Mycoviruses as Triggers and Targets of RNA Silencing in White Mold Fungus Sclerotinia sclerotiorum

Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strains and Culture Conditions
2.2. Construction of dcl-1, dcl-2 and dcl-1/dcl-2 Null Alleles
2.3. Fungal Transformation
2.4. Complementation of dcl-1
2.5. Phenotypic Characterization of Gene Deletion Mutants
2.6. Virulence Assay of Gene Deletion Mutants
2.7. Transfection of Mutants with In Vitro Transcripts of SsHV2
2.8. Construction of An Infectious Clone of SsHADV-1 and Transfection of Mutants with SsHADV-1
2.9. Preparation of Small RNA Libraries and Sequencing Analysis
3. Results
3.1. Generation of Disruption Mutants for Dicer Genes
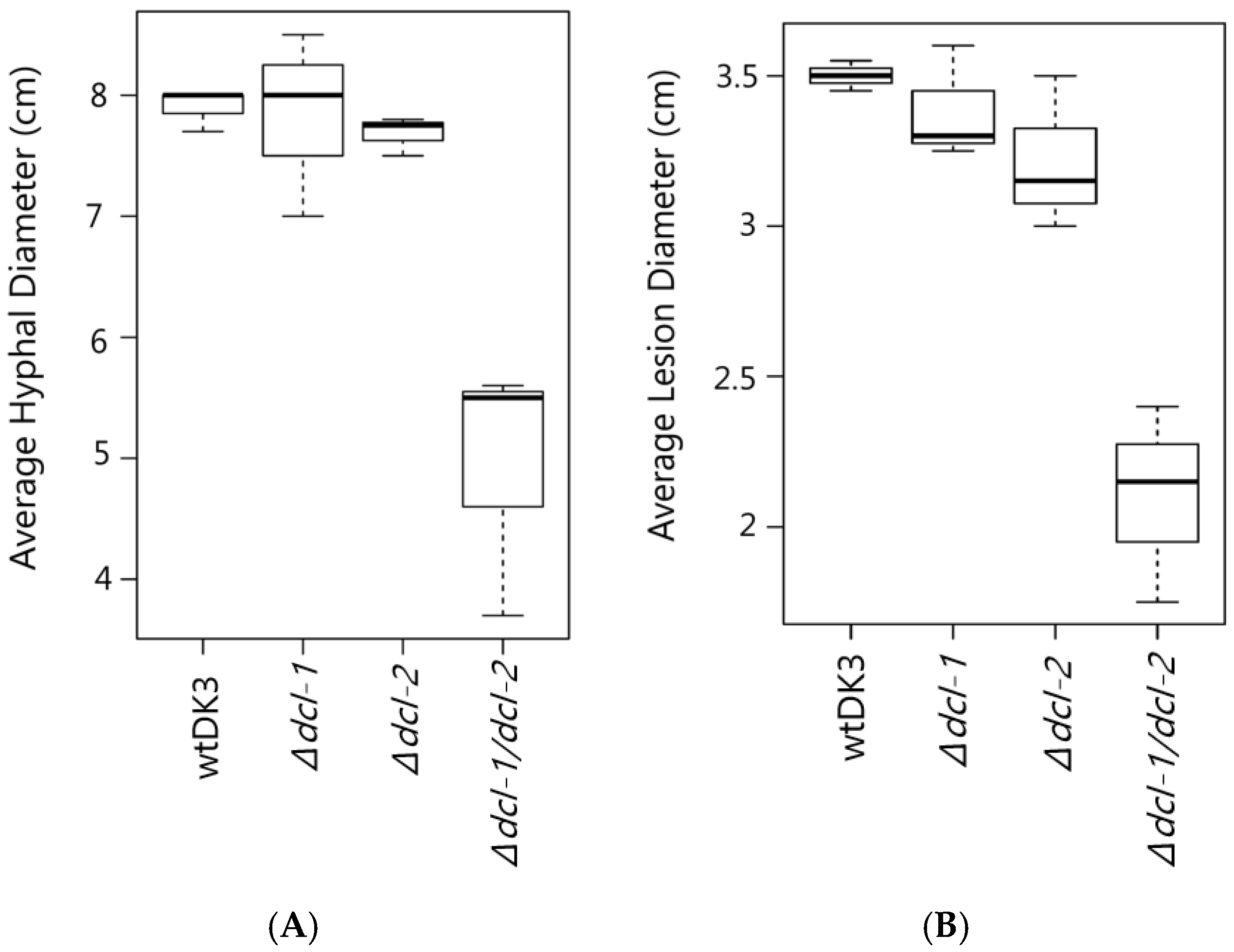
3.2. Effect of Dicer Gene Disruption on S. sclerotiorum Phenotype
3.3. Effects of Dicer Gene Disruptions on S. sclerotiorum Pathogenicity
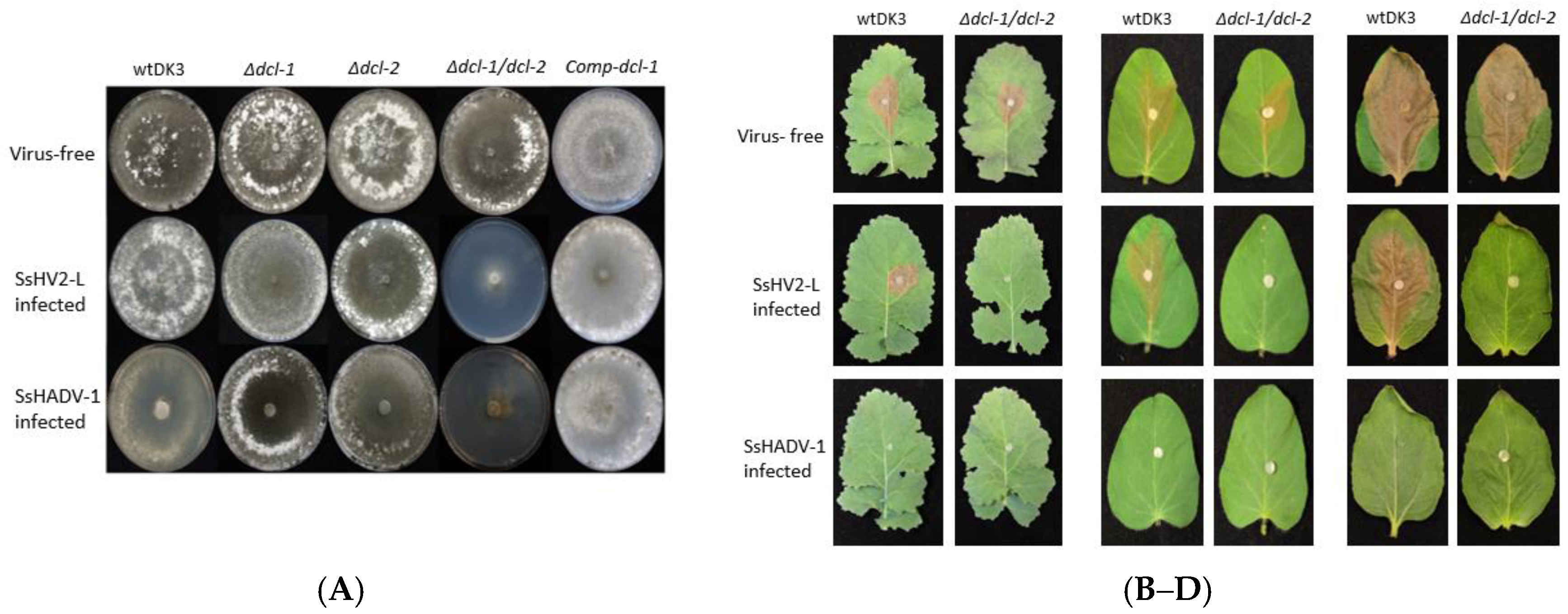
3.4. Transfection of Dicer Gene Deletion Mutants with SsHV2-L or SsHADV-1 Viruses Consistently Results in Severe Debilitation in the Δdcl-1/dcl-2 Mutant
3.5. Infectious Clone of SsHADV-1 Causes Severe Debilitation and Significantly Reduced Virulence in wtDK3 at Lower Temperatures
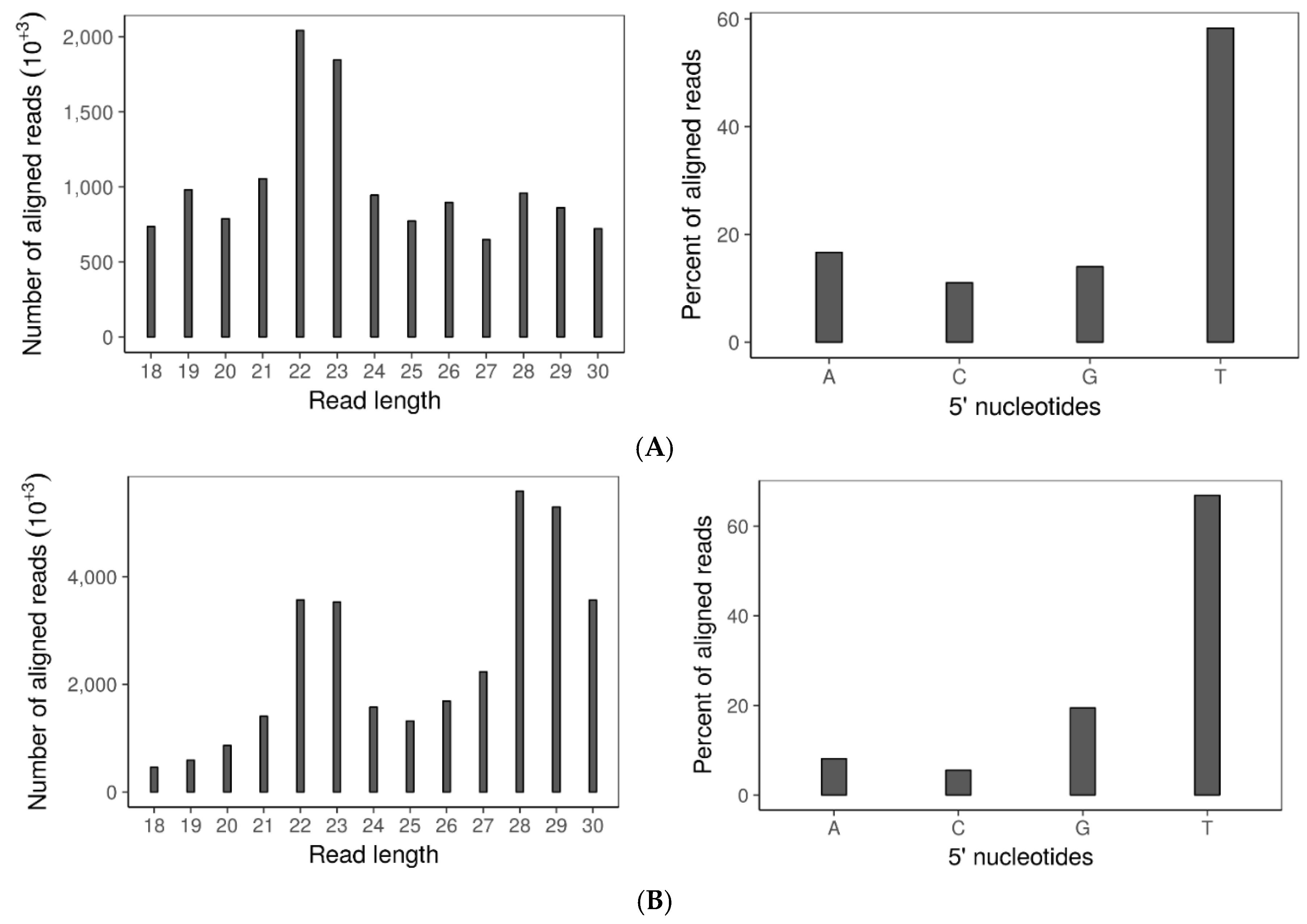
3.6. Double Dicer Disruption Mutant Has Reduced 21–24 nt sRNA Accumulation
3.7. SsHADV-1 and SsHV2-L Are both Processed by Virus-Infected wtDK3
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baulcombe, D. RNA silencing. Trends Biochem. Sci. 2005, 30, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.M.; Wang, M.B.; Lough, T. Gene silencing as an adaptive defence against viruses. Nature 2001, 411, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Baulcombe, D. RNA silencing in plants. Nature 2004, 431, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Campo, S.; Gilbert, K.B.; Carrington, J.C. Small RNA-based antiviral defense in the phytopathogenic fungus Colletotrichum higginsianum. PLoS Pathog. 2016, 12, e1005640. [Google Scholar] [CrossRef] [PubMed]
