Mouse Model of Cytomegalovirus Disease and Immunotherapy in the Immunocompromised Host: Predictions for Medical Translation that Survived the “Test of Time”



Abstract
1. Introduction
2. The Importance of “Model Design” for Fitting a Clinical Correlate
- (i)
- Except in the case of congenital hCMV infection, transmission occurs under birth or at any time after birth, most frequently via mucosal exposure to virus-containing saliva in early childhood. This often happens in daycare centers or in more-child families where multiple contacts increase the probability of transmission. In immunocompetent humans, even in newborns, primary hCMV infection is associated only with unspecific feverish symptoms rarely-to-never diagnosed as hCMV infection. Primary infection can be associated with a mild mononucleosis, which is detected only by sheer chance in a routine hemogram. However, even then, hCMV is rarely taken into consideration as the causative agent. As an important consequence, the time of primary hCMV infection of an individual in the hCMV-seropositive but healthy part of the population is, in general, unknown. Likewise, the viral dose during the first encounter is an unknown parameter. Presumably, the initial dose is very low rather than high. This lack of information is a critical issue since the mouse model, by comparing infection of newborn versus adult mice, predicts that the time of primary infection and the viral dose determine the viral intra-host spread and the extent of amplification by viral replication. This defines the viral load/genome number present during viral latency after resolution of productive primary infection, which, in consequence, determines the probability for reactivation from latency to recurrent infection [39] and associated long-term effects on the immune system including “memory inflation” and immuno-senescence [40].Conclusion: Primary mCMV infection of immunocompetent mice with doses of infection or via routes of infection that result in severe morbidity or even lethality with organ manifestations disqualifies as a model for the natural biology of CMVs.
- (ii)
- hCMV infection of immunocompromised HCT recipients in the period between hematoablative anti-leukemia therapy and immunological reconstitution by HCT develops into multiple-organ CMV disease with the lungs as a major manifestation site clinically presenting as interstitial pneumonia with an often lethal outcome if left untreated (reviewed in Reference [41]).Conclusion: mCMV strains of low virulence that fail to cause disease, including interstitial pneumonia, in immunocompromised mice disqualify for a model agent. As mentioned above, attenuated laboratory strains of hCMV would also fail in this respect in “humanized” mice with human tissue implants.
- (iii)
- hCMV disease in humans implies that the individual has no genetic resistance to hCMV infection. If resistance by genetically-determined intrinsic defense mechanisms that prevent virus replication in host cells exist, the mechanisms would be of interest but would not apply to individuals who are at risk of becoming CMV patients.Conclusion: Only genetically-susceptible host genotypes such as strains of mice susceptible to mCMV infection qualify for use as a model for hCMV disease.
- (iv)
- It is a hallmark of hCMV infection of humans that resolution of productive primary infection results in a state of “latency” that is defined by the absence of infectious virions despite maintained presence of viral genomes from which reactivation to recurrent productive infection can occur by triggering mechanisms that are still under investigation.Conclusion: A model system must reproduce resolution of productive infection, establishment of latency, and reactivation to productive recurrent infection.
3. The BALB/c Mouse Model of CMV Disease
- (i)
- Intraplantar infection of young-adult BALB/c mice at an age of 8–10 weeks with moderate doses of the Smith strain of mCMV, namely 105 PFU, led to a strong immune response in the draining regional lymph node [42]. Viral spread to vital host organs was very limited and, in accordance with the absence of detectable histopathology, morbidity was not observed and all mice survived long-term without any single exception [43]. Thus, in fulfillment of condition (i) defined by hCMV infection, mCMV is not pathogenic in the chosen immunocompetent model host.
- (ii)
- When the very same route and dose of infection were used for BALB/c mice immunocompromised by a hemato-ablative, sub-lethal dose of total-body γ-irradiation prior to infection, which is a regimen used in the human correlate for leukemia therapy preceding HCT, the virus was spreading to essentially all organs. This caused an extensive viral histopathology with an invariably lethal outcome [43]. Importantly, the lungs were found to represent a major manifestation site of CMV disease in this model. Thus, in fulfillment of condition (ii), mCMV proved to be highly pathogenic and caused lethal disease with a histopathology and cell-type tropism resembling hCMV disease in humans, interstitial pneumonia included.
- (iii)
- From the viewpoint of the host, the approach described for (ii) simultaneously also proved that the BALB/c strain of mice is not genetically resistant to mCMV infection by cell-intrinsic antiviral defense mechanisms. Thus, in fulfillment of condition (iii), BALB/c mice are susceptible to mCMV infection and, therefore, qualify as a model host. Besides BALB/c, the mouse inbred strain C57BL/6 is frequently used for studying mCMV infection since it has the advantage that most of the available knock-out, knock-in, and transgenic mice are based on the C57BL/6 genetic background (for an overview, see Reference [44]). A comparison between these two strains with respect to the course of mCMV infection has been comprehensively reviewed by the group of A.B. Hill [45]. C57BL/6 is considered to be a “resistant” strain, which raised the question if mCMV infection of C57BL/6 mice and C57BL/6-based mouse mutants has a clinical correlation that qualifies them as model hosts for studying CMV disease. Investigations into the mechanism of resistance identified Ly49H+ natural killer (NK) cells, which do not exist in BALB/c mice, as the cellular mediators of resistance that become activated via the virally-encoded ligand m157 [46,47,48,49]. This means that the resistance results from a mouse strain-specific branch of innate immunity and not from a cell-intrinsic antiviral defense mechanism. Accordingly, mCMV replicates and causes disease in C57BL/6 mice that are immunocompromised by hemato-ablative treatment [50] and the complication imposed by m157 ligation of the activatory NK cell receptor Ly49H can be avoided by infection with an mCMV mutant in which the m157 encoding gene is deleted [49]. Thus, with this in mind, C57BL/6 and mouse mutants derived thereof also qualify as model hosts, and other mouse strains may do as well.
