
Synthesis and In Vitro Activity Assessment of Novel Silicon Oxycarbide-Based Bioactive Glasses




 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
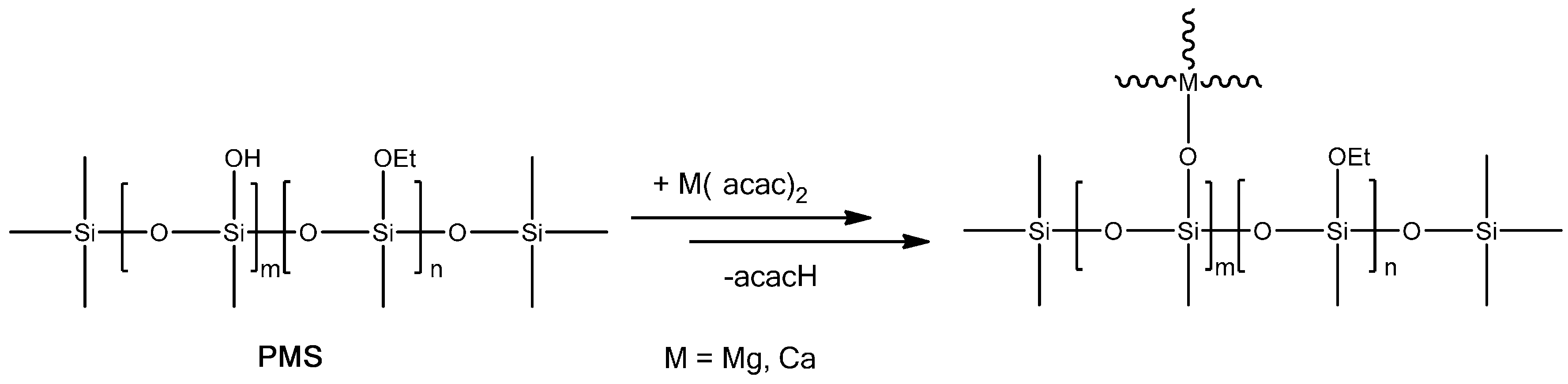
2.1. SiOC and SiCaMgOC Preparation
2.2. Structural Characterization of SiOC and SiCaMgOC Powders
2.3. In Vitro Acellular Assessment of the Bioactivity of the SiOC and SiCaMgOC Powders: Cytotoxicity Tests
3. Results and Discussion
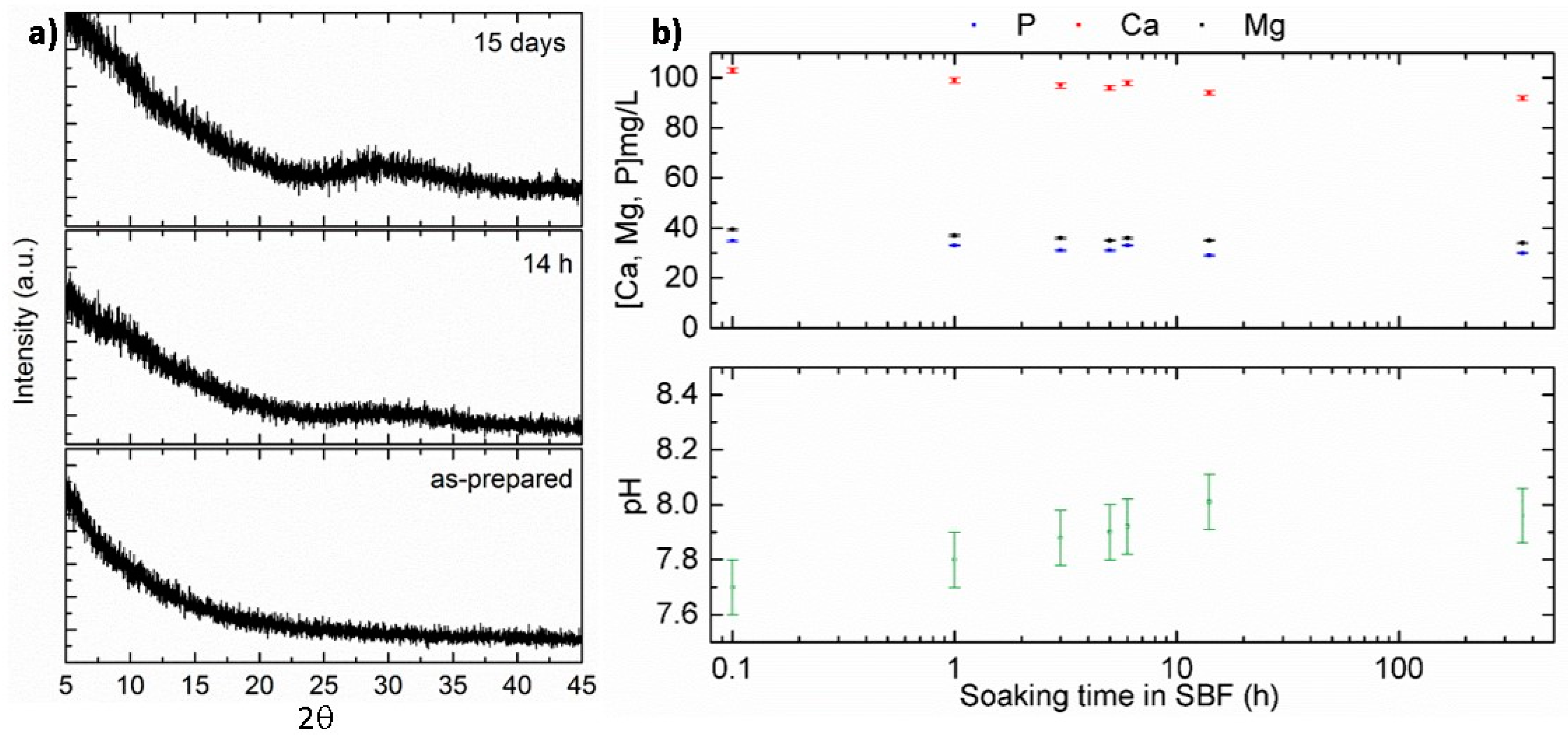
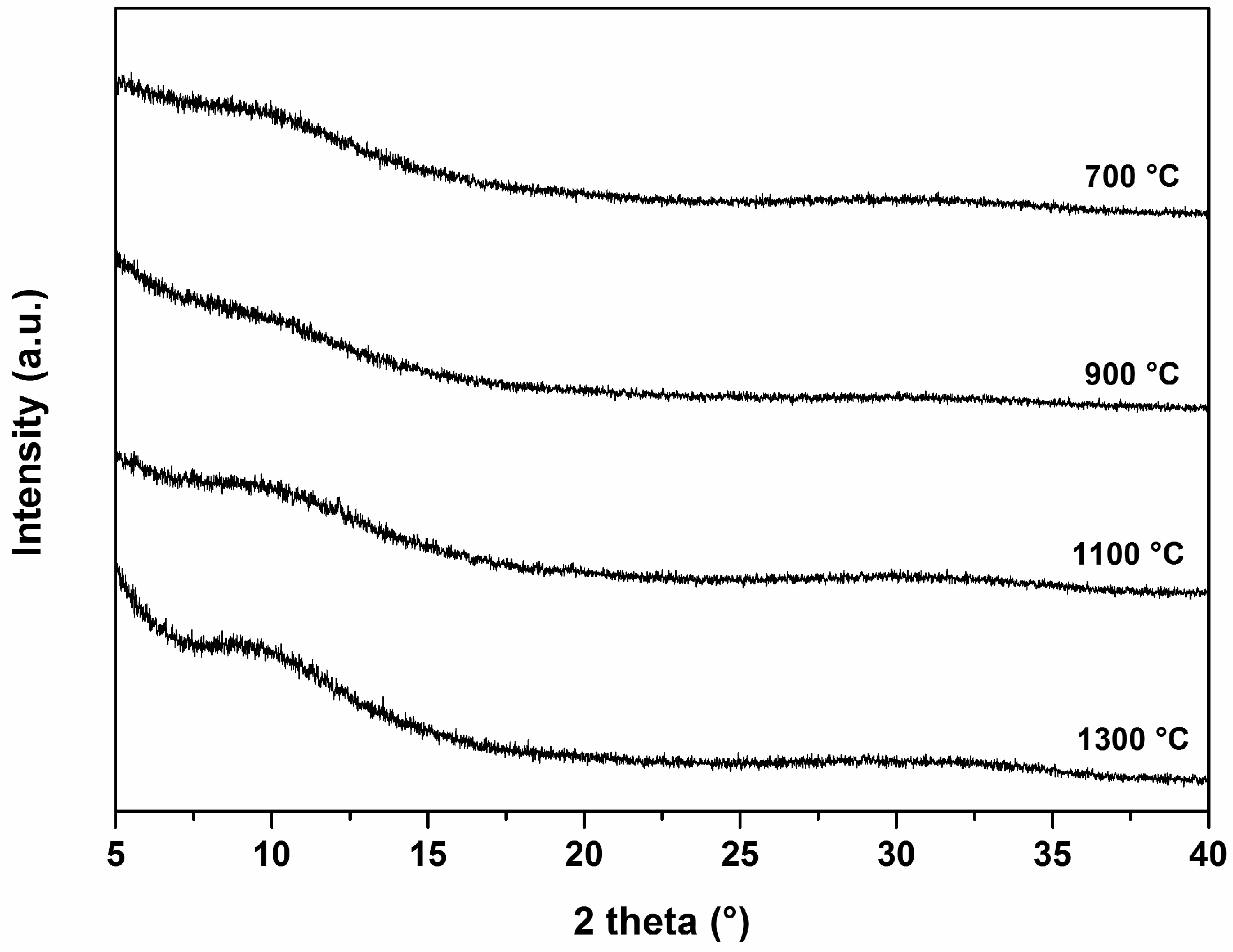
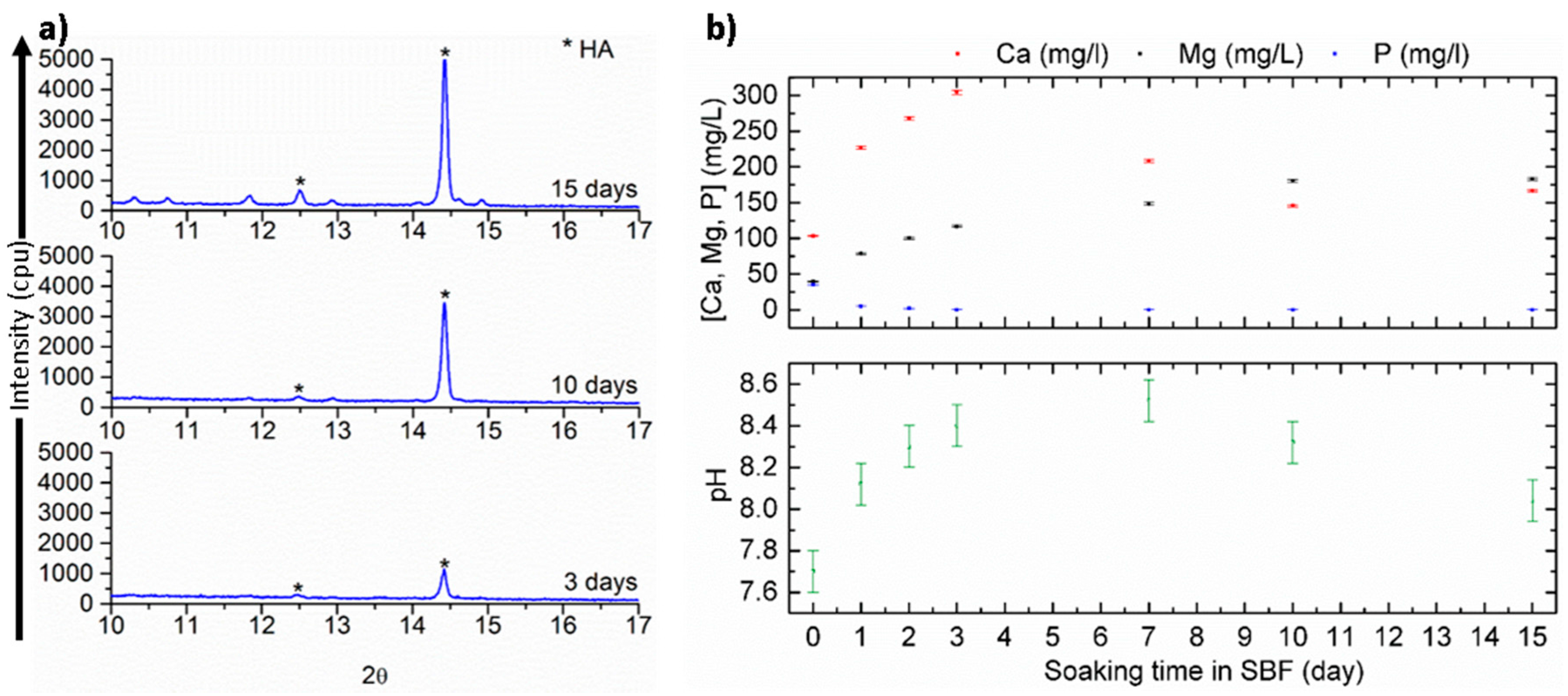
3.1. Material Characterization and Acellular Bioactivity
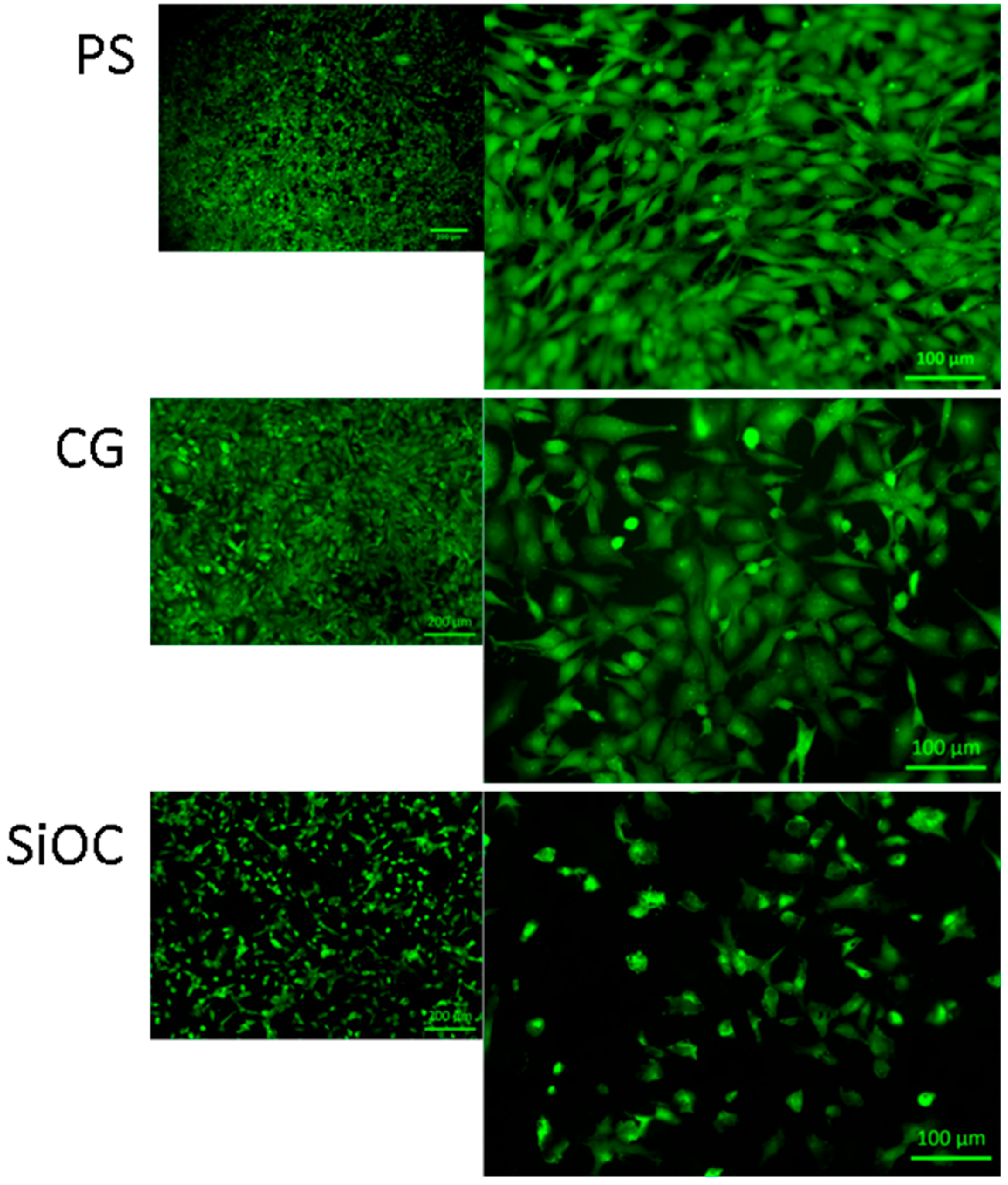
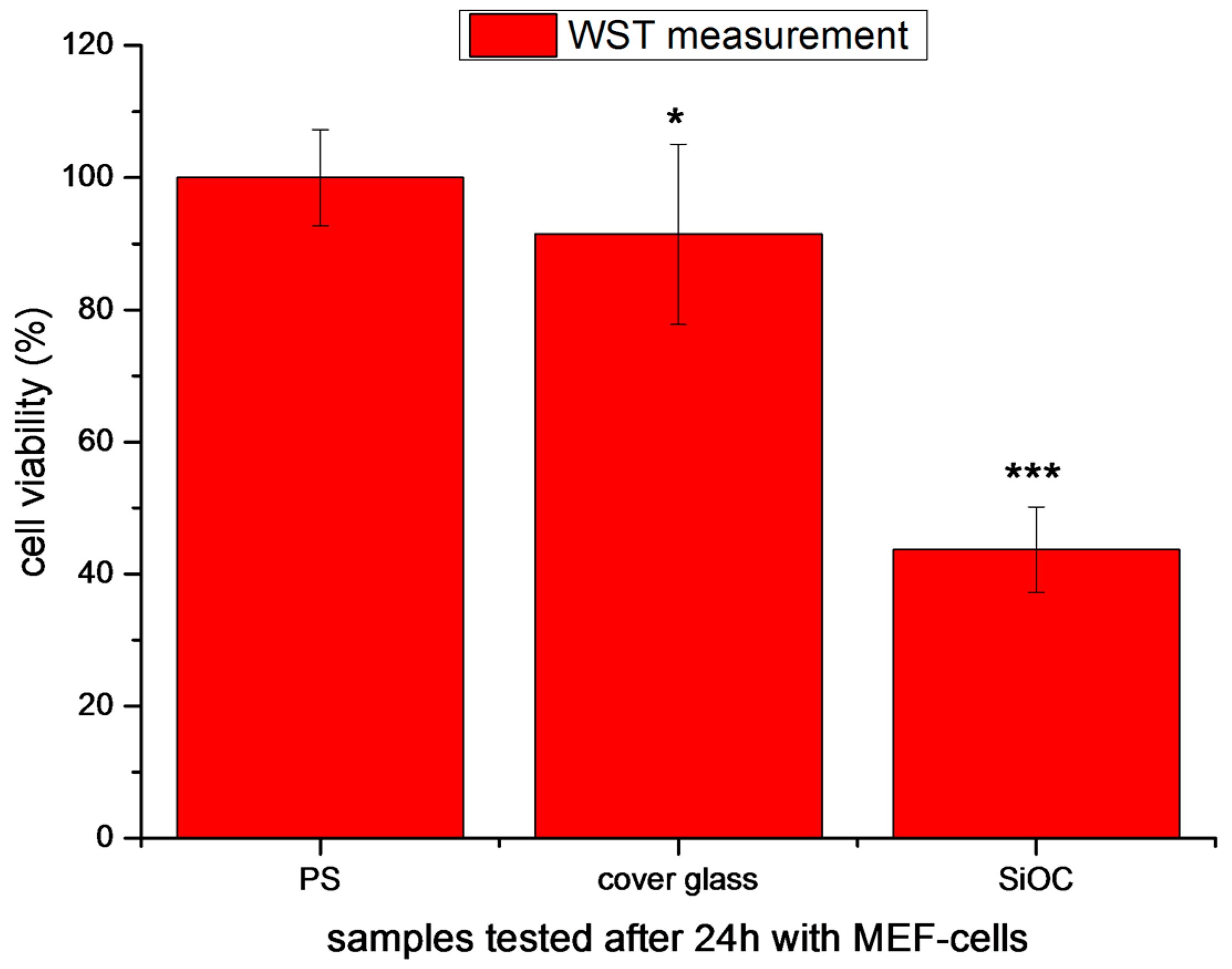
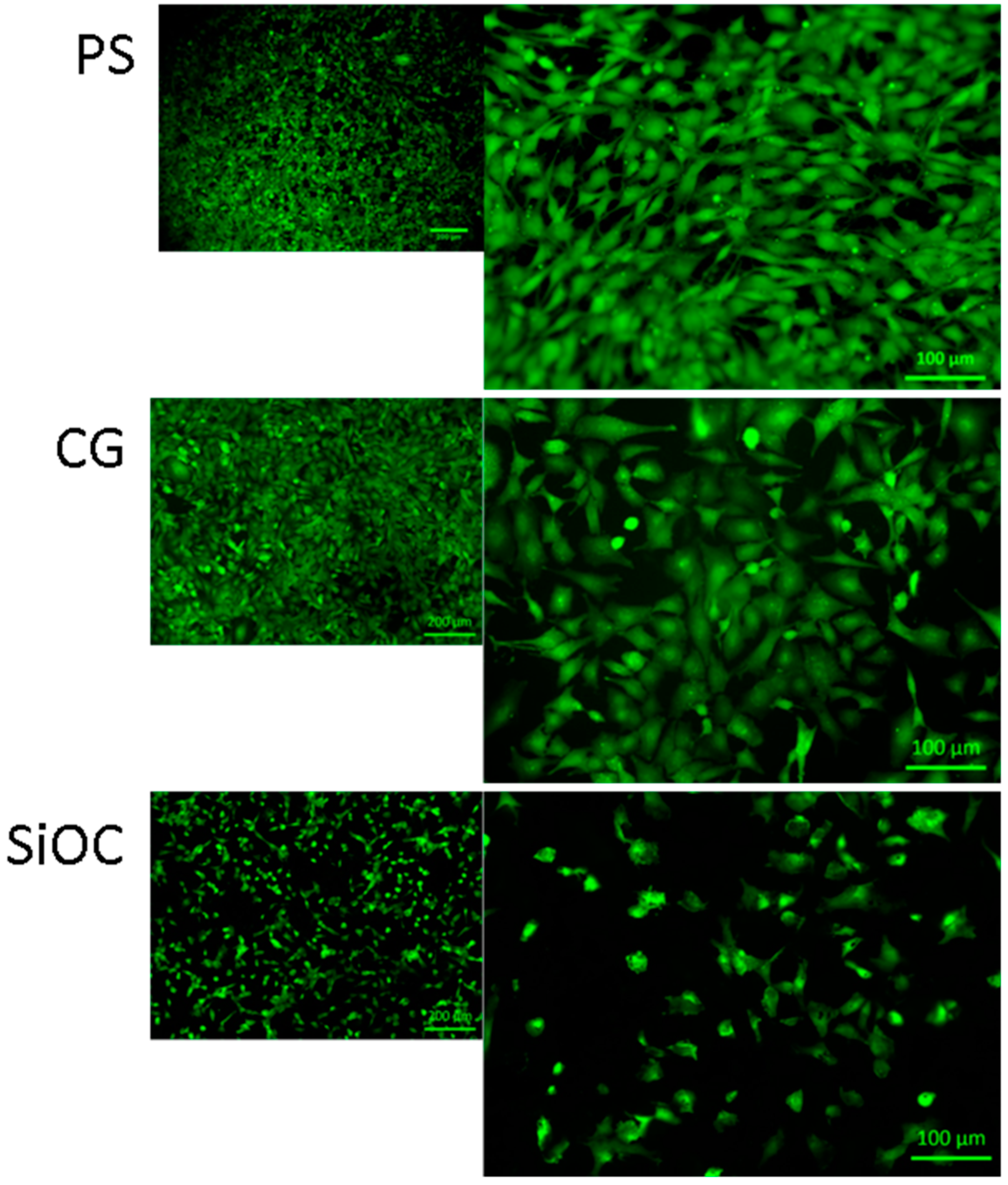
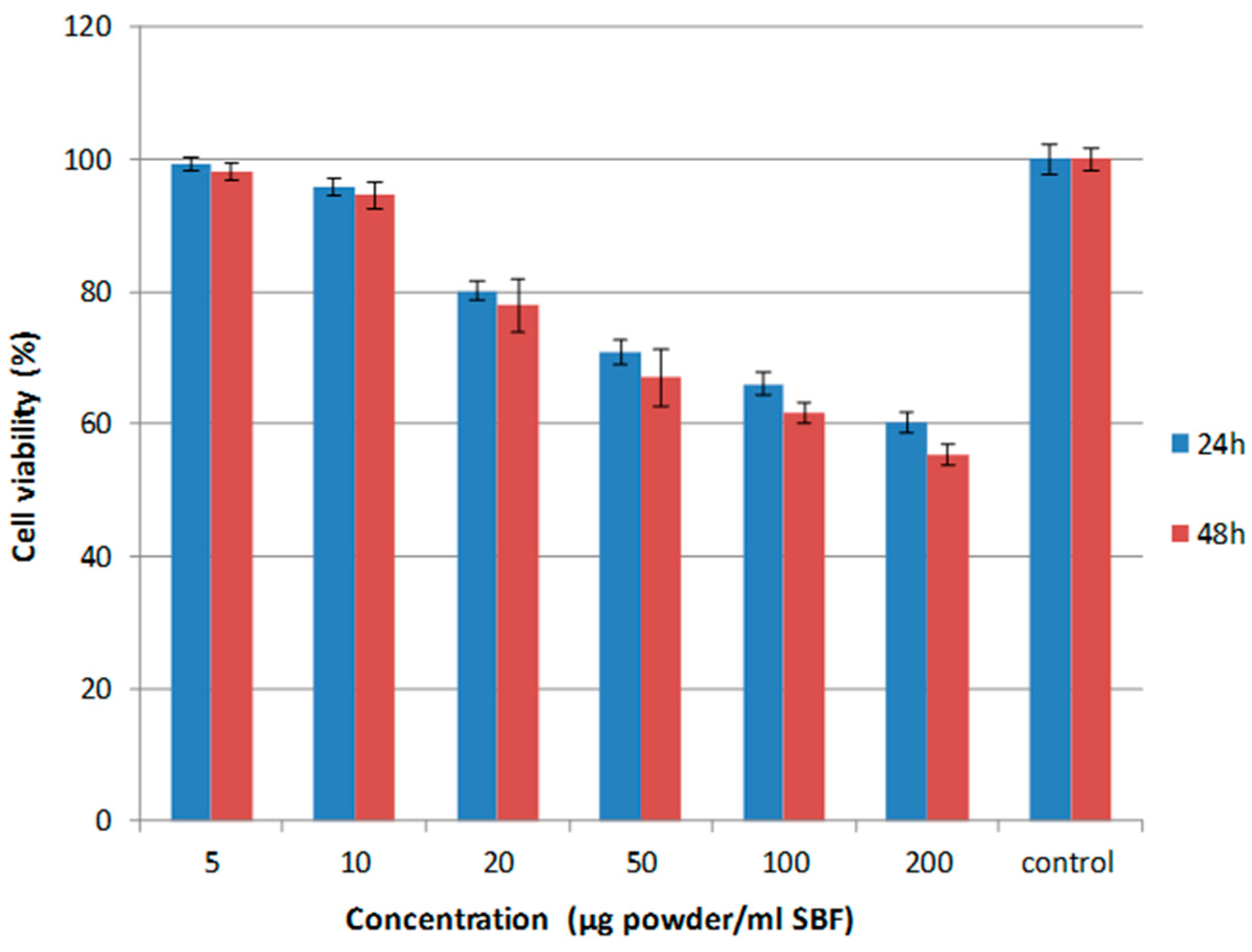
3.2. Cytotoxicity Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hench, L.L. The story of bioglass. J. Mater. Sci. Mater. Med. 2006, 17, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Best, S.M.; Bonfield, W.; Gibson, I.R.; Hing, K.A.; Damien, E.; Revell, P.A. A comparative study on the in vivo behavior of hydroxyapatite and silicon substituted hydroxyapatite granules. J. Mater. Sci. Mater. Med. 2002, 13, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Andersson, Ö.H.; Kangasniemi, I. Calcium