1. Introduction
The porcine epidemic diarrhea virus (PEDV) is an enveloped, positive-sense and single-stranded RNA virus, belonging to the order
Nidovirales, family
Coronaviridae, and genus
Alphacoronavirus [
1]. The genome of PEDV is about 28 kilobase pairs in length and comprises of seven open reading frames (ORF), including ORF1a and b genes that constitute the 5′ two thirds of the genome and encode the replication complex; the spike (S) gene that governs viral entry; the envelop (E), membrane (M), and nucleocapsid (N) genes that are responsible for virion assembly; and the accessory ORF3 gene with an undetermined function [
2].
The porcine epidemic diarrhea virus is the causative agent of porcine epidemic diarrhea (PED), a historic, highly contagious enteric swine disease characterized by diarrhea, dehydration, and the growth retardation in pigs of all ages [
1]. In late 2010, new and highly virulent PEDV strains arose in China and spread rapidly worldwide by late 2013, resulting in nearly 100% mortality in the affected nursing piglets [
3,
4,
5]. To date, there are still indelible endemics and considerable economic losses in the global swine market [
6]. Besides, the protection conferred by currently available vaccines is, unfortunately, unsatisfactory [
6,
7]. Based on the nucleotide identity of the S gene, PEDVs are categorized into four genogroups (Gs), namely G1a, G1b, G2a, and G2b [
8]. Among these, the G2b PEDV strains that predominate the field in Asia and North America, show higher pathogenicity [
9] and appear to elicit broader protection across different genogroups [
10,
11,
12]. Although the increased virulence of new PEDV strains was ascribed to several mutations in the S gene through viral escape from antibody neutralization induced by traditional vaccines [
13,
14], the detailed mechanism remains elusive.
Previously, we generated an attenuated G2b Taiwan PEDV strain, PEDV Pintung 52 passage 96 (PEDVPT-P96) virus, by serial passage of the parental PEDVPT strain in Vero cells [
15]. Despite the high potential of PEDVPT-P96 as a future vaccine candidate against PEDV as indicated by its reduced pathogenicity and robust host immune response in our 5-week-old piglet model [
15], the difficulty in PEDV isolation and subsequent lengthy passage process rendered the PEDVPT-P96 unable to promptly respond to the vast outbreak in late 2013. This year, Zhou et al. [
16] reported a new disastrous swine disease outbreak in China in 2016 caused by an HKU2-related coronavirus of bat origin, again highlighting the potential burden of the interspecies jumping of coronavirus, and a pressing need for a readily applicable vaccine platform for new emergences.
The reverse genetics system has been widely used to study viral pathogenesis and novel vaccine design. At present, the reported PEDV infectious cDNA clones were exclusively constructed based on sequences of the representative wild-type PEDV isolates [
17,
18,
19,
20,
21]. Consequently, they are highly pathogenic and fatal in suckling piglets, comparable to their parental viruses. With regard to vaccine use, further genetic editing is necessary to attenuate these recombinant viruses. Alternatively, a complementary approach exploiting the attenuated phenotype to address the safety concerns has not been described. In the present study, a full-length cDNA clone of the attenuated PEDVPT-P96, iPEDVPT-P96, was generated. In addition, the pathogenicity, immunogenicity, and protection against virulent PEDVPT-P5 challenge by iPEDVPT-P96 were evaluated in the 5-week-old piglet model. Our data suggest that the iPEDVPT-P96 virus was more attenuated but able to elicit similar immunogenicity and immunoprotection against the autologous virulent PEDVPT-P5 challenge compared to the parental PEDVPT-P96 virus. This iPEDVPT-P96 cDNA clone is expected to allow for the manipulation of the viral genome to study viral pathogenesis. On the other hand, it can serve as a vaccine platform, for example, by directly replacing the S gene with that of the (re)emerging swine coronaviruses.
2. Materials and Methods
2.1. Cells and Viruses
Vero C1008 cells (ATCC No. CRL-1586) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine protein (FBS), 250 ng/mL Amphotericin B, 100 U/mL Penicillin, and 100 μg/mL Streptomycin. Viral stock of PEDVPT-P96 in post-inoculation medium (PI medium) containing DMEM supplemented with 0.3% tryptose phosphate broth (TBP), 0.02% yeast extract (0.02%), and 10 μg/mL trypsin, as prepared in our previous study [
15], was used for generating the infectious cDNA clone and as the control for in vivo and in vitro studies, whereas the virulent PEDVPT-P5 virus was used for animal challenge.
