B.; Althage, A.; Chisari, F.V. Clearance of hepatitis B virus from the liver of transgenic mice by short hairpin RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Wooddell, C.I.; Chavez, D.; Oropeza, C.; Chu, Q.; Hamilton, H.L.; McLachlan, A.; Given, B.; Anzalone, C.R.; Lewis, D.L. ARC-520 RNAi therapeutic reduces hepatitis B virus DNA, S antigen and e antigen in a chimpanzee with a very high viral titer. Hepatology 2013, 58, 1305. [Google Scholar]
- Stray, S.J.; Bourne, C.R.; Punna, S.; Lewis, W.G.; Finn, M.G.; Zlotnick, A. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 8138–8143. [Google Scholar] [CrossRef] [PubMed]
- Bourne, C.; Lee, S.; Venkataiah, B.; Lee, A.; Korba, B.; Finn, M.G.; Zlotnick, A. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J. Virol. 2008, 82, 10262–10270. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.B.; Han, J.C.; Wei, L.; Peng, D.D.; Gao, Y. Subcellular distribution and translocation of hepatitis B virus core protein in HepG2.2.15 cells. Zhonghua Gan Zang Bing Za Zhi 2008, 16, 29–32. [Google Scholar] [PubMed]
- Haryanto, A.; Wijayanti, N.; Kann, M. Effect of the HBV capsid assembly inhibitor Bayer 41–4109 on the intracellular localization of EGFP-core fusion proteins. Indones. J. Biotechnol. 2007, 12, 998–1004. [Google Scholar] [CrossRef]
- Wang, X.Y.; Wei, Z.M.; Wu, G.Y.; Wang, J.H.; Zhang, Y.J.; Li, J.; Zhang, H.H.; Xie, X.W.; Wang, X.; Wang, Z.H.; et al. In vitro inhibition of HBV replication by a novel compound, GLS4, and its efficacy against adefovirdipivoxil-resistant HBV mutations. Antivir Ther. 2012, 17, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.G. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr. Opin. Pharmacol. 2016, 30, 131–137. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, W.A.; Ramiz, M.M.; Abdel-Rahman, A.A. Anti-hepatitis B virus activity of new N4-beta-d-glycoside Pyrazolo [3,4-d]pyrimidine derivatives. Z. Naturforsch. C 2009, 64, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Kawakami, H.; Inoue, T.; Katsukura, Y.; Obuchi, W.; Uchida, Y.; Kamiie, J.; Horie, T. Validation of uPA/SCID mouse with humanized liver as a human liver model: Protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases by LC–MS/MS. Drug Metab. Dispos. 2014, 42, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Park, J.O.; Stephen, Z.; Sun, C.; Veiseh, O.; Kievit, F.M.; Fang, C.; Leung, M.; Mok, H.; Zhang, M. Glypican-3 targeting of liver cancer cells using multifunctional nanoparticles. Mol. Imaging 2011, 10, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Dekhtyar, Y.; Romanova, M.; Kachanovska, A.; Skrastina, D.; Reinhofa, R.; Pumpens, P.; Patmalnieks, A. Inorganic nanoparticle as a carrier for hepatitis b viral capsids. In Technological Innovations in Sensing and Detection of Chemical, Biological, Radiological, Nuclear Threats and Ecological Terrorism; Vaseashta, A., Braman, E., Susmann, P., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 221–225. [Google Scholar]
- Yang, W.; Mou, T.; Shao, G.; Wang, F.; Zhang, X.; Liu, B. Copolymer-based hepatocyte asialoglycoprotein receptor targeting agent for SPECT. J. Nucl. Med. 2011, 52, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Su, J.; Cai, W.; Lu, P.; Yuan, L.; Jin, T.; Chen, S.; Sheng, J. Hepatocyte-targeting gene transfer mediated by galactosylated poly(ethylene glycol)-graft-polyethylenimine derivative. Drug Des. Dev. Ther. 2013, 7, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Giri, N.; Tomar, P.; Karwasara, V.S.; Pandey, R.S.; Dixit, V.K. Targeted novel surface-modified nanoparticles for interferon delivery for the treatment of hepatitis B. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Rawat, A.; Hope-Weeks, L.; Ahsan, F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol. Pharm. 2011, 8, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Xu, Y.; Zeng, C.; Ren, H.; Peng, M. Chitosan-modified poly(d,l-lactide-co-glycolide) nanospheres for plasmid DNA delivery and HBV gene-silencing. Int. J. Pharm. 2011, 415, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Du, Y.Z.; Yuan, H.; Zhang, X.G.; Miao, J.; Cui, F.D.; Hu, F.Q. Synthesis of lamivudine stearate and antiviral activity of stearic acid-g-chitosan oligosaccharide polymeric micelles delivery system. Eur. J. Pharm. Sci. 2010, 41, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.T.; Du, Y.Z.; Yuan, H.; Zhang, X.G.; Miao, J.; Cui, F.D.; Hu, F.Q. Synthesis and anti-hepatitis B virus activity of acyclovir conjugated stearic acid-g-chitosan oligosaccharide micelle. Carbohydr. Polym. 2011, 83, 1715–1722. [Google Scholar] [CrossRef]