phosphate formation at the surface of bioactive glass in vitro. J. Biomed. Mater. Res. 1991, 25, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, C.; Kushitani, H.; Kokubo, T.; Kotani, S.; Yamamuro, T. Apatite formation on the surface of ceravital-type glass–ceramic in the body. J. Biomed. Mater. Res. 1991, 25, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Neo, M.; Nakamura, T.; Ohtsuki, C.; Kokubo, T.; Yamamuro, T. Apatite formation on three kinds of bioactive material at an early stage in vivo: A comparative study by transmission electron microscopy. J. Biomed. Mater. Res. 1993, 27, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Hench, L.L. Bioactive materials. Ceram. Int. 1996, 22, 493–507. [Google Scholar] [CrossRef]
- Jones, J.R.; Ehrenfried, L.M.; Hench, L.L. Optimising bioactive glass scaffolds for bone tissue engineering. Biomaterials 2006, 27, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Z.; Thompson, I.D.; Boccaccini, A.R. 45s5 bioglass-derived glass–ceramic scaffolds for bone tissue engineering. Biomaterials 2006, 27, 2414–2425. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, P.; Jones, J.R.; Hench, L.L. Bioactive sol-gel foams for tissue repair. J. Biomed. Mater. Res. 2002, 59, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.R. Review of bioactive glass: From hench to hybrids. Acta Biomater. 2013, 9, 4457–4486. [Google Scholar] [CrossRef] [PubMed]