2.2. Generation of the Full-Length cDNA Clone, iPEDVPT-P96
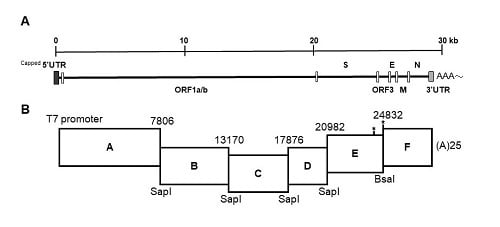
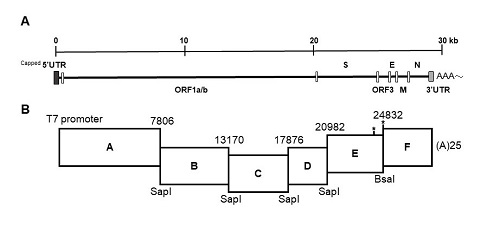
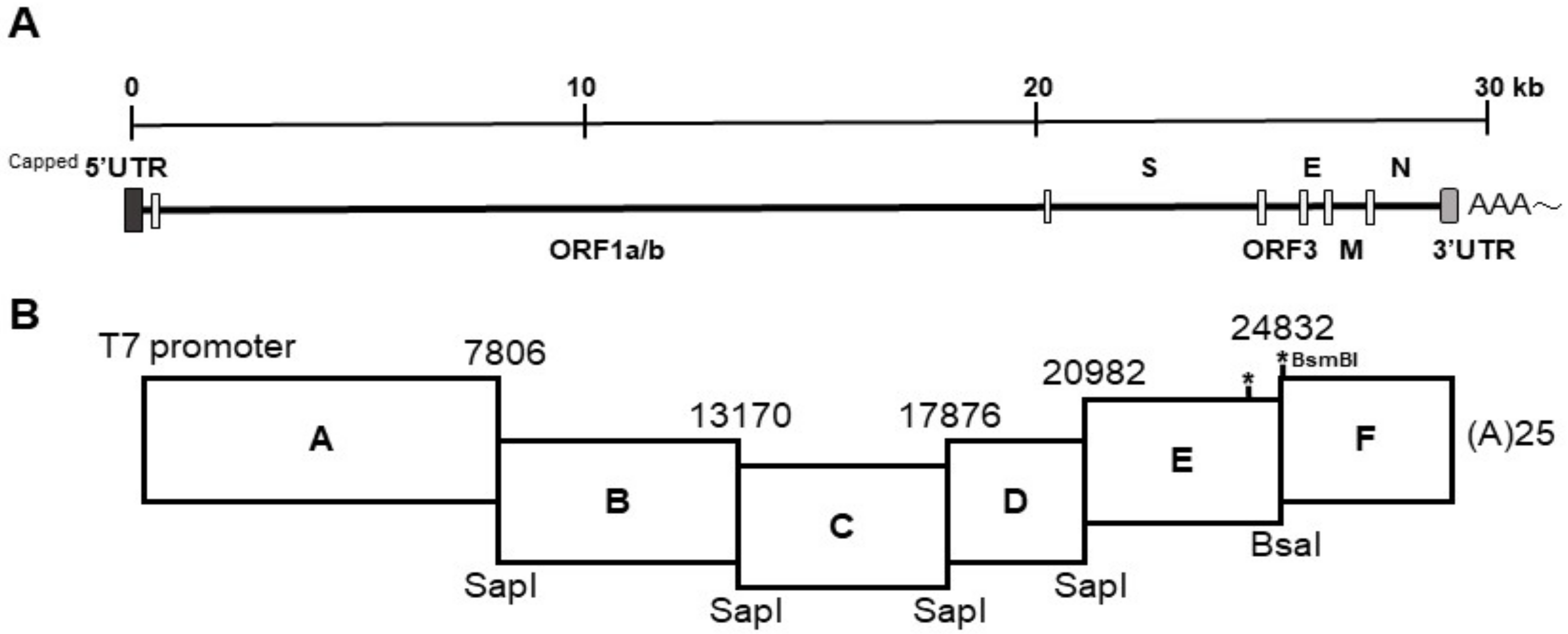
The strategy used to engineer the full-length cDNA clone of PEDVPT-P96, namely iPEDVPT-P96, was modified according to a previously published method [
22]. Briefly, the complete genome of PEDVPT-P96 (Genbank accession No. KY929406) was divided into six fragments by PCR amplification using primer pairs (
Table 1) incorporated with specific type-IIS restriction enzyme sites for seamless ligation. Fragment A contained a T7 promoter sequence at its 5′ end to allow in vitro transcription and the sequence was designed following the article published previously [
19]; a 25-adenosine sequence was added to the 3′ end of fragment F to simulate polyadenylation. A naturally occurring
BsaI site in fragment E was removed by introducing a silent mutation (C24341T) by site-directed mutagenesis according to the previously described protocol [
23]. An additional synonymous mutation (T24841C) was generated near the junction of fragments E and F, to create a novel
BsmBI recognition site. PCR amplicons of all fragments were subcloned into plasmid vectors (pJET1.2; Thermo Fisher Scientific, Waltham, MA, USA). Each subclone was digested with the corresponding type-IIS restriction enzymes as indicated in
Figure 1 and gel-purified using a QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Assembly of the full-length cDNA was conducted by employing T4 ligase (NEB, Ipswich, MA, USA) overnight at 4 °C. The ligated full-length cDNA was phenol-chloroform extracted and in vitro transcribed to full-length RNA transcripts using a mMessage mMachine T7 transcription kit (Ambion, Austin, CA, USA) following the manufacturer’s instructions. The cap analog to GTP ratio was adjusted to 1:1 to increase the yield of full-length transcripts. To facilitate viral recovery, nucleocapsid (N) transcripts were also generated from amplicons flanking the entire N gene with the addition of the T7 promoter sequence and poly-A tail at the 3′ and 5′ ends, respectively. The N transcripts were precipitated using lithium chloride (Ambion, Austin, CA, USA) and purified with ethanol.
2.3. Recovery of the Full-Length cDNA Clone of iPEDVPT-P96
The reaction mixture of full-length transcripts (30 μL) and 5 μg of N transcripts were mixed thoroughly and electroporated into Vero cells in a suspension of 800 μL with 107 cells/mL in RNase-free phosphate buffered saline (PBS) using a Gene Pulser Xcell™ Electroporation System (Bio-Rad, Hercules, CA, USA), with four pulses at 450 V, 50 μF and 2–3 s rest between each pulse. After electroporation, the cells were initially incubated at room temperature for 15 min and then resuspended in DMEM supplemented with 10% FBS in a 75-cm2 flask overnight at 37 °C to allow for recovery. On the next day, the cells were washed twice with Dulbecco's phosphate-buffered saline (DPBS) and maintained in PI medium for an additional 2–3 days until cytopathic effects (CPE) characterized by cell fusion, syncytial cell formation, and cell detachment were observed. The whole flask was subjected to one freeze-and-thaw cycle and the rescued virus was passaged once to generate a viral stock of iPEDVPT-P96 for further use. The viral stock was titrated in Vero cells in a 96-well plate to determine viral titer.