- (iv)
- Numerous studies early on demonstrated latent mCMV infection in immunocompetent mice and reactivation to recurrent infection by diverse immunosuppressive experimental regimes. For reviews of pioneering work on mCMV latency, see References [51,52]. For more recent reviews on mCMV latency and reactivation in HCT, kidney-SOT, and sepsis models, see References [53,54,55,56]. Importantly, in a study performed with mice infected as neonates, the lungs, which is a major organ site of hCMV disease after HCT [41], proved to be a predilection site of mCMV infection not only in the acute primary infection but also for viral latency and reactivation [57]. The same conclusion applied to a study on the establishment of mCMV latency in the lungs after experimental HCT [58]. As mentioned above, the time of primary infection is decisive in that infection of immunocompetent adult mice in comparison to neonatal mice results in limited viral spread to organs and limited viral replication within tissues. This accordingly results in a lower number of latent viral genomes harbored in tissues and, thus, also a lower incidence of reactivation to “recurrence” upon immunosuppressive treatment [39].Conclusion: The pathobiology of mCMV in the mouse model resembles its hCMV-human counterpart in critical parameters including the main organ site of disease manifestation even though virus-host co-speciation definitively must have generated distinct differences between the model host and human infection in details. In particular, one must keep in mind that “biological convergence” has created analogous functions for non-homologous genes and their gene products, which resulted in different molecular mechanisms. As a consequence, the mouse model can predict principles, but not, or at least rarely, the precise molecular mechanisms that are effective in hCMV infection.
4. Why Are Models Needed at All?
5. Why a Mouse Model?
6. Are “Humanized” Mice the Future in CMV Research?
7. Immunotherapy of CMV Disease by Adoptive Immune Cell Transfer: An Early Prediction from the BALB/c Mouse Model that Went to Clinical Application
8. Challenging the Validity of the Model: CD8+ T Cells Are Dispensable for Controlling CMV
9. Challenging the Validity of the Model: Antigen-Specificity of Protective CD8+ T Cells
10. Immune Evasion of CMVs Correctly Predicted by mCMV
11. Results from the Mouse Model that Await Clinical Investigation
11.1. Verifying the Cellular Site of Latent Infection
11.2. Intravital Imaging: An Advantage of the Mouse Model
11.3. Role for Mast Cells in Controlling CMV Infection
11.4. Role for Alternative Viral gH/gL Entry Complexes in the Initial Host Organ Colonization and Intra-Tissue Spread
11.5. Improved Anti-Viral Efficacy of CD8+ T Cells by “Cognate Help” from MHC-I Redirected CD4+ T Helper Cells
11.6. Preventing Graft-versus-Host Disease in Allogeneic HCT by Regulatory T Cells
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davison, A.J.; Holton, M.; Dolan, A.; Dargan, D.J.; Gatherer, D.; Hayward, G.S. Comparative genomics of primate cytomegaloviruses. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 1–22. [Google Scholar]
- Ho, M. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 2008, 197, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Boppana, S.B.; Britt, W.J. Synopsis of clinical aspects of human cytomegalovirus disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 1–25. [Google Scholar]
- Cannon, M.J.; Grosse, S.D.; Fowler, K.B. The epidemiology and public health impact of congenital cytomegalovirus infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 26–48. [Google Scholar]
- Redwood, A.J.; Shellam, G.R.; Smith, L.M. Molecular evolution of murine cytomegalovirus genomes. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 23–37. [Google Scholar]
- Smith, M.G. Propagation in tissue cultures of a cytopathogenic virus from human salivary gland virus (SGV) disease. Proc. Soc. Exp. Biol. Med. 1956, 92, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J. Margaret Gladys Smith, mother of cytomegalovirus: 60th anniversary of cytomegalovirus isolation. Med. Microbiol. Immunol. 2015, 204, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, E.; Pawletko, K.; Indenbirken, D.; Schumacher, U.; Brune, W. Stepwise adaptation of murine cytomegalovirus to cells of a foreign host for identification of host range determinants. Med. Microbiol. Immunol. 2015, 204, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.; Früh, K. Rhesus CMV: An emerging animal model for human CMV. Med. Microbiol. Immunol. 2008, 197, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Wang, Z.; Abel, K.; Li, J.; Strelow, L.; Mandarino, A.; Eberhardt, M.K.; Schmidt, K.A.; Diamond, D.J.; Barry, P.A. Evaluation of recombinant modified vaccinia Ankara virus-based rhesus cytomegalovirus vaccines in rhesus macaques. Med. Microbiol. Immunol. 2008, 197, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Früh, K.; Malouli, D.; Oxford, K.L.; Barry, P.A. Non-human-primate models of cytomegalovirus infection, prevention, and therapy. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 461–494. [Google Scholar]
- Deere, J.D.; Barry, P.A. Using the nonhuman primate model of HCMV to guide vaccine development. Viruses 2014, 6, 1483–1501. [Google Scholar] [CrossRef]