- Midha, S.; van den Bergh, W.; Kim, T.B.; Lee, P.D.; Jones, J.R.; Mitchell, C.A. Bioactive glass foam scaffolds are remodelled by osteoclasts and support the formation of mineralized matrix and vascular networks in vitro. Adv. Healthcare Mater. 2013, 2, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Baino, F.; Fiorilli, S.; Vitale-Brovarone, C. Bioactive glass-based materials with hierarchical porosity for medical applications: Review of recent advances. Acta Biomater. 2016, 42, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Brink, M. The influence of alkali and alkaline earths on the working range for bioactive glasses. J. Biomed. Mater. Res. 1997, 36, 109–117. [Google Scholar] [CrossRef]
- Brink, M.; Turunen, T.; Happonen, R.-P.; Yli-Urpo, A. Compositional dependence of bioactivity of glasses in the system Na2O-K2O-MgO-CaO-B2O3-P2O5-SiO2. J. Biomed. Mater. Res. 1997, 37, 114–121. [Google Scholar] [CrossRef]
- Brown, R.F.; Day, D.E.; Day, T.E.; Jung, S.; Rahaman, M.N.; Fu, Q. Growth and differentiation of osteoblastic cells on 13–93 bioactive glass fibers and scaffolds. Acta Biomater. 