- Segers, G.C.; Zhang, X.; Deng, F.; Sun, Q.; Nuss, D.L. Evidence that RNA silencing functions as an antiviral defense mechanism in fungi. Proc. Natl. Acad. Sci. USA 2007, 104, 12902–12906. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Pallotta, M.; ReFalo, P.; Sachs, M.S.; Vayssie, L.; Macino, G.; Cogoni, C. Redundancy of the two dicer genes in transgene-induced posttranscriptional gene silencing in Neurospora crassa. Mol. Cell. Biol. 2004, 24, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Schmitt, M.J. Yeast Killer Toxin K28: Biology and Unique Strategy of Host Cell Intoxication and Killing. Toxins 2017, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Laurie, J.D.; Ali, S.; Linning, R.; Mannhaupt, G.; Wong, P.; Güldener, U.; Münsterkötter, M.; Moore, R.; Kahmann, R.; Bakkeren, G.; et al. Genome comparison of barley and maize smut fungi reveals targeted loss of RNA silencing components and species-specific presence of transposable elements. Plant Cell 2012, 24, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W. RNAi: The nuts and bolts of the RISC machine. Cell 2005, 122, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Amselem, J.; Cuomo, C.A.; Van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; De Vries, R.P.; Dyer, P.S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Ghabrial, S.A.; Jiang, D.; Varsani, A. Genomoviridae: A new family of widespread single-stranded DNA viruses. Arch. Virol. 2016, 161, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Marzano, S.Y.L.; Hobbs, H.A.; Nelson, B.D.; Hartman, G.L.; Eastburn, D.M.; McCoppin, N.K.; Domier, L.L. Transfection of Sclerotinia sclerotiorum with in vitro transcripts of a naturally occurring interspecific recombinant of Sclerotinia sclerotiorum hypovirus 2 significantly reduces virulence of the fungus. J. Virol. 2015, 89, 5060–5071. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weiberg, A.; Lin, F.M.; Thomma, B.P.; Huang, H.D.; Jin, H. Bidirectional cross-kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nat. Plants 2016, 2, 16151. [Google Scholar] [CrossRef] [PubMed]