- Itell, H.L.; Kaur, A.; Deere, J.D.; Barry, P.A.; Permar, S.R. Rhesus monkeys for a nonhuman primate model of cytomegalovirus infections. Curr. Opin. Virol. 2017, 25, 126–133. [Google Scholar] [CrossRef]
- Früh, K.; Picker, L. CD8+ T cell programming by cytomegalovirus vectors: Applications in prophylactic and therapeutic vaccination. Curr. Opin. Immunol. 2017, 47, 52–56. [Google Scholar] [CrossRef]
- Wilkinson, G.W.; Davison, A.J.; Tomasec, P.; Fielding, C.A.; Aicheler, R.; Murrell, I.; Seirafian, S.; Wang, E.C.; Weekes, M.; Lehner, P.J.; et al. Human cytomegalovirus: Taking the strain. Med. Microbiol. Immunol. 2015, 204, 273–284. [Google Scholar] [CrossRef]
- Renzette, N.; Gibson, L.; Bhattacharjee, B.; Fisher, D.; Schleiss, M.R.; Jensen, J.D.; Kowalik, T.F. Rapid intrahost evolution of human cytomegalovirus is shaped by demography and positive selection. PLoS Genet. 2013, 9, e1003735. [Google Scholar] [CrossRef]
- Renzette, N.; Gibson, L.; Jensen, J.D.; Kowalik, T.F. Human cytomegalovirus intrahost evolution—A new avenue for understanding and controlling herpesvirus infections. Curr. Opin. Virol. 2014, 8, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Pokalyuk, C.; Gibson, L.; Bhattacharjee, B.; Schleiss, M.R.; Hamprecht, K.; Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Britt, W.J.; Jensen, J.D.; et al. Limits and patterns of cytomegalovirus genomic diversity in humans. Proc. Natl. Acad. Sci. USA 2015, 112, E4120–E4128. [Google Scholar] [CrossRef]
- Brown, J.M.; Kaneshima, H.; Mocarski, E.S. Dramatic interstrain differences in the replication of human cytomegalovirus in SCID-hu mice. J. Infect. Dis. 1995, 171, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Fleming, P.; Davis-Poynter, N.; Degli-Esposti, M.; Densley, E.; Papadimitriou, J.; Shellam, G.; Farrell, H. The murine cytomegalovirus chemokine homolog, m131/129, is a determinant of viral pathogenicity. J. Virol. 1999, 73, 6800–6809. [Google Scholar] [PubMed]
- Saederup, N.; Aguirre, S.A.; Sparer, T.E.; Bouley, D.M.; Mocarski, E.S. Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J. Virol. 2001, 75, 9966–9976. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.M.; Brizic, I.; Prager, A.; Trsan, T.; Arapovic, M.; Lemmermann, N.A.; Podlech, J.; Reddehase, M.J.; Lemnitzer, F.; Bosse, J.B.; et al. The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog. 2013, 9, e1003493. [Google Scholar] [CrossRef]
- Manning, W.C.; Stoddart, C.A.; Lagenaur, L.A.; Abenes, G.B.; Mocarski, E.S. Cytomegalovirus determinant of replication in salivary glands. J. Virol. 1992, 66, 3794–3802. [Google Scholar]
- Lagenaur, L.A.; Manning, W.C.; Vieira, J.; Martens, C.L.; Mocarski, E.S. Structure and function of the murine cytomegalovirus sgg1 gene: A determinant of viral growth in salivary gland acinar cells. J. Virol. 1994, 68, 7717–7727. [Google Scholar]
- Jordan, S.; Krause, J.; Prager, A.; Mitrovic, M.; Jonjic, S.; Koszinowski, U.H.; Adler, B. Virus progeny of murine cytomegalovirus bacterial artificial chromosome pSM3fr show reduced growth in salivary glands due to a fixed mutation of MCK-2. J. Virol. 2011, 85, 10346–10353. [Google Scholar] [CrossRef]
- Daley-Bauer, L.P.; Roback, L.J.; Wynn, G.M.; Mocarski, E.S. Cytomegalovirus hijacks CX3CR1(hi) patrolling monocytes as immune-privileged vehicles for dissemination in mice. Cell Host Microbe 2014, 15, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Lemmermann, N.A.; Krmpotic, A.; Podlech, J.; Brizic, I.; Prager, A.; Adler, H.; Karbach, A.; Wu, Y.; Jonjic, S.; Reddehase, M.J.; et al. Non-redundant and redundant roles of cytomegalovirus gH/gL complexes in host organ entry and intra-tissue spread. PLoS Pathog. 2015, 11, e1004640. [Google Scholar] [CrossRef]
- Adler, B.; Sinzger, C. Cytomegalovirus inter-strain variance in cell-type tropism. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 297–321. [Google Scholar]
- Ruzsics, Z.; Borst, E.M.; Bosse, J.B.; Brune, W.; Messerle, M. Manipulating CMV genomes by BAC mutagenesis: Strategies and applications. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 38–58. [Google Scholar]
- Lemmermann, N.A.; Kropp, K.A.; Seckert, C.K.; Grzimek, N.K.; Reddehase, M.J. Reverse genetics modification of cytomegalovirus antigenicity and immunogenicity by CD8 T-cell epitope deletion and insertion. J. Biomed. Biotechnol. 2011, 2011, 812742. [Google Scholar] [CrossRef]
- Podlech, J.; Holtappels, R.; Wirtz, N.; Steffens, H.P.; Reddehase, M.J. Reconstitution of CD8 T cells is essential for the prevention of multiple-organ cytomegalovirus histopathology after bone marrow transplantation. J. Gen. Virol. 1998, 79, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Sacher, T.; Podlech, J.; Mohr, C.A.; Jordan, S.; Ruzsics, Z.; Reddehase, M.J.; Koszinowski, U.H. The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 2008, 3, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Sacher, T.; Andrassy, J.; Kalnins, A.; Dölken, L.; Jordan, S.; Podlech, J.; Ruzsics, Z.; Jauch, K.W.; Reddehase, M.J.; Koszinowski, U.H. Shedding light on the elusive role of endothelial cells in cytomegalovirus dissemination. PLoS Pathog. 2011, 7, e1002366. [Google Scholar] [CrossRef] [PubMed]