2008, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Kushitani, H.; Sakka, S.; Kitsugi, T.; Yamamuro, T. Solutions able to reproduce in vivo surface-structure changes in bioactive glass–ceramic a-w3. J. Biomed. Mater. Res. 1990, 24, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Koudehi, M.F.; Fooladi, A.A.I.; Mansoori, K.; Jamalpoor, Z.; Amiri, A.; Nourani, M.R. Preparation and evaluation of novel nano-bioglass/gelatin conduit for peripheral nerve regeneration. J. Mater. Sci.-Mater. Med. 2014, 25, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, E.; Linck, C.; Fasel, C.; Müller, M.; Kleebe, H.J.; Riedel, R. Polymer-derived SiOC/ZrO2 ceramic nanocomposites with excellent high-temperature stability. J. Am. Ceram. Soc. 2010, 93, 241–250. [Google Scholar] [CrossRef]
- Ionescu, E.; Papendorf, B.; Kleebe, H.-J.; Riedel, R. Polymer-derived silicon oxycarbide/hafnia ceramic nanocomposites. Part II: Stability toward decomposition and microstructure evolution at t ≫ 1000 °C. J. Am. Ceram. Soc. 2010, 93, 1783–1789. [Google Scholar] [CrossRef]
- Ionescu, E.; Kleebe, H.-J.; Riedel, R. Silicon-containing polymer-derived ceramic nanocomposites (pdc-ncs): Preparative approaches and properties. Chem. Soc. Rev. 2012, 41, 5032–5052. