
Stainless Steel 304 and Carbon Mild Steel A36 Activity in Chloride-Containing Hybrid Pumice-Portland Cement Extract Pore Environment



Abstract
1. Introduction
2. Materials and Methods
2.1. Steel Samples
2.2. Hybrid Cement HB1 and HB1 + 5 g L−1 NaCl
2.3. Immersion Tests
2.4. Electrochemical Measurements
3. Results
3.1. Change in pH of HB1 + Cl Cement Extract and Steel Corrosion Potential
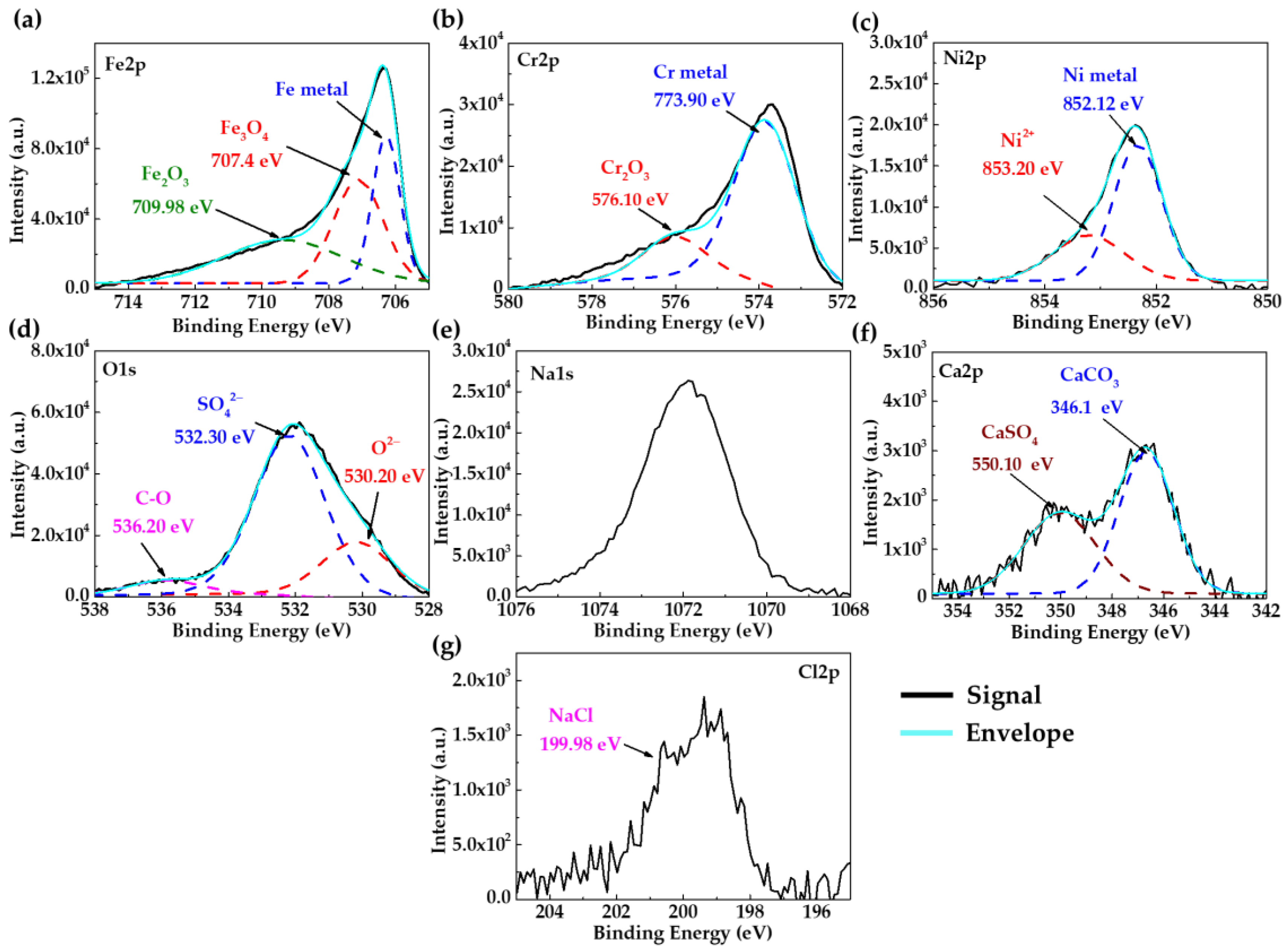
3.2