- Seckert, C.K.; Renzaho, A.; Tervo, H.M.; Krause, C.; Deegen, P.; Kühnapfel, B.; Reddehase, M.J.; Grzimek, N.K. Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J. Virol. 2009, 83, 8869–8884. [Google Scholar] [CrossRef]
- Plachter, B.; Sinzger, C.; Jahn, G. Cell types involved in replication and distribution of human cytomegalovirus. Adv. Virus Res. 1996, 46, 195–261. [Google Scholar]
- Cicin-Sain, L.; Podlech, J.; Messerle, M.; Reddehase, M.J.; Koszinowski, U.H. Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J. Virol. 2005, 79, 9492–9502. [Google Scholar] [CrossRef]
- Picarda, G.; Benedict, C.A. Cytomegalovirus: Shape-shifting the immune system. J. Immunol. 2018, 200, 3881–3889. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Balthesen, M.; Rapp, M.; Jonjić, S.; Pavić, I.; Koszinowski, U.H. The conditions of primary infection define the load of latent viral genome in organs and the risk of recurrent cytomegalovirus disease. J. Exp. Med. 1994, 179, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Redeker, A.; Remmerswaal, E.B.; van der Gracht, E.T.; Weltenm, S.P.; Hölltm, T.; Koning, F.; Cicin-Sain, L.; Nikolich-Žugich, J.; Ten Berge, I.J.; van Lier, R.A.; et al. The contribution of cytomegalovirus infection to immune senescence is set by the infectious dose. Front. Immunol. 2018, 8, 1953. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Boeckh, M. Clinical cytomegalovirus research: Haematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 335–351. [Google Scholar]
- Reddehase, M.J.; Keil, G.M.; Koszinowski, U.H. The cytolytic T lymphocyte response to the murine cytomegalovirus. I. Distinct maturation stages of cytolytic T lymphocytes constitute the cellular immune response during acute infection of mice with the murine cytomegalovirus. J. Immunol. 1984, 132, 482–489. [Google Scholar] [PubMed]
- Reddehase, M.J.; Weiland, F.; Münch, K.; Jonjic, S.; Lüske, A.; Koszinowski, U.H. Interstitial murine cytomegalovirus pneumonia after irradiation: Characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 1985, 55, 264–273. [Google Scholar] [PubMed]
- Benedict, C.A.; Crozat, K.; Degli-Esposti, M.; Dalod, M. Host genetic models in cytomegalovirus immunology. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 310–334. [Google Scholar]
- Doom, C.M.; Hill, A.B. MHC class I immune evasion in MCMV infection. Med. Microbiol. Immunol. 2008, 197, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Arase, H.; Mocarski, E.S.; Campbell, A.E.; Hill, A.B.; Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002, 296, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.R.; Heusel, J.W.; Mehta, I.K.; Kim, S.; Dorner, B.G.; Naidenko, O.V.; Iizuka, K.; Furukawa, H.; Beckman, D.L.; Pingel, J.T.; et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 8826–8831. [Google Scholar] [CrossRef]
- Voigt, V.; Forbes, C.A.; Tonkin, J.N.; Degli-Esposti, M.A.; Smith, H.R.; Yokoyama, W.M.; Scalzo, A.A. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13483–13488. [Google Scholar] [CrossRef]
- Bubić, I.; Wagner, M.; Krmpotić, A.; Saulig, T.; Kim, S.; Yokoyama, W.M.; Jonjić, S.; Koszinowski, U.H. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J. Virol. 2004, 78, 7536–7544. [Google Scholar] [CrossRef]
- Holtappels, R.; Podlech, J.; Pahl-Seibert, M.; Jülch, M.; Thomas, D.; Simon, C.O.; Wagner, M.; Reddehase, M.J. Cytomegalovirus misleads its host by priming of CD8 T cells specific for an epitope not presented in infected tissues. J. Exp. Med. 2004, 199, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Podlech, J.; Grzimek, N.K. Mouse models of cytomegalovirus latency: Overview. J. Clin. Virol. 2002, 25 (Suppl. 2), S23–S36. [Google Scholar] [CrossRef]
- Hummel, M.; Abecassis, M.M. A model for reactivation of CMV from latency. J. Clin. Virol. 2002, 25, S123–S136. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Simon, C.O.; Seckert, C.K.; Lemmermann, N.; Grzimek, N.K. Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 2008, 325, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Seckert, C.K.; Griessl, M.; Büttner, J.K.; Scheller, S.; Simon, C.O.; Kropp, K.A.; Renzaho, A.; Kühnapfel, B.; Grzimek, N.K.; Reddehase, M.J. Viral latency drives “memory inflation”: A unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Seckert, C.K.; Griessl, M.; Buttner, J.K.; Freitag, K.; Lemmermann, N.A.; Hummel, M.A.; Liu, X.F.; Abecassis, M.I.; Angulo, A.; Messerle, M.; et al. Immune surveillance of cytomegalovirus latency and reactivation in murine models: Link to “memory inflation”. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 374–416. [Google Scholar]
- Liu, X.F.; Wang, X.; Yan, S.; Zhang, Z.; Abecassis, M.; Hummel, M. Epigenetic control of cytomegalovirus latency and reactivation. Viruses 2013, 5, 1325–1345. [Google Scholar] [CrossRef]
- Balthesen, M.; Messerle, M.; Reddehase, M.J. Lungs are a major organ site of cytomegalovirus latency and recurrence. J. Virol. 1993, 67, 5360–5366. [Google Scholar]
- Kurz, S.; Steffens, H.; Mayer, A.; Harris, J.; Reddehase, M. Latency versus persistence or intermittent recurrences: Evidence for a latent state of murine cytomegalovirus in the lungs. J. Virol. 1997, 71, 2980–2987. [Google Scholar]
- Georgiades, P.; Ferguson-Smith, A.C.; Burton, G.J. Comparative developmental anatomy of the murine and human definitive placentae. Placenta 2002, 23, 3–19. [Google Scholar] [CrossRef]