2.4. In Vitro Characterization of the Full-Length cDNA Clone of iPEDVPT-P96
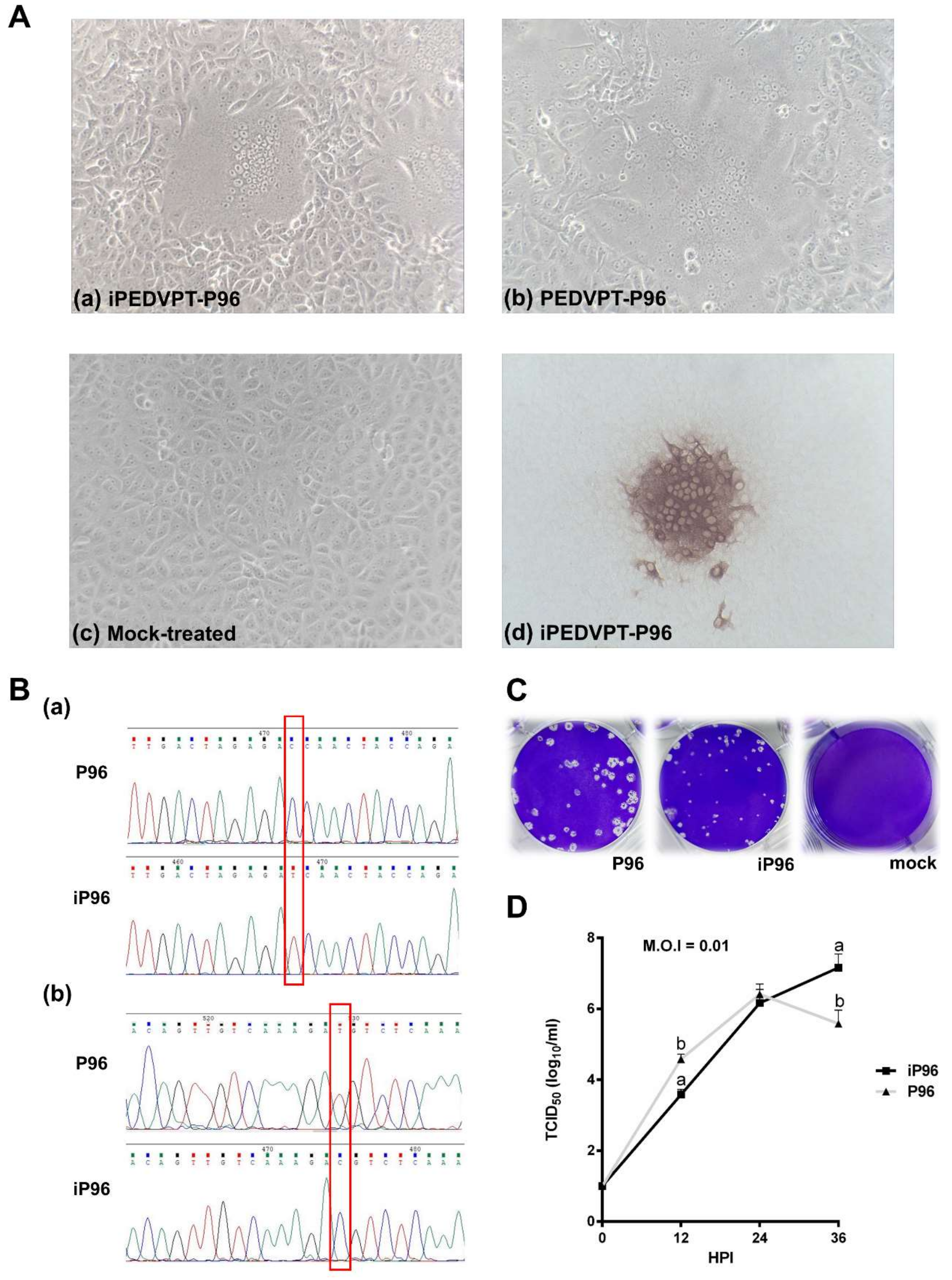
2.4.1. Immunocytochemistry
To detect the PEDV antigen, immunocytochemistry (ICC) was performed as described previously [
15]. Briefly, Vero cells infected with iPEDVPT-P96 showing typical CPE were fixed with 80% ice-cold acetone, air-dried, and incubated with an in-house anti-PEDV N antibody at room temperature (RT) for 1 h. After washing three times with PBS, a polyclonal anti-rabbit/mouse immunoglobulin, EnVision-DAB
+ system (Dako, Carpinteria, CA, USA), was applied for 1 h at RT. Following three washes with PBS, the cells were incubated with 3, 3′-diaminobenzidine (DAB) chromogen from a peroxidase DAB substrate kit (Dako, Carpinteria, CA, USA) according to the manufacturer’s instructions. Positive signals were visualized under an inverted light microscope (Nikon, Tokyo, Japan).
2.4.2. Sequence Analysis
Sequence analysis was conducted as described previously [
15] and a primer pair (SF5 and N-4 listed in
Table 1) targeting the genome between nucleotides 23037 to 25574 was used to identify the presence of marker mutations, C24341T and T24841C, in the iPEDVPT-P96 viral stock.
2.4.3. Comparison of Growth Kinetics and Plaque Morphologies between PEDVPT-P96 and iPEDVPT-P96
The growth characteristics of iPEDVPT-P96 and PEDVPT-P96 in Vero cells were evaluated and compared by examining the growth kinetics and plaque morphologies. Confluent monolayers of Vero cells were prepared on 6-well plates and infected with each virus at the multiplicity of infection (MOI) 0.01 for 1 h at 37 °C in triplicate. To determine the growth kinetics, cells were washed twice with DPBS and then maintained in the PI medium. The supernatants at indicated time points, 0, 12, 24, and 36 h post-inoculation (HPI), were collected and subjected to titration in Vero cells seeded in 96-well plates. Plaque assays were performed to characterize the plaque morphologies. After the adsorption of PEDVs at MOI 0.01, Vero cells were washed twice with DPBS and overlaid with PI medium containing 1% agarose (Invitrogen, Carlsbad, USA) pre-warmed to 42 °C. Upon solidification of the overlays, the plates were incubated for 3 days at 37 °C to allow PEDV-infected Vero cells to produce distinct plaques. The cells were fixed in 3.16% neutral formalin for 1 h at RT. The semisolid overlays were then removed manually and the cells were stained with 1% crystal violet in 20% ethanol and distilled water for 1 min. The viral plaques were inspected after washing off the crystal violet, rinsing the plates with water, and air-drying at RT. The diameters of representative plaques for each virus were measured and compared.