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, E.; Terzioglu, C.; Linck, C.; Kaspar, J.; Navrotsky, A.; Riedel, R. Thermodynamic control of phase composition and crystallization of metal-modified silicon oxycarbides. J. Am. Ceram. Soc. 2013, 96, 1899–1903. [Google Scholar] [CrossRef]
- Depardieu, M.E.C.; Henry, D. Marking Coating. Patent WO2013154893 A1, 2013. [Google Scholar]
- Maeno, S.; Niki, Y.; Matsumoto, H.; Morioka, H.; Yatabe, T.; Funayama, A.; Toyama, Y.; Taguchi, T.; Tanaka, J. The effect of calcium ion concentration on osteoblast viability, proliferation and differentiation in monolayer and 3D culture. Biomaterials 2005, 26, 4847–4855. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J. The calcium-sensing receptor in bone cells: A potential therapeutic target in osteoporosis. Bone 2010, 46, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Valerio, P.; Pereira, M.M.; Goes, A.M.; Leite, M.F. Effects of extracellular calcium concentration on the glutamate release by bioactive glass (bg60s) preincubated osteoblasts. Biomed. Mater. 2009, 4, 045011. [Google Scholar] [CrossRef] [PubMed]
- Zreiqat, H.; Howlett, C.R.; Zannettino, A.; Evans, P.; Schulze-Tanzil, G.; Knabe, C.; Shakibaei, M. Mechanisms of magnesium-stimulated adhesion of osteoblastic cells to commonly used orthopaedic implants. J. Biomed. Mater. Res. 