- McGregor, A.; McVoy, M.A.; Schleiss, M.R. The Guinea pig model of congenital cytomegalovirus infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 88–118. [Google Scholar]
- Pereira, L.; Tabata, T.; Petitt, M.; Fang-Hoover, J. Cytomegalovirus replication in the developing human placenta. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 74–87. [Google Scholar]
- Slavuljica, I.; Kveštak, D.; Huszthy, P.C.; Kosmac, K.; Britt, W.J.; Jonjić, S. Immunobiology of congenital cytomegalovirus infection of the central nervous system—The murine cytomegalovirus model. Cell. Mol. Immunol. 2015, 12, 180–191. [Google Scholar] [CrossRef]
- Brizić, I.; Hiršl, L.; Britt, W.J.; Krmpotić, A.; Jonjić, S. Immune responses to congenital cytomegalovirus infection. Microbes Infect. 2018, 20, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Voigt, S.; Ettinger, J.; Streblow, D.N. The rat model of cytomegalovirus infection and vascular disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 312–336. [Google Scholar]
- Kim, S.J.; Varghese, T.K.; Zhang, Z.; Zhao, L.C.; Thomas, G.; Hummel, M.; Abecassis, M. Renal ischemia/reperfusion injury activates the enhancer domain of the human cytomegalovirus major immediate early promoter. Am. J. Transplant. 2005, 5, 1606–1613. [Google Scholar] [CrossRef]
- Smith, M.S.; Streblow, D.N.; Caposio, P.; Nelson, J.A. Humanized mouse models of cytomegalovirus pathogenesis and latency. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume I, pp. 417–436. [Google Scholar]
- Crawford, L.B.; Streblow, D.N.; Hakki, M.; Nelson, J.A.; Caposio, P. Humanized mouse models of human cytomegalovirus infection. Curr. Opin. Virol. 2015, 13, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Steffens, H.P.; Kurz, S.; Holtappels, R.; Reddehase, M.J. Preemptive CD8 T-cell immunotherapy of acute cytomegalovirus infection prevents lethal disease, limits the burden of latent viral genomes, and reduces the risk of virus recurrence. J. Virol. 1998, 72, 1797–1804. [Google Scholar] [PubMed]
- Holtappels, R.; Böhm, V.; Podlech, J.; Reddehase, M.J. CD8 T-cell-based immunotherapy of cytomegalovirus infection: “proof of concept” provided by the murine model. Med. Microbiol. Immunol. 2008, 197, 125–134. [Google Scholar] [CrossRef]
- Holtappels, R.; Ebert, S.; Podlech, J.; Fink, A.; Bohm, V.; Lemmermann, N.A.; Freitag, K.; Renzaho, A.; Thomas, D.; Reddehase, M.J. Murine model for cytoimmunotherapy of CMV disease after haematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Wymondham, Norfolk, UK, 2013; Volume II, pp. 354–381. [Google Scholar]
- Ebert, S.; Podlech, J.; Gillert-Marien, D.; Gergely, K.M.; Büttner, J.K.; Fink, A.; Freitag, K.; Thomas, D.; Reddehase, M.J.; Holtappels, R. Parameters determining the efficacy of adoptive CD8 T-cell therapy of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 527–539. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Jonjić, S.; Weiland, F.; Mutter, W.; Koszinowski, U.H. Adoptive immunotherapy of murine cytomegalovirus adrenalitis in the immunocompromised host: CD4-helper-independent antiviral function of CD8-positive memory T lymphocytes derived from latently infected donors. J. Virol. 1988, 62, 1061–1065. [Google Scholar]
- Nauerth, M.; Weißbrich, B.; Knall, R.; Franz, T.; Dössinger, G.; Bet, J.; Paszkiewicz, P.J.; Pfeifer, L.; Uckert, M.; Holtappels, R.; et al. TCR-ligand koff rate correlates with the protective capacity of antigen-specific CD8+ T cells for adoptive transfer. Sci. Transl. Med. 2013, 5, 192ra87. [Google Scholar] [CrossRef]
- Holtappels, R.; Simon, C.O.; Munks, M.W.; Thomas, D.; Deegen, P.; Kühnapfel, B.; Däubner, T.; Emde, S.; Podlech, J.; Grzimek, N.K.; et al. Subdominant CD8 T-cell epitopes account for protection against cytomegalovirus independent of immunodomination. J. Virol. 2008, 82, 5781–5796. [Google Scholar] [CrossRef]
- Ebert, S.; Lemmermann, N.A.; Thomas, D.; Renzaho, A.; Reddehase, M.J.; Holtappels, R. Immune control in the absence of immunodominant epitopes: Implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med. Microbiol. Immunol. 2012, 201, 541–550. [Google Scholar] [CrossRef]
- Holtappels, R.; Lemmermann, N.A.; Podlech, J.; Ebert, S.; Reddehase, M.J. Reconstitution of CD8 T cells protective against cytomegalovirus in a mouse model of hematopoietic cell transplantation: Dynamics and inessentiality of epitope immunodominance. Front. Immunol. 2016, 7, 232. [Google Scholar] [CrossRef] [PubMed]
- Pahl-Seibert, M.F.; Juelch, M.; Podlech, J.; Thomas, D.; Deegen, P.; Reddehase, M.J.; Holtappels, R. Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 T cells purified by T-cell receptor-based cell sorting. J. Virol. 2005, 79, 5400–5413. [Google Scholar] [CrossRef] [PubMed]
- Böhm, V.; Podlech, J.; Thomas, D.; Deegen, P.; Pahl-Seibert, M.; Lemmermann, N.A.; Grzimek, N.K.; Oehrlein-Karpi, S.A.; Reddehase, M.J.; Holtappels, R. Epitope-specific in vivo protection against cytomegalovirus disease by CD8 T cells in the murine model of preemptive immunotherapy. Med. Microbiol. Immunol. 2008, 197, 135–144. [Google Scholar] [CrossRef]