2.5. Animal Experiment
Fifteen, 4-week-old, Large White × Duroc, crossbred piglets that were PEDV-seronegative and PEDV-shedding negative were selected from a conventional pig farm with no history of a G2b PEDV strain infection. These piglets were randomly assigned to three groups, including the PEDVPT-P96 group (n = 5), iPEDVPT-P96 group (n = 5), and mock group (n = 5), and acclimated for one week prior to inoculation. At 5 weeks of age, the piglets in each group were orally inoculated with 4 mL of 5 × 105 TCID50/mL of PEDVPT-P96, 5 × 105 TCID50/mL of the PEDVPT-P96 virus, or PI medium, respectively. To evaluate the protective efficacy induced by iPEDVPT-P96, piglets at 9 weeks of age in all groups were orally challenged with 5 mL 105 TCID50/mL of PEDVPT-P5. The animal experimental procedure was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the National Taiwan University (Taiwan, Republic of China) with approval No.: NTU105EL-00160.
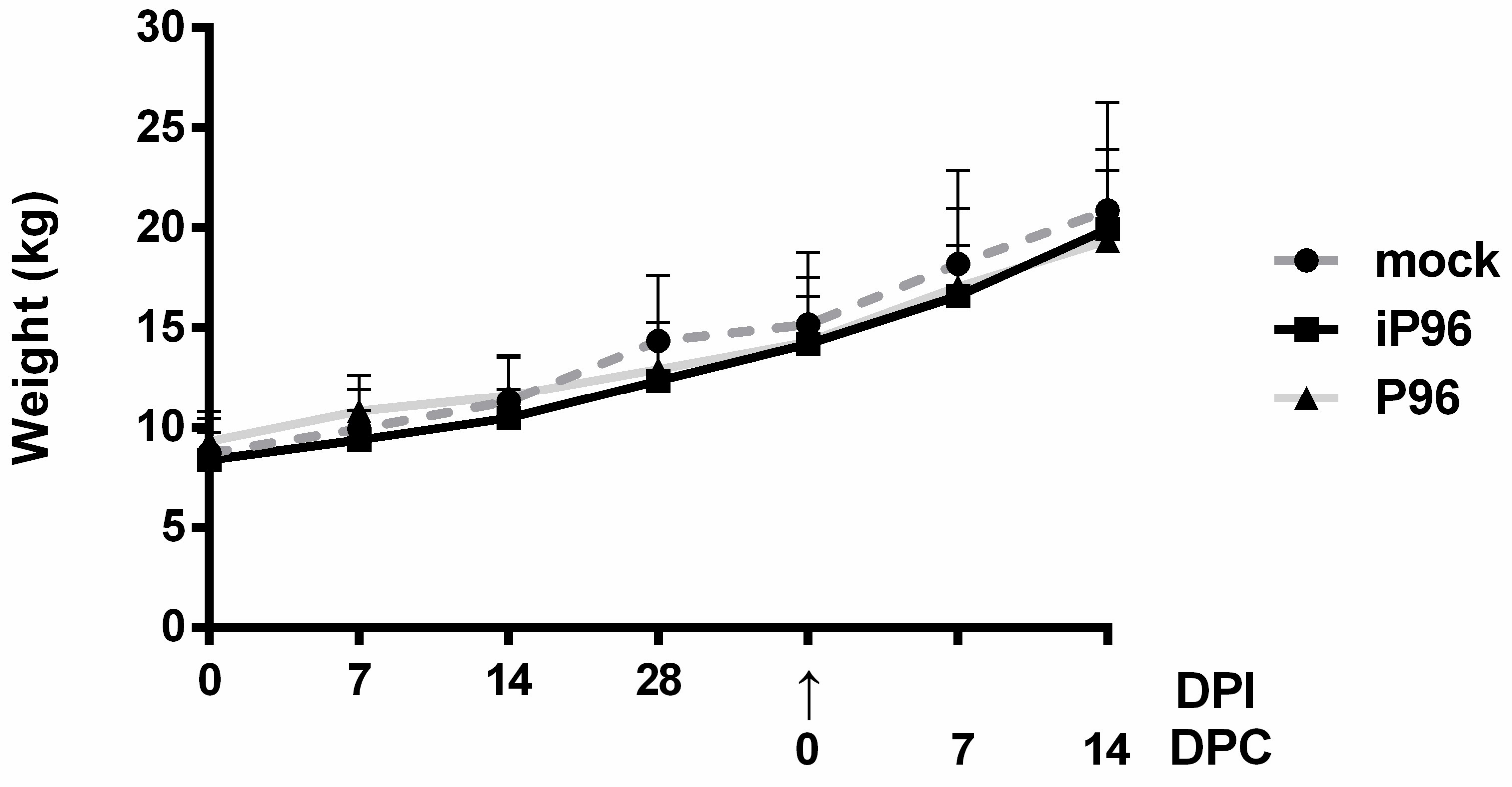
2.5.1. Clinical Signs and Body Weight
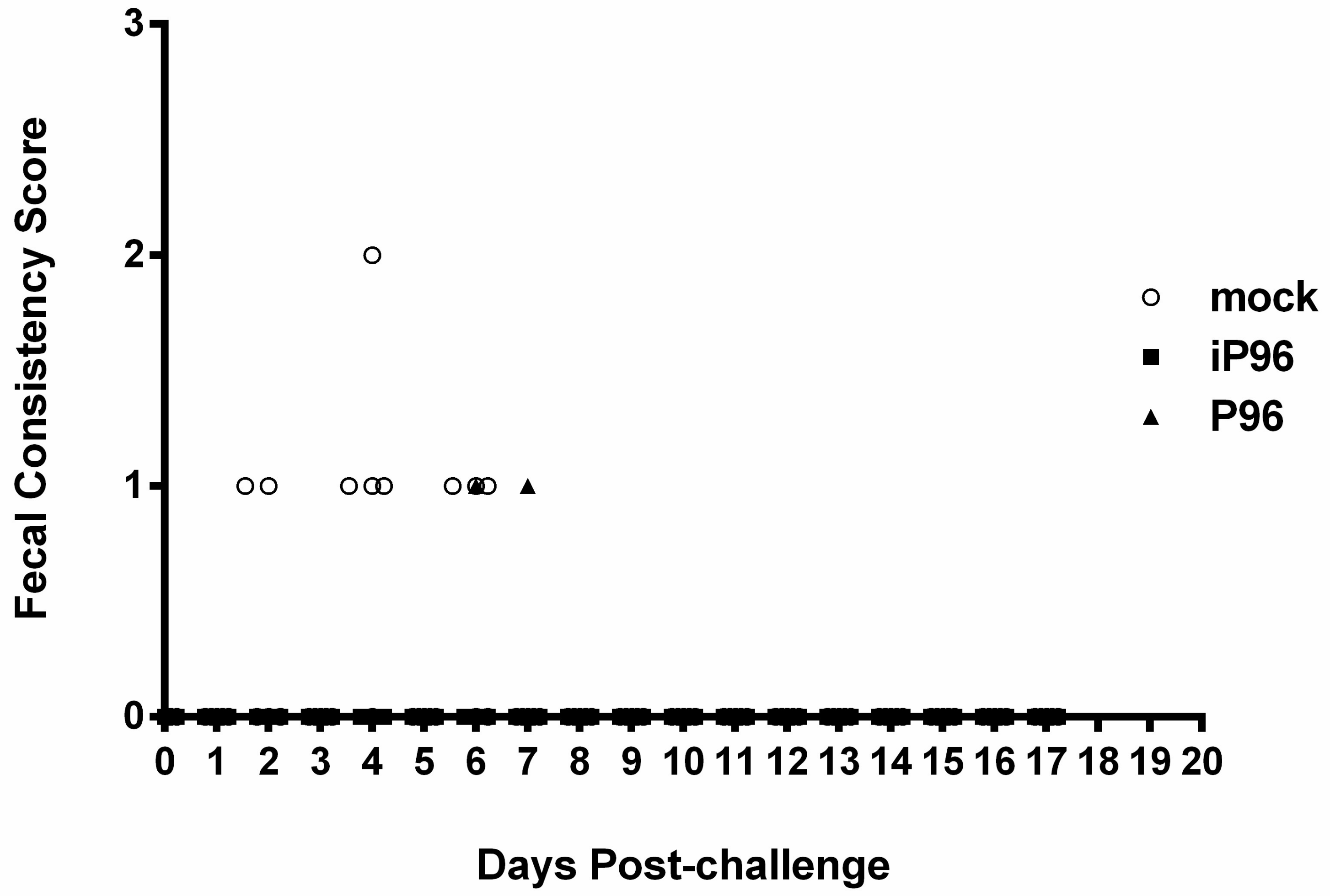
Fecal consistency was monitored daily and scored visually as 0 = normal, 1 = loose, 2 = semi-fluid, and 3 = watery, as described previously [
15]. The body weight of each piglet was measured weekly.