- Weiberg, A.; Wang, M.; Lin, F.M.; Zhao, H.; Zhang, Z.; Kaloshian, I.; Huang, H.D.; Jin, H. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 2013, 342, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.X.; Domier, L.L.; Radwan, O.; Yendrek, C.R.; Hudson, M.E.; Hartman, G.L. Identification of multiple phytotoxins produced by Fusarium virguliforme including a phytotoxic effector (FvNIS1) associated with sudden death syndrome foliar symptoms. Mol. Plant-Microbe Interact. 2016, 29, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Staben, C.; Jensen, B.; Singer, M.; Pollock, J.; Schechtman, M.; Kinsey, J.; Selker, E. Use of a bacterial hygromycin B resistance gene as a dominant selectable marker in Neurospora crassa transformation. Fungal Genet. Rep. 1989, 36, 79. [Google Scholar] [CrossRef]
- Nelson, M.D.; Fitch, D.H. Overlap extension PCR: An efficient method for transgene construction. In Molecular Methods for Evolutionary Genetics; Springer: Berlin, Germany, 2012; pp. 459–470. [Google Scholar]
- Ge, C.Y.; Duan, Y.B.; Zhou, M.G.; Chen, C.J. A Protoplast Transformation System for Gene Deletion and Complementation in Sclerotinia sclerotiorum. J. Phytopathol. 2013, 161, 800–806. [Google Scholar] [CrossRef]
- Rollins, J.A. Sclerotinia sclerotiorum pac1 Gene Is Required for Sclerotial Development and Virulence. Mol. Plant-Microbe Interact. 2003, 16, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.R.; Lee, M.H. Split-Marker-Mediated Transformation and Targeted Gene Disruption in Filamentous Fungi. In Genetic Transformation Systems in Fungi; Springer: Berlin, Germany, 2015; Volume 2, pp. 175–180. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu, Z.; Li, Y.; Wu, J. Biogenesis, function, and applications of virus-derived small RNAs in plants. Front. Microbiol. 2015, 6, 1237. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Csorba, T.; Kontra, L.; Burgyán, J. Viral silencing suppressors: Tools forged to fine-tune host-pathogen coexistence. Virology 2015, 479, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Segers, G.C.; Sun, Q.; Deng, F.; Nuss, D.L. Characterization of hypovirus-derived small RNAs generated in the chestnut blight fungus by an inducible DCL-2-dependent pathway. J. Virol. 2008, 82, 2613–2619. [Google Scholar] [CrossRef] [PubMed]
- Shahid, S.; Kim, G.; Johnson, N.R.; Wafula, E.; Wang, F.; Coruh, C.; Bernal-Galeano, V.; Phifer, T.; Westwood, J.H.; Axtell, M.J. MicroRNAs from the parasitic plant Cuscuta campestris target host messenger RNAs. Nature 2018, 553, 82. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Tripp, J.; Lozoya-Gloria, E.; Rivera-Bustamante, R.F. Symptom remission and specific resistance of pepper plants after infection by Pepper golden mosaic virus. Phytopathology 2007, 97, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Guo, W.; Xie, Y.; Xie, Q.; Fan, L.; Zhou, X. Characterization of small interfering RNAs derived from the geminivirus/betasatellite complex using deep sequencing. PLoS ONE 2011, 6, e16928. [Google Scholar] [CrossRef] [PubMed]
- Kurzynska-Kokorniak, A.; Koralewska, N.; Pokornowska, M.; Urbanowicz, A.; Tworak, A.; Mickiewicz, A.; Figlerowicz, M. The many faces of Dicer: The complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res. 2015, 43, 4365–4380. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Niwa, M.; Walter, P.; Silverman, R.H. Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. RNA 2001, 7, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Halary, S.; Duraisamy, R.; Fancello, L.; Monteil-Bouchard, S.; Jardot, P.; Biagini, P.; Gouriet, F.; Raoult, D.; Desnues, C. Novel single-stranded DNA circular viruses in pericardial fluid of patient with recurrent pericarditis. Emerg. Infect. Dis. 2016, 22, 1839. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M. Networks of evolutionary interactions underlying the polyphyletic origin of ssDNA viruses. Curr. Opin. Virol. 2013, 3, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xie, J.; Cheng, J.; Li, B.; Chen, T.; Fu, Y.; Li, G.; Wang, M.; Jin, H.; Wan, H.; et al. Fungal DNA virus infects a mycophagous insect and utilizes it as a transmission vector. Proc. Natl. Acad. Sci. USA 2016, 113, 12803–12808. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, B.; Hussain, M.; Asgari, S. RNA interference as a cellular defense mechanism against the DNA virus baculovirus. J. Virol. 2012, 86, 13729–13734. [Google Scholar] [CrossRef] [PubMed]
- Hanley-Bowdoin, L.; Bejarano, E.R.; Robertson, D.; Mansoor, S. Geminiviruses: Masters at redirecting and reprogramming plant processes. Nat. Rev. Microbiol. 2013, 11, 777. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, J.; Han, Y.; Fan, X.; Ding, S.W. RNA interference functions as an antiviral immunity mechanism in mammals. Science 2013, 342, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Xu, Y.; Zhang, Y.; Zhou, H.; Deng, Y.Q.; Li, X.F.; Miao, M.; Zhang, Q.; Zhong, B.; Hu, Y. Human virus-derived small RNAs can confer antiviral immunity in mammals. Immunity 2017, 46, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Hocquemiller, M.; Giersch, L.; Audrain, M.; Parker, S.; Cartier, N. Adeno-associated virus-based gene therapy for CNS diseases. Hum. Gene Ther. 2016, 27, 478–496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhang, S.; Gong, Q.; Hao, A. A novel gemycircularvirus in an unexplained case of child encephalitis. Virol. J. 2015, 12, 197. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Messacar, K.; Dominguez, S.R.; Da Costa, A.C.; Deng, X.; Delwart, E. A new densovirus in cerebrospinal fluid from a case of anti-NMDA-receptor encephalitis. Arch. Virol. 2016, 161, 3231–3235. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SsHADV-1 | 5′-terminal mismatch (%) | 3′-terminal mismatch (%) | ||||||