- Thomas, S.; Klobuch, S.; Podlech, J.; Plachter, B.; Hoffmann, P.; Renzaho, A.; Theobald, M.; Reddehase, M.J.; Herr, W.; Lemmermann, N.A. Evaluating human T-cell therapy of cytomegalovirus organ disease in HLA-transgenic mice. PLoS Pathog. 2015, 11, e1005049. [Google Scholar] [CrossRef] [PubMed]
- Riddell, S.R.; Watanabe, K.S.; Goodrich, J.M.; Li, C.R.; Agha, M.E.; Greenberg, P.D. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992, 2257, 238–241. [Google Scholar] [CrossRef]
- Einsele, H.; Roosnek, E.; Rufer, N.; Sinzger, C.; Riegler, S.; Löffler, J.; Grigoleit, U.; Moris, A.; Rammensee, H.G.; Kanz, L.; et al. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 2002, 99, 3916–3922. [Google Scholar] [CrossRef]
- Peggs, K.S.; Verfuerth, S.; Pizzey, A.; Khan, N.; Guiver, M.; Moss, P.A.; Mackinnon, S. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet 2003, 362, 1375–1377. [Google Scholar] [CrossRef]
- Cobbold, M.; Khan, N.; Pourgheysari, B.; Tauro, S.; McDonald, D.; Osman, H.; Assenmacher, M.; Billingham, L.; Steward, C.; Crawley, C.; et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J. Exp. Med. 2005, 202, 379–386. [Google Scholar] [CrossRef]
- Feuchtinger, T.; Opherk, K.; Bethge, W.A.; Topp, M.S.; Schuster, F.R.; Weissinger, E.M.; Mohty, M.; Or, R.; Maschan, M.; Schumm, M.; et al. Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood 2010, 116, 4360–4367. [Google Scholar] [CrossRef]
- Schmitt, A.; Tonn, T.; Busch, D.H.; Grigoleit, G.U.; Einsele, H.; Odendahl, M.; Germeroth, L.; Ringhoffer, M.; Ringhoffer, S.; Wiesneth, M.; et al. Adoptive transfer and selective reconstitution of streptamer-selected cytomegalovirus-specific CD8+ T cells leads to virus clearance in patients after allogeneic peripheral blood stem cell transplantation. Transfusion 2011, 51, 591–599. [Google Scholar] [CrossRef]
- Odendahl, M.; Grigoleit, G.U.; Bönig, H.; Neuenhahn, M.; Albrecht, J.; Anderl, F.; Germeroth, L.; Schmitz, M.; Bornhäuser, M.; Einsele, H.; et al. Clinical-scale isolation of “minimally manipulated” cytomegalovirus-specific donor lymphocytes for the treatment of refractory cytomegalovirus disease. Cytotherapy 2014, 16, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Stemberger, C.; Graef, P.; Odendahl, M.; Albrecht, J.; Dössinger, G.; Anderl, F.; Buchholz, V.R.; Gasteiger, G.; Schiemann, M.; Grigoleit, G.U.; et al. Lowest numbers of primary CD8(+) T cells can reconstitute protective immunity upon adoptive immunotherapy. Blood 2014, 124, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Jonjić, S.; Pavić, I.; Lucin, P.; Rukavina, D.; Koszinowski, U.H. Efficacious control of cytomegalovirus infection after long-term depletion of CD8+ T lymphocytes. J. Virol. 1990, 64, 5457–5464. [Google Scholar] [PubMed]
- Polić, B.; Jonjić, S.; Pavić, I.; Crnković, I.; Zorica, I.; Hengel, H.; Lucin, P.; Koszinowski, U.H. Lack of MHC class I complex expression has no effect on spread and control of cytomegalovirus infection in vivo. J. Gen. Virol. 1996, 77, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Podlech, J.; Holtappels, R.; Pahl-Seibert, M.F.; Steffens, H.P.; Reddehase, M.J. Murine model of interstitial cytomegalovirus pneumonia in syngeneic bone marrow transplantation: Persistence of protective pulmonary CD8-T-cell infiltrates after clearance of acute infection. J. Virol. 2000, 74, 7496–7507. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, J.F.; Warner, J.F.; Dennert, G.; Welsh, R.M. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J. Exp. Med. 1985, 161, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Klenovsek, K.; Weisel, F.; Schneider, A.; Appelt, U.; Jonjic, S.; Messerle, M.; Bradel-Tretheway, B.; Winkler, T.H.; Mach, M. Protection from CMV infection in immunodeficient hosts by adoptive transfer of memory B cells. Blood 2007, 110, 3472–3479. [Google Scholar] [CrossRef]
- Jeitziner, S.M.; Walton, S.M.; Torti, N.; Oxenius, A. Adoptive transfer of cytomegalovirus-specific effector CD4+ T cells provides antiviral protection from murine CMV infection. Eur. J. Immunol. 2013, 43, 2886–2895. [Google Scholar] [CrossRef]
- Sell, S.; Dietz, M.; Schneider, A.; Holtappels, R.; Mach, M.; Winkler, T.H. Control of murine cytomegalovirus infection by γδ T cells. PLoS Pathog. 2015, 11, e1004481. [Google Scholar] [CrossRef]
- Khairallah, C.; Netzer, S.; Villacreces, A.; Juzan, M.; Rousseau, B.; Dulanto, S.; Giese, A.; Costet, P.; Praloran, V.; Moreau, J.F.; et al. γδ T cells confer protection against murine cytomegalovirus (MCMV). PLoS Pathog. 2015, 11, e1004702. [Google Scholar] [CrossRef]
- Stinski, M.F.; Isomura, H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 2008, 197, 223–321. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, E.A.; Spector, D.H. Regulation of human cytomegalovirus gene expression. Adv. Virus Res. 1999, 54, 61–128. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Keil, G.M.; Koszinowski, U.H. The cytolytic T lymphocyte response to the murine cytomegalovirus. II. Detection of virus replication stage-specific antigens by separate populations of in vivo active cytolytic T lymphocyte precursors. Eur. J. Immunol. 1984, 14, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Koszinowski, U.H. Significance of herpesvirus immediate early gene expression in cellular immunity to cytomegalovirus infection. Nature 1984, 312, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Rothbard, J.B.; Koszinowski, U.H. A pentapeptide as minimal antigenic determinant for MHC class I-restricted T lymphocytes. Nature 1989, 337, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J. Antigens and immunoevasins: Opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002, 2, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Mutter, W.; Münch, K.; Bühring, H.J.; Koszinowski, U.H. CD8-positive T lymphocytes specific for murine cytomegalovirus immediate-early antigens mediate protective immunity. J. Virol. 1987, 61, 3102–3218. [Google Scholar]