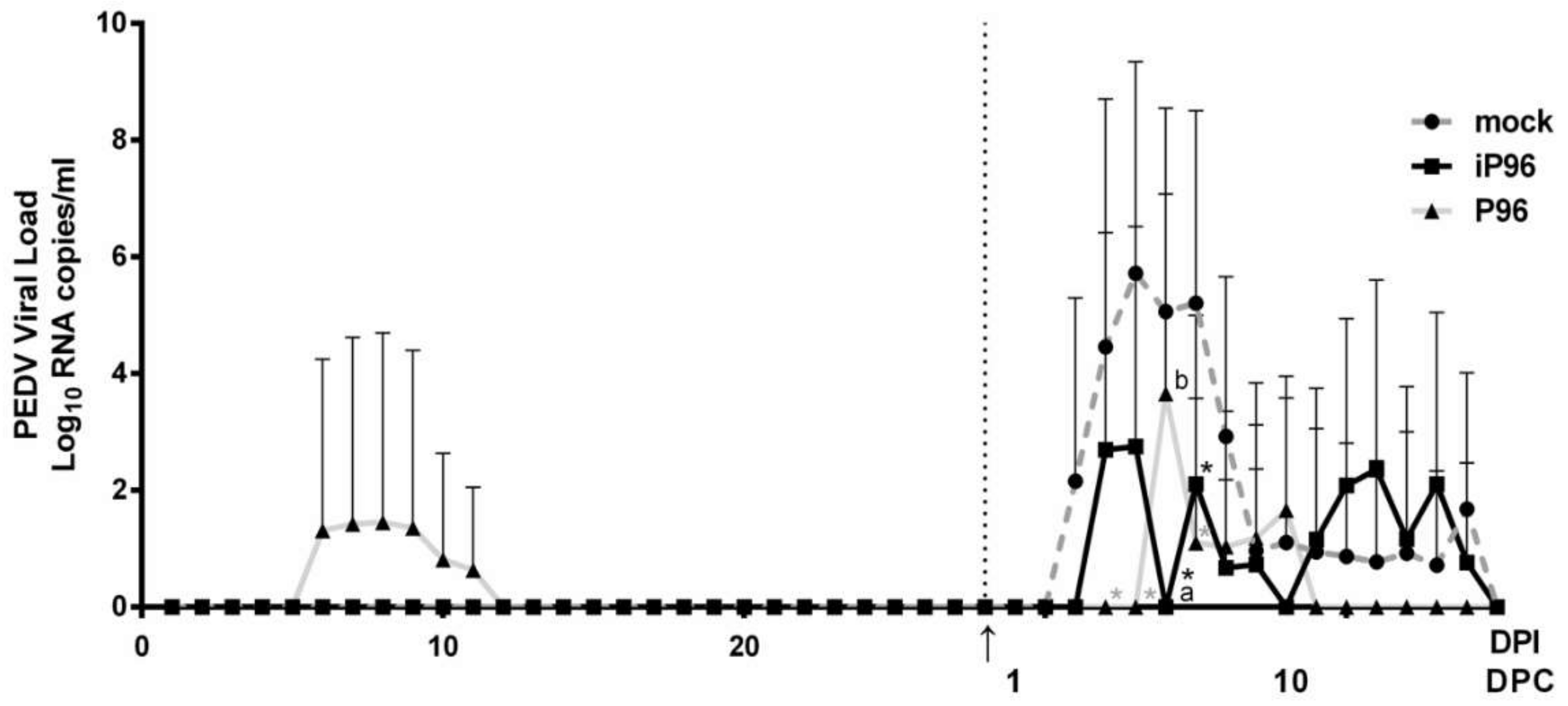
2.5.2. Detection of Fecal PEDV Viral Load
To quantitate the viral RNA in stools, fecal samples collected from rectal swabs were resuspended in 1000 μL DPBS, pulse-vortexed for 5 s and precipitated by centrifugation at 13,000 rpm for 5 min. RNA was extracted automatically from 200 μL of stool suspension on a QIAcube using the Cador Pathogen 96 QIAcube HT Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. cDNA was reverse-transcribed using a QuantiNova Reverse Transcription Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocols and it was used for quantitative real-time PCR analysis (qPCR). qPCR was conducted using the previously published primer-probe set [
4] and a QuantiNova
® Probe PCR Kit (Qiagen, Hilden, Germany) on a CFX96 Thermal Cycler (Bio-Rad, Hercules, CA, USA). The thermal profile comprised an initial denaturation at 95 °C for 2 min followed by 45 cycles of 95 °C for 15 s followed by 60 °C for 15 s. The detection limit of the assay was determined by generating standard curves from serial 10-fold dilutions of known amounts of in vitro transcribed RNA followed by reverse transcription and qPCR quantification as described above. The detection limit was calculated as 100 RNA copies per mL.