2002, 62, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Yoshida, Y.; Okazaki, M.; Shimazu, A.; Uchida, T.; Kubo, T.; Akagawa, Y.; Hamada, Y.; Takahashi, J.; Matsuura, N. Synthesis of functionally graded MgCO3 apatite accelerating osteoblast adhesion. J. Biomed. Mater. Res. 2002, 62, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Maçon, A.L.B.; Kim, T.B.; Valliant, E.M.; Goetschius, K.; Brow, R.K.; Day, D.E.; Hoppe, A.; Boccaccini, A.R.; Kim, I.Y.; Ohtsuki, C.; et al. A unified in vitro evaluation for apatite-forming ability of bioactive glasses and their variants. J. Mater. Sci. Mater. Med. 2015, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Crespiera, S.; Ionescu, E.; Kleebe, H.-J.; Riedel, R. Pressureless synthesis of fully dense and crack-free sioc bulk ceramics via photo-crosslinking and pyrolysis of a polysiloxane. J. Eur. Ceram. Soc. 2011, 31, 913–919. [Google Scholar] [CrossRef]
- Karlsson, K.H.; Fröberg, K.; Ringbom, T. A structural approach to bone adhering of bioactive glasses. J. Non-Cryst. Solids 1989, 112, 69–72. [Google Scholar] [CrossRef]
- Kokubo, T. Bioactive glass ceramics: Properties and applications. Biomaterials 1991, 12, 155–163. [Google Scholar] [CrossRef]