| vsiRNA Sequence length | A | C | G | T | A | C | G | T |
| 18 | 16.9 | 1.9 | 1.1 | 0.8 | 18.2 | 5.0 | 3.0 | 14.6 |
| 19 | 4.2 | 1.1 | 1.0 | 2.8 | 21.0 | 7.3 | 3.9 | 19.6 |
| 20 | 10.3 | 0.8 | 0.8 | 1.1 | 24.5 | 4.8 | 3.2 | 22.4 |
| 21 | 5.0 | 0.6 | 0.8 | 1.6 | 27.9 | 3.2 | 4.7 | 22.4 |
| 22 | 26.8 | 0.8 | 0.8 | 1.0 | 20.2 | 3.0 | 2.7 | 12.3 |
| 23 | 46.1 | 0.6 | 0.9 | 0.7 | 12.5 | 2.5 | 1.5 | 9.1 |
| 24 | 5.9 | 1.7 | 2.0 | 0.6 | 28.0 | 3.4 | 2.0 | 24.4 |
| SsHV2-L | 5′-terminal mismatch (%) | 3′-terminal mismatch (%) | ||||||
| vsiRNA Sequence length | A | C | G | T | A | C | G | T |
| 18 | 1.1 | 0.4 | 1.4 | 0.5 | 16.6 | 3.0 | 6.6 | 23.1 |
| 19 | 1.2 | 0.6 | 1.0 | 0.3 | 18.5 | 3.0 | 6.1 | 26.9 |
| 20 | 0.6 | 0.5 | 1.1 | 0.3 | 21.1 | 2.6 | 5.0 | 26.9 |
| 21 | 0.7 | 0.5 | 1.0 | 0.4 | 17.1 | 2.6 | 5.1 | 20.6 |
| 22 | 2.3 | 0.4 | 1.0 | 0.4 | 11.7 | 3.2 | 4.5 | 17.1 |
| 23 | 0.8 | 0.7 | 1.7 | 0.3 | 14.1 | 2.5 | 4.6 | 19.0 |
| 24 | 0.2 | 1.3 | 2.8 | 0.5 | 19.6 | 1.9 | 5.2 | 22.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mochama, P.; Jadhav, P.; Neupane, A.; Lee Marzano, S.-Y. Mycoviruses as Triggers and Targets of RNA Silencing in White Mold Fungus Sclerotinia sclerotiorum. Viruses 2018, 10, 214. https://doi.org/10.3390/v10040214
Mochama P, Jadhav P, Neupane A, Lee Marzano S-Y. Mycoviruses as Triggers and Targets of RNA Silencing in White Mold Fungus Sclerotinia sclerotiorum. Viruses. 2018; 10(4):214. https://doi.org/10.3390/v10040214
Chicago/Turabian StyleMochama, Pauline, Prajakta Jadhav, Achal Neupane, and Shin-Yi Lee Marzano. 2018. "Mycoviruses as Triggers and Targets of RNA Silencing in White Mold Fungus Sclerotinia sclerotiorum" Viruses 10, no. 4: 214. https://doi.org/10.3390/v10040214
APA StyleMochama, P., Jadhav, P., Neupane, A., & Lee Marzano, S.-Y. (2018). Mycoviruses as Triggers and Targets of RNA Silencing in White Mold Fungus Sclerotinia sclerotiorum. Viruses, 10(4), 214. https://doi.org/10.3390/v10040214