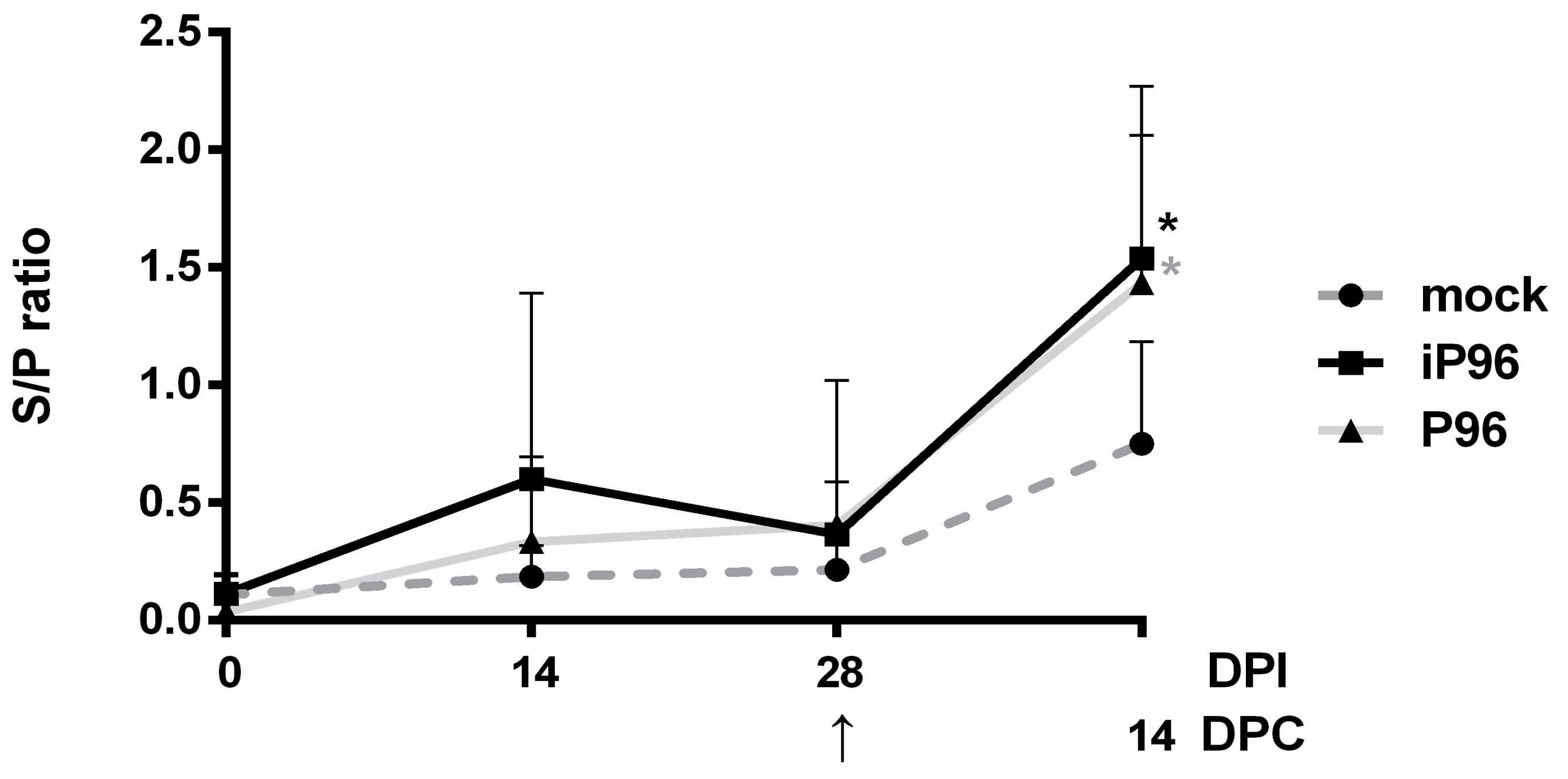
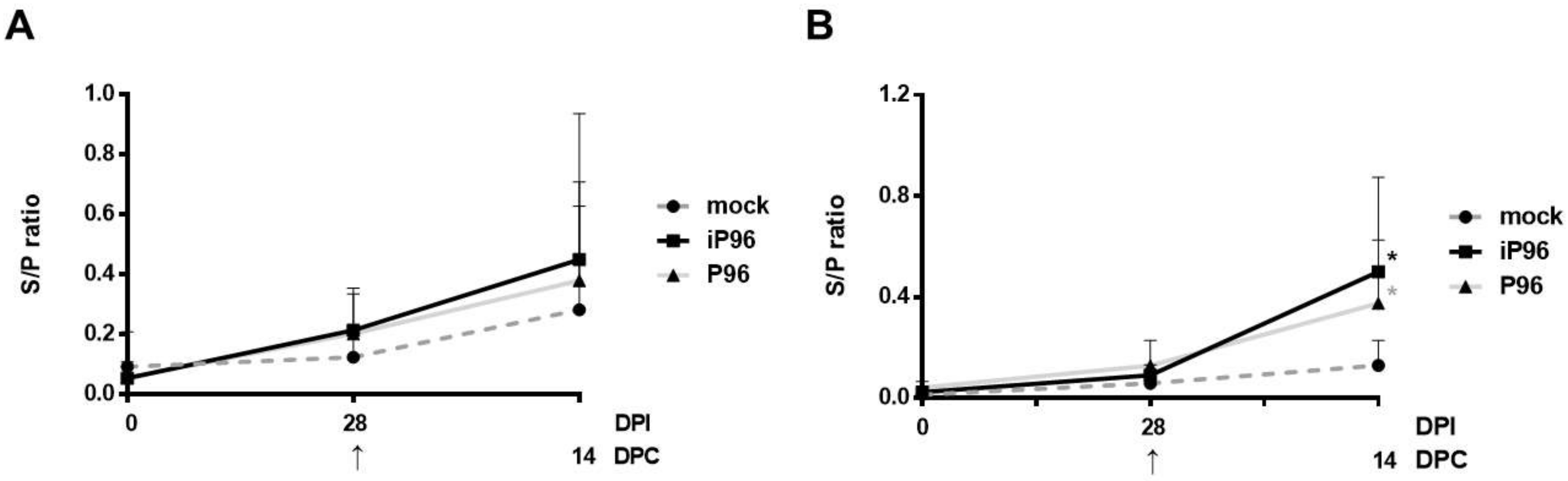
2.5.3. Detection of PEDV-Spike Specific Plasma IgG and Fecal and Salivary IgA
To detect PEDV specific plasma IgG and fecal and salivary IgA, an in-house, PEDV S protein based indirect enzyme-linked immunosorbent assay (ELISA) was conducted as described in the previous study [
24]. In brief, 96-well, flat-bottom microtiter plates (Nunc, Roskilde, Denmark) were coated with 2 μg/mL purified recombinant PEDVPT S protein (200 ng/well) and incubated overnight at 4 °C. The plates were washed six times with 100 μL of PBST (PBS containing 0.05% Tween 20) and then blocked with 300 μL of blocking buffer (1% bovine serum albumin in PBS) at RT for 1 h. For the detection of plasma IgG, 100 μL of 40-fold diluted plasma samples in blocking buffer were added following six washes and incubated at RT for 1 h. For fecal and salivary IgA, 100 μL of eluted fecal suspension and saliva at 1:2 dilution in blocking buffer were added following six washes and kept overnight at 4 °C. After incubation, the samples were discarded and the plates were washed six times. To detect plasma IgG, and the fecal and salivary IgA, 100 μL of either horseradish peroxidase (HRP) conjugated goat anti-pig IgG (Kirkegaard & Perry Laboratories, Milford, MA, USA) at 1:1000 dilution, or goat-anti-pig IgA (Abcam, Cambridge, UK) diluted 1:10,000 were added, respectively, and incubated at RT for 1 h. Following a wash step, 50 μL of tetramethylbenzidine (TMB) substrate solution (Kirkegaard and Perry Laboratories) was added to allow color development at RT for 10 min. The reactions were terminated by adding 50 μL of TMB stop solution (Kirkegaard and Perry Laboratories) to each well. The optical density (OD) at 405 nm was measured on an ELISA reader (Molecular Devices, Sunnyvale, CA, USA). The antibody titers were expressed as sample-to-positive control ratio (S/P ratio) values.
2.5.4. Viral Neutralization (VN) Assay