- Del Val, M.; Schlicht, H.J.; Volkmer, H.; Messerle, M.; Reddehase, M.J.; Koszinowski, U.H. Protection against lethal cytomegalovirus infection by a recombinant vaccine containing a single nonameric T-cell epitope. J. Virol. 1991, 65, 3641–3646. [Google Scholar]
- Simon, C.O.; Holtappels, R.; Tervo, H.-M.; Böhm, V.; Däubner, T.; Oehrlein-Karpi, S.A.; Kühnapfel, B.; Renzaho, A.; Strand, D.; Podlech, J.; et al. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J. Virol. 2006, 80, 10436–10456. [Google Scholar] [CrossRef]
- Holtappels, R.; Pahl-Seibert, M.F.; Thomas, D.; Reddehase, M.J. Enrichment of immediate-early 1 (m123/pp89) peptide-specific CD8 T cells in a pulmonary CD62L(lo) memory-effector cell pool during latent murine cytomegalovirus infection of the lungs. J. Virol. 2000, 74, 11495–11503. [Google Scholar] [CrossRef]
- Snyder, C.M. Buffered memory: A hypothesis for the maintenance of functional, virus-specific CD8+ T cells during cytomegalovirus infection. Immunol. Res. 2011, 51, 195–204. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, G.A.; Welten, S.P.; Klenerman, P.; Arens, R. Memory T cell inflation: Understanding cause and effect. Trends Immunol. 2012, 33, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Taylor, E.; Pande, H.; Forman, S.J.; Tanamachi, B.; Li, C.R.; Zaia, J.A.; Greenberg, P.D.; Riddell, S.R. Identification of the major late human cytomegalovirus matrix protein pp65 as a target antigen for CD8+ virus-specific cytotoxic T lymphocytes. J. Med. Virol. 1994, 43, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Frankenberg, N.; Lischka, P.; Pepperl-Klindworth, S.; Stamminger, T.; Plachter, B. Nucleocytoplasmic shuttling and CRM1-dependent MHC class I peptide presentation of human cytomegalovirus pp65. Med. Microbiol. Immunol. 2012, 201, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Sissons, J.G.; Wills, M.R. How understanding immunology contributes to managing CMV disease in immunosuppressed patients: Now and in future. Med. Microbiol. Immunol. 2015, 204, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Kern, F.; Surel, I.P.; Faulhaber, N.; Frömmel, C.; Schneider-Mergener, J.; Schönemann, C.; Reinke, P.; Volk, H.D. Target structures of the CD8(+)-T-cell response to human cytomegalovirus: The 72-kilodalton major immediate-early protein revisited. J. Virol. 1999, 73, 8179–8184. [Google Scholar]
- Khan, N.; Cobbold, M.; Keenan, R.; Moss, P.A. Comparative analysis of CD8+ T cell responses against human cytomegalovirus proteins pp65 and immediate early 1 shows similarities in precursor frequency, oligoclonality, and phenotype. J. Infect. Dis. 2002, 185, 1025–1034. [Google Scholar] [CrossRef]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef]
- Bunde, T.; Kirchner, A.; Hoffmeister, B.; Habedank, D.; Hetzer, R.; Cherepnev, G.; Proesch, S.; Reinke, P.; Volk, H.D.; Lehmkuhl, H.; et al. Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J. Exp. Med. 2005, 201, 1031–1036. [Google Scholar] [CrossRef]
- Büscher, N.; Paulus, C.; Nevels, M.; Tenzer, S.; Plachter, B. The proteome of human cytomegalovirus virions and dense bodies is conserved across different strains. Med. Microbiol. Immunol. 2015, 204, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Munks, M.W.; Gold, M.C.; Zajac, A.L.; Doom, C.M.; Morello, C.S.; Spector, D.H.; Hill, A.B. Genome-wide analysis reveals a highly diverse CD8 T cell response to murine cytomegalovirus. J. Immunol. 2006, 176, 3760–3766. [Google Scholar] [CrossRef] [PubMed]
- Adams, N.M.; Sun, J.C. Spatial and temporal coordination of antiviral responses by group 1 ILCs. Immunol. Rev. 2018, 286, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Powers, C.J.; Richards, R.; Ventura, A.B.; Ford, J.C.; Siess, D.; Axthelm, M.K.; Nelson, J.A.; Jarvis, M.A.; Picker, L.J.; et al. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 2010, 328, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Krmpotic, A.; Messerle, M.; Crnkovic-Mertens, I.; Polic, B.; Jonjic, S.; Koszinowski, U.H. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 1999, 190, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Krmpotić, A.; Busch, D.H.; Bubić, I.; Gebhardt, F.; Hengel, H.; Hasan, M.; Scalzo, A.A.; Koszinowski, U.H.; Jonjić, S. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 2002, 3, 529–535. [Google Scholar] [CrossRef]
- Fink, A.; Renzaho, A.; Reddehase, M.J.; Lemmermann, N.A. The p36 isoform of murine cytomegalovirus m152 protein suffices for mediating innate and adaptive immune evasion. Viruses 2013, 5, 3171–3191. [Google Scholar] [CrossRef]