- Tian, Q.; Wang, X.H.; Wang, W.; Zhang, C.N.; Wang, P.; Yuan, Z. Self-assembly and liver targeting of sulfated chitosan nanoparticles functionalized with glycyrrhetinic acid. Nanomedicine 2012, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Mével, M.; Kamaly, N.; Carmona, S.; Oliver, M.H.; Jorgensen, M.R.; Crowther, C.; Salazar, F.H.; Marion, P.L.; Fujino, M.; Natori, Y.; et al. DODAG; a versatile new cationic lipid that mediates efficient delivery of pDNA and siRNA. J. Controll. Release 2010, 143, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Morrey, J.D.; Motter, N.E.; Chang, S.; Fairman, J. Breaking B and T cell tolerance using cationic lipid--DNA complexes (CLDC) as a vaccine adjuvant with hepatitis B virus (HBV) surface antigen in transgenic mice expressing HBV. Antivir. Res. 2011, 90, 227–230. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.; Zhang, Q.; Peng, Q.; Zhou, J.; Liao, L.; Sun, X.; Zhang, L.; Gong, T. Hepatitis B virus preS1-derived lipopeptide functionalized liposomes for targeting of hepatic cells. Biomaterials 2014, 35, 6130–6141. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.; Korsholm, K.S.; Andersen, P.; Agger, E.M. Cationic liposomes as vaccine adjuvants. Expert Rev. Vaccines 2011, 10, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, K.; Nakashima, Y.; Aramaki, Y.; Hirayama, F.; Uekama, K.; Arima, H. In vitro gene delivery mediated by asialofetuin-appended cationic liposomes associated with γ-cyclodextrin into hepatocytes. J. Drug Deliv. 2011, 476137. [Google Scholar] [CrossRef]
- Uhl, P.; Helm, F.; Hofhaus, G.; Brings, S.; Kaufman, C.; Leotta, K.; Urban, S.; Haberkorn, U.; Mier, W.; Fricker, G. A liposomal formulation for the oral application of the investigational hepatitis B drug Myrcludex B. Eur. J. Pharm. Biopharm. 2016, 103, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Hean, J.; Crowther, C.; Ely, A.; Ul Islam, R.; Barichievy, S.; Bloom, K.; Weinberg, M.S.; van Otterlo, W.A.; de Koning, C.B.; Salazar, F.; et al. Inhibition of hepatitis B virus replication in vivo using lipoplexes containing altritol-modified antiviral siRNAs. Artif. DNA PNA XNA 2010, 1, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Marimani, M.D.; Ely, A.; Buff, M.C.R.; Bernhardt, S.; Engels, J.W.; Scherman, D.; Escriou, V.; Arbuthnot, P. Inhibition of replication of hepatitis B virus in transgenic mice following administration of hepatotropic lipoplexes containing guanidinopropyl-modified siRNAs. J. Controll. Release 2015, 209, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Rui, M.; Guo, W.; Ding, Q.; Wei, X.; Xu, J.; Xu, Y. Recombinant high-density lipoprotein nanoparticles containing gadolinium-labeled cholesterol for morphologic and functional magnetic resonance imaging of the liver. Int. J. Nanomed. 2012, 7, 3751–3768. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, W.; Wang, B.; Zhu, H.; Zhang, B.; Feng, M. Delivery of hydrophilic drug doxorubicin hydrochloride-targeted liver using apoAI as carrier. J. Drug Target. 2013, 21, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.; Mishra, D.; Mishra, P.K.; Nahar, M.; Dubey, V.; Jain, N.K. Evaluation of solid lipid nanoparticles as carriers for delivery of hepatitis B surface antigen for vaccination using subcutaneous route. J. Pharm. Pharm. Sci. 2010, 13, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.H.; Park, K.; Lee, M.Y.; Lee, H.; Sung, D.K.; Hahn, S.K. Cationic solid lipid nanoparticles derived from apolipoprotein-free LDLs for target specific systemic treatment of liver fibrosis. Biomaterials 2013, 34, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Xun, Y.; Pan, Q.; Tang, Z.; Chen, X.; Yu, Y.; Xi, M.; Zang, G. Intracellular-delivery of a single-chain antibody against hepatitis B core protein via cell-penetrating peptide inhibits hepatitis B virus replication in vitro. Int. J. Mol. Med. 2013, 31, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.B.; Wei, L.; Han, J.C.; Ma, H.; Deng, K.; Cong, X. Artificial recombinant cell-penetrating peptides interfere with envelopment of hepatitis B virus nucleocapsid and viral production. Antivir. Res. 2011, 89, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Montrose, K.; Yang, Y.; Krissansen, G.W. X-pep, a novel cell-penetrating peptide motif derived from the hepatitis B virus. Biochem. Biophys. Res. Commun. 