Plasma samples of piglets were heated at 56 °C for 30 min to inactivate complement prior to use. For each well, mixtures containing 50 μL of PEDVPT-P5 virus (50 viral particles) and 50 μL of 2-fold diluted plasma samples in PI medium were incubated at 37 °C for 1 h before applying to Vero cells (2 × 104/well). After incubation with the virus-plasma mixture for 1 h, Vero cells were washed twice and maintained in PI medium for 24 h. Cytopathic effects were detected using inverted light microscopy (Nikon, Tokyo, Japan). The neutralizing titer was defined as the highest dilution without CPE.
2.6. Statistical Analysis
The results of body weight, antibody titers, viral titer of growth kinetics at each time point, and fecal viral shedding were analyzed statistically on GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA) with Two-way ANOVA by time. A p value less than 0.05 was considered statistically significant.
4. Discussion
In the present study, we described the first development of an infectious cDNA clone of iPEDVPT-P96, and evaluated it’s in vitro and in vivo characteristics. Compared to the parental PEDVPT-P96 virus, iPEDVPT-P96 replicated more slowly in the beginning and reached a similar peak viral titer with similar but more uniform plaque sizes, suggesting that the composition of viral quasispecies in iPEDVPT-P96 is less complex than that in the original PEDVPT-P96 stock. Moreover, neither fecal PEDV RNA shedding nor a PEDV-associated clinical illness was detected in conventional 5-week-old piglets inoculated with the iPEDVPT-P96 virus, indicating a further attenuated phenotype in vivo. Importantly, piglets in the iPEDVPT-P96-inoculated group showed comparable levels of anti-PEDV specific plasma IgG, fecal/salivary IgA, plasma neutralizing antibody titers, and a weakened but modest immunoprotection against the virulent PEDVPT-P5 challenge compared to the parental PEDVPT-P96-inoculated piglets. Taken together, our results suggest that iPEDVPT-P96 is immunogenic in piglets and could be a potential safe viral vector candidate for vaccine development.