- Li, P.; Zhang, F. The electrochemistry of a glass surface and its application to bioactive glass in solution. J. Non-Cryst. Solids 1990, 119, 112–118. [Google Scholar] [CrossRef]
- Li, P.; Ohtsuki, C.; Kokubo, T.; Nakanishi, K.; Soga, N.; de Groot, K. The role of hydrated silica, titania, and alumina in inducing apatite on implants. J. Biomed. Mater. Res. 1994, 28, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Smeets, R.; Kolk, A.; Gerressen, M.; Driemel, O.; Maciejewski, O.; Hermanns-Sachweh, B.; Riediger, D.; Stein, J.M. A new biphasic osteoinductive calcium composite material with a negative zeta potential for bone augmentation. Head Face Med. 2009, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Society for Biomaterials. Biomaterials: The Enabling Technology. In Proceedings of the 31st Annual Meeting: Society for Biomaterials, Pittsburgh, PA, USA, 22–26 April 2006.
- Teng, N.C.; Nakamura, S.; Takagi, Y.; Yamashita, Y.; Ohgaki, M.; Yamashita, K. A new approach to enhancement of bone formation by electrically polarized hydroxyapatite. J. Dent. Res. 2001, 80, 1925–1929. [Google Scholar] [CrossRef] [PubMed]
- Hench, L.L. Bioceramics: From concept to clinic. J. Am. Ceram. Soc. 1991, 74, 1487–1510. [Google Scholar] [CrossRef]