- Smith, M.S.; Goldman, D.C.; Bailey, A.S.; Pfaffle, D.L.; Kreklywich, C.N.; Spencer, D.B.; Othieno, F.A.; Streblow, D.N.; Garcia, J.V.; Fleming, W.H.; et al. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 2010, 8, 284–291. [Google Scholar] [CrossRef]
- Seckert, C.K.; Renzaho, A.; Reddehase, M.J.; Grzimek, N.K. Hematopoietic stem cell transplantation with latently infected donors does not transmit virus to immunocompromised recipients in the murine model of cytomegalovirus infection. Med. Microbiol. Immunol. 2008, 197, 251–259. [Google Scholar] [CrossRef]
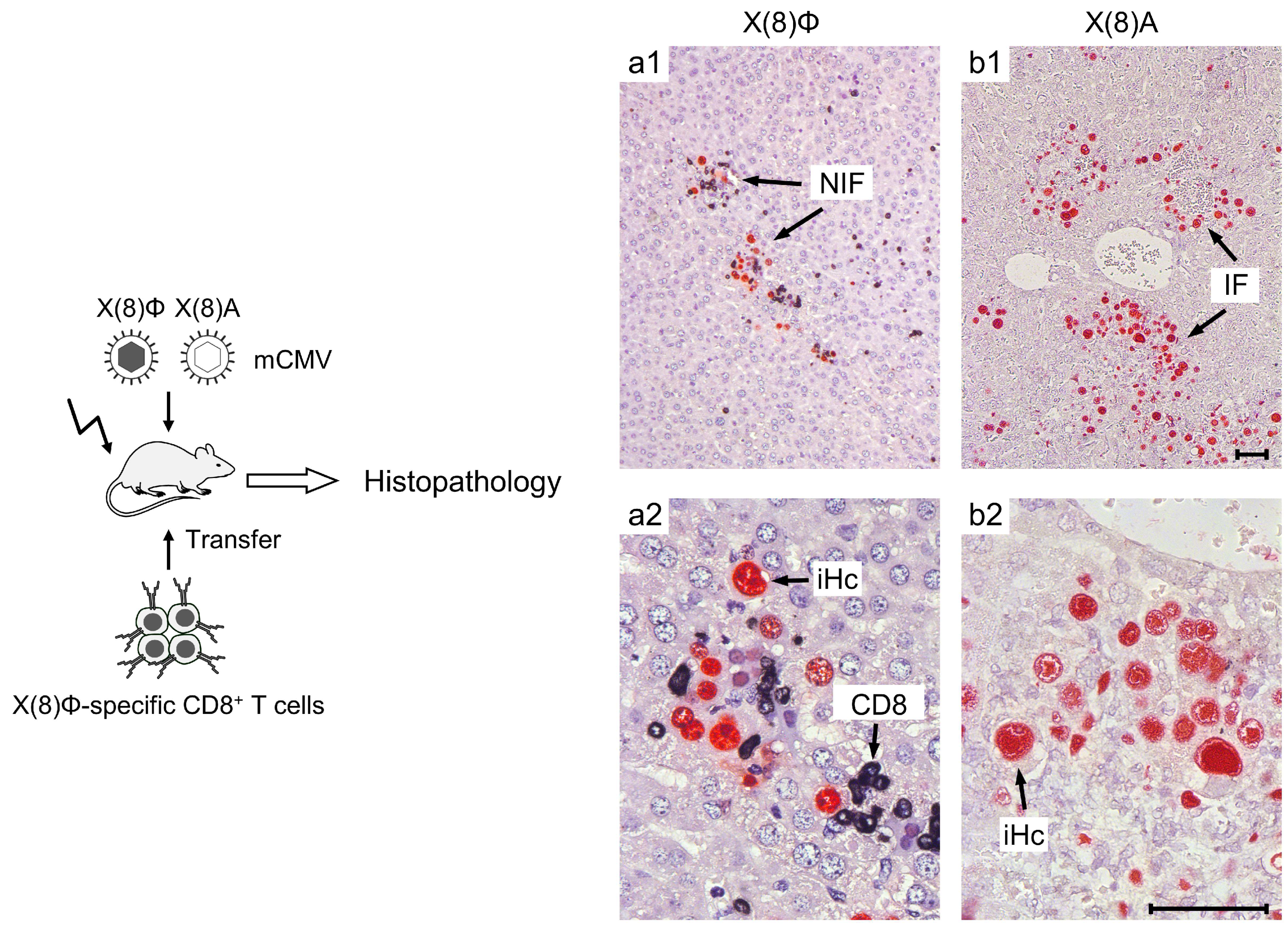
- Stahl, F.R.; Heller, K.; Halle, S.; Keyser, K.A.; Busche, A.; Marquardt, A.; Wagner, K.; Boelter, J.; Bischoff, Y.; Kremmer, E.; et al. Nodular inflammatory foci are sites of T cell priming and control of murine cytomegalovirus infection in the neonatal lung. PLoS Pathog. 2013, 9, e1003828. [Google Scholar] [CrossRef]
- Halle, S.; Keyser, K.A.; Stahl, F.R.; Busche, A.; Marquardt, A.; Zheng, X.; Galla, M.; Heissmeyer, V.; Heller, K.; Boelter, J.; et al. In vivo killing capacity of cytotoxic T cells is limited and involves dynamic interactions and T cell cooperativity. Immunity 2016, 44, 233–245. [Google Scholar] [CrossRef]
- Lueder, Y.; Heller, K.; Ritter, C.; Keyser, K.A.; Wagner, K.; Liu, X.; Messerle, M.; Stahl, F.R.; Halle, S.; Förster, R. Control of primary mouse cytomegalovirus infection in lung nodular inflammatory foci by cooperation of interferon-gamma expressing CD4 and CD8 T cells. PLoS Pathog. 2018, 14, e1007252. [Google Scholar] [CrossRef]
- Podlech, J.; Ebert, S.; Becker, M.; Reddehase, M.J.; Stassen, M.; Lemmermann, N.A. Mast cells: Innate attractors recruiting protective CD8 T cells to sites of cytomegalovirus infection. Med. Microbiol. Immunol. 2015, 204, 327–334. [Google Scholar] [CrossRef]
- Ebert, S.; Becker, M.; Lemmermann, N.A.; Büttner, J.K.; Michel, A.; Taube, C.; Podlech, J.; Böhm, V.; Freitag, K.; Thomas, D.; et al. Mast cells expedite control of pulmonary murine cytomegalovirus infection by enhancing the recruitment of protective CD8 T cells to the lungs. PLoS Pathog. 2014, 10, e1004100. [Google Scholar] [CrossRef]
- Lemmermann, N.A.; Reddehase, M.J. Refining human T-cell immunotherapy of cytomegalovirus disease: A mouse model with “humanized” antigen presentation as a new preclinical study tool. Med. Microbiol. Immunol. 2016, 205, 549–561. [Google Scholar] [CrossRef]
- Nguyen, V.H.; Shashidhar, S.; Chang, D.S.; Ho, L.; Kambham, N.; Bachmann, M.; Brown, J.M.; Negrin, R.S. The impact of regulatory T cells on T-cell immunity following hematopoietic cell transplantation. Blood 2008, 111, 945–953. [Google Scholar] [CrossRef]
- Chopra, M.; Biehl, M.; Steinfatt, T.; Brandl, A.; Kums, J.; Amich, J.; Vaeth, M.; Kuen, J.; Holtappels, R.; Podlech, J.; et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med. 2016, 213, 1881–1900. [Google Scholar] [CrossRef]
- Reddehase, M.J. Mutual interference between cytomegalovirus and reconstitution of protective immunity after hematopoietic cell transplantation. Front. Immunol. 2016, 7, 294. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Parameter | Conventional Mouse Model | Humanized Mouse Model |
|---|---|---|
| Human virus | No | Yes |
| Human host | No | Chimeric |
| Virus-host adaptation | Yes | Partial |
| Ethical concerns | Moderate | Donor consent required |
| Technical demands | Low | High |
| Statistical demands | Easy to fulfill | Difficult to fulfill |
| Morbidity-mortality studies | Yes | No |
| Comprehensive organ disease | Yes | No |
| Viral histopathology | Yes | Restricted to implants |
| Intra-host virus spread | Yes | Limited |
| Host-to-host transmission | Yes | No |
| Cytokine signaling | Intact | Partially disturbed |
| Host genetic variance | Yes, strains or targeted mutation | Limited to donor typing |
| Virus genetic variance | Yes, isolates or targeted mutation | Yes, isolates or targeted mutation |
| Co- and super-infection | Yes | Yes |
| Test and control cohort identity | Yes | Limited by donor material |
| Immunotherapy | Yes | Restricted to implants |
| Testing of antivirals | Doubtful | Yes, though with caution |
| Intravital imaging | Yes | Yes |
| Model for fetal brain infection | Yes | Unrealistic |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddehase, M.J.; Lemmermann, N.A.W. Mouse Model of Cytomegalovirus Disease and Immunotherapy in the Immunocompromised Host: Predictions for Medical Translation that Survived the “Test of Time”. Viruses 2018, 10, 693. https://doi.org/10.3390/v10120693
Reddehase MJ, Lemmermann NAW. Mouse Model of Cytomegalovirus Disease and Immunotherapy in the Immunocompromised Host: Predictions for Medical Translation that Survived the “Test of Time”. Viruses. 2018; 10(12):693. https://doi.org/10.3390/v10120693
Chicago/Turabian StyleReddehase, Matthias J., and Niels A. W. Lemmermann. 2018. "Mouse Model of Cytomegalovirus Disease and Immunotherapy in the Immunocompromised Host: Predictions for Medical Translation that Survived the “Test of Time”" Viruses 10, no. 12: 693. https://doi.org/10.3390/v10120693
APA StyleReddehase, M. J., & Lemmermann, N. A. W. (2018). Mouse Model of Cytomegalovirus Disease and Immunotherapy in the Immunocompromised Host: Predictions for Medical Translation that Survived the “Test of Time”. Viruses, 10(12), 693. https://doi.org/10.3390/v10120693