While inoculation with iPEDVPT-P96 was demonstrated to completely protect the piglets from developing diarrhea after challenge with the virulent PEDVPT-P5, the iPEDVPT-P96-inoculated group showed an earlier onset and longer duration of fecal PEDV RNA shedding with a higher peak value than that of the parental PEDVPT-P96-inoculated group. These data suggested that iPEDVPT-P96 conferred a relatively weakened protection than that of the parental PEDVPT-P96. Considering that the major variation between the PEDVPT-P96 and iPEDVPT-P96 viruses should be the heterogeneity of viral population as noted in plaque assays wherein the iPEDVPT-P96 virus produced more uniform plaques than those of the parental PEDVPT-P96 virus in Vero cells, we speculate that the decrease in quasispecies diversity in iPEDVPT-P96 may partly contribute to its further attenuation in vivo. To investigate the speculation, we conducted the next generation sequencing (NGS) to determine the quasispecies diversity of both viruses by exploring the recovered variants against our previous published sequence of PEDVPT-P96 generated by Sanger sequence (see
Table S1). The results clearly demonstrated that the PEDVPT-P96 carried a greater sequence diversity than that of the iPEDVPT-P96, including 23 single nucleotide variants (SNV) and resultant 16 amino acid substitutions; of interest, seven SNV were found in the spike gene and 3 of them were the same as the virulent PEDVPT-P5. For iPEDVPT-P96, excluding the artificially introduced marker mutations and those derived from the SNV of PEDVPT-P96 upon the initial construction of subclones of iPEDVPT-P96, only 3 SNV were uncovered. Indeed, generating viruses with high-fidelity replication has been proposed as a rational strategy to develop genetically stable and safe attenuated vaccines [
25,
26]. However, for attenuated viruses, the in vivo fitness and antigenicity are already abated after serial cell culture passage. On the basis of quasispecies theory that the cooperative interplay between different variants determines viral characteristics including virulence [
27,
28], it is possible that the consensus sequence used for constructing iPEDVPT-P96 no longer sustained the original affinity of virus-host interaction or viral replication in enterocytes, which therefore impaired viral entry and/or limited the viral infection. Furthermore, the limited diversity of the S gene in the iPEDVPT-P96 viral stock might also diminish the potency and protective broadness of the induced antibody responses explaining the comparable level of systemic and mucosal antibody response but weakened protection in the iPEDVPT-P96-inoculated group. This speculation could be tested by comparing the tissue tropism and the quantity of PEDV antigens of both PEDVPT-P96 and iPEDVPT-P96 in enterocytes using a 7-day-old piglet model. Other explanation for the attenuation of iPEDVPT-P96 might be attributed to the possible addition of the five nucleotides “GGAGA” at the very extreme of the 5′UTR based on our primer design (see
Table 1). Considering the location that these additional nucleotides sequence should neither alter the secondary structures of the SL1 stem-loop, that serve as
cis-acting elements required for driving subgenomic RNA synthesis nor change any consensus transcription regulatory sequences 5′-XUA(A/G)AC-3′, we assumed that the effect of these additional nucleotides in the rescued iPEDVPT-P96 virus on replication might be minimal. Nonetheless, further studies are required to determine the potential effect of these five additional nucleotides on iPEDVPT-P96 replication.
In the present study, the profile of fecal PEDV RNA shedding and the pattern of antibody responses after inoculation with PEDVPT-P96 appeared milder than those observed in our previous findings despite the fact that the same viral stock was used and that the conventional piglets used in this study were purchased from the same pig farm and housed in the same animal facility as described previously [
15]. That is, it seemed that the conventional piglets used in the current study were more resistant to the PEDVPT-P96 infection. Several host factors such as genetic variation, type of feed, gut microflora, and immune status among different litters at the time of inoculation may play an important role in the varying severity of clinical outcomes and immune responses [
9]. Ideally, the discrepancy between the previous experiment might be minimized by expanding the size of groups, particularly if we chose conventional piglets as our target animals. Nevertheless, due to the difficulty in collecting large numbers of PEDV-negative piglets in Taiwan where PED has become endemic, we decided to use five piglets per treatment to reach a statistical effect practice. Although the use of conventional pigs often magnifies those variables and results in higher variation in the experimental results, to mimic the pathogenicity and immunoprotection of PEDV in field conditions, the conventional pig model is still a preferred as a preclinical trial model.
Since the sudden appearance of the severe acute respiratory syndrome-coronavirus (SARS-CoV) in the early 21st century, the emergence and re-emergence of coronaviruses continually threatens the global public and animal health by causing severe illnesses, by having a high potential of zoonosis, and by causing great economic losses, indicating a desperate need for an effective and readily responsive vaccine platform. The large genome size of coronaviruses warrants a great tolerance for foreign genes and subsequent expression of heterologous antigens [
29,
30]. In addition, the insertion of other antigens by replacing the accessory ORF3 gene or creating a novel expression cassette is also an attractive approach to design multivalent vaccines [
17,
18,
21]. Under this concept, iPEDVPT-P96 could be a potential safe vaccine backbone that facilitates the prompt generation of chimeric vaccines with the induction of mucosal immunity. For instance, viral antigenicity can be manipulated by replacing the iPEDVPT-P96 S gene with that of other PEDV or emerging swine coronaviruses although the potential loss of attenuation due to the S substitution requires further clarification. However, it is noteworthy that the inherent genetic instability of coronaviruses due to high recombination and mutation frequency is also a matter of concern as it raises the possibility of virulence reversion and acquisition of new tropism [
31]. Besides, the intrinsic characteristics of the gene of interest, the targeted locus within the coronavirus genome, may also affect the expression level and stability of the recombinant viruses [
30]. Accordingly, further characterization of the virulence variation and genetic stability of iPEDVPT-P96 after different genetic modifications needs to be conducted.
In this article, we described the first successful construction of an attenuated G2b PEDV infectious cDNA clone of the iPEDVPT-P96 virus and demonstrated the maintenance of fitness in vitro along with further attenuation in vivo. We also proposed that the initial low quasispecies diversity and the additional 5 nucleotides at the 5′ end of iPEDVPT-P96 may contribute to a further attenuated phenotype and potentially less effective immunoprotection against the virulent strain challenge. Based on the results of antibody response, a prime-boost strategy or further optimization of iPEDVPT-P96 antigenicity is essential to induce sufficient protective immunity in all recipients. Together, the full-length cDNA clone of iPEDVPT-P96 generated herein is expected to provide an access to study the attenuation determinants of PEDVPT-P96 and establish a PEDVPT-P96-based recombinant vector as a vaccine platform for developing multivalent vaccines for PEDV and other porcine pathogens.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}