- Cerruti, M.; Greenspan, D.; Powers, K. Effect of ph and ionic strength on the reactivity of bioglass® 45s5. Biomaterials 2005, 26, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Wallin, R.F.; Arscott, E.F. A practical guide to iso 10993-5: Cytotoxicity. Med. Device Diagn. Ind. Mag. 1998, 20, 96–98. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalo-Juan, I.; Detsch, R.; Mathur, S.; Ionescu, E.; Boccaccini, A.R.; Riedel, R. Synthesis and In Vitro Activity Assessment of Novel Silicon Oxycarbide-Based Bioactive Glasses. Materials 2016, 9, 959. https://doi.org/10.3390/ma9120959
Gonzalo-Juan I, Detsch R, Mathur S, Ionescu E, Boccaccini AR, Riedel R. Synthesis and In Vitro Activity Assessment of Novel Silicon Oxycarbide-Based Bioactive Glasses. Materials. 2016; 9(12):959. https://doi.org/10.3390/ma9120959
Chicago/Turabian StyleGonzalo-Juan, Isabel, Rainer Detsch, Sanjay Mathur, Emanuel Ionescu, Aldo R. Boccaccini, and Ralf Riedel. 2016. "Synthesis and In Vitro Activity Assessment of Novel Silicon Oxycarbide-Based Bioactive Glasses" Materials 9, no. 12: 959. https://doi.org/10.3390/ma9120959
APA StyleGonzalo-Juan, I., Detsch, R., Mathur, S., Ionescu, E., Boccaccini, A. R., & Riedel, R. (2016). Synthesis and In Vitro Activity Assessment of Novel Silicon Oxycarbide-Based Bioactive Glasses. Materials, 9(12), 959. https://doi.org/10.3390/ma9120959