2014, 1, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Urban, S. Inhibitors of Hepatitis B Virus Attachment and Entry. Intervirology 2014, 57, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.; Allweiss, L.; Ben, M.; Barek, M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Chotiyaputta, W.; Lok, A.S.F. Endpoints of hepatitis B treatment. J. Viral Hepat. 2010, 17, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.K.; Lo, A.S.F. Antiviral options for the treatment of chronic hepatitis B. J. Antimicrob. Chemother. 2006, 57, 1030–1034. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agent | Effectiveness as Anti-HBV Therapy | Reference |
|---|---|---|
| IFN-α/-PEG/-λ | Suppressed HBV viral copying, restored T-helper lymphocyte responses/immunoregulatory, allows for anti-HbeAg seroconversion, prolonged decline in the progression of HBV, prolonged diminished growth of HCC, sustained clinical subsidence and HBsAG seroconversion, subdued HCV replication | [20,21,22,23,24] |
| LMV | T-helper lymphocyte response restored, HBV DNA level decline, rapid HBeAg removal, decreased levels of serum ALT, minimal risk of developing liver cirrhosis and HCC, improved liver histology, sustained HBeAg responses after treatment | [26,30,31,32,33] |
| ADV | Decreased HBV DNA levels, improved serum liver histology, treatment of LMV resistance, decreased ALT, increased HBeAg seroconversion, decreased HBsAg and HBeAg levels, prolonged therapeutic efficacy | [34,35,36,37] |
| ETV | Potent HBV inhibition, minimal drug-resistance progression, effective long-term, precipitates virological responses, favorable side-effect profile | [26,38,39] |
| TDF | Potent HBV inhibition, decreased HBV viral load, more effective therapy compared to ADV, sustained virologic responses, favorable safety profile, prolonged liver cirrhosis reversal | [26,36,40] |
| Telbivudine | Highest HBeAg seroconversion rate, immunomodulation, rapid decrease in HBsAG levels, possible complete removal of HBsAg | [41,42] |
| Therapeutic Agent | Effectiveness as Anti-HBV Therapy | Reference |
|---|---|---|
| Thiazolides | Potent and selective HBV replication inhibition, HBeAg and HBsAg seroconversion, decreased HBV DNA levels, treatment in LMV- and ADV-resistance, decreased Hep2.2.15 cell HBV proteins, no HBV RNA transcription effect | [43,44,45,46,48,49] |
| siRNAs | Gene expression inhibited, viral antigen inhibition, HBV transcript inhibition, decreased serum HBV DNA and RNA levels, suppressed HBV replication markers, mRNA cleavage, polymerase and precore region targeting, effective continuance of treatment | [50,51,54,60] |
| HAPs | Potent HBV capsid maturing inhibition, HBV core protein degradation, increased potency compared to LMV, potent antigen inhibition effect, misassembly of HBV capsid proteins, dose-dependent decrease in HBV DNA levels, decreased HBcAg | [5,62,63,64,65,68] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, L.; Indermun, S.; Govender, M.; Kumar, P.; Du Toit, L.C.; Choonara, Y.E.; Pillay, V. Drug Delivery Strategies for Antivirals against Hepatitis B Virus. Viruses 2018, 10, 267. https://doi.org/10.3390/v10050267
Singh L, Indermun S, Govender M, Kumar P, Du Toit LC, Choonara YE, Pillay V. Drug Delivery Strategies for Antivirals against Hepatitis B Virus. Viruses. 2018; 10(5):267. https://doi.org/10.3390/v10050267
Chicago/Turabian StyleSingh, Latavia, Sunaina Indermun, Mershen Govender, Pradeep Kumar, Lisa C. Du Toit, Yahya E. Choonara, and Viness Pillay. 2018. "Drug Delivery Strategies for Antivirals against Hepatitis B Virus" Viruses 10, no. 5: 267. https://doi.org/10.3390/v10050267
APA StyleSingh, L., Indermun, S., Govender, M., Kumar, P., Du Toit, L. C., Choonara, Y. E., & Pillay, V. (2018). Drug Delivery Strategies for Antivirals against Hepatitis B Virus. Viruses, 10(5), 267. https://doi.org/10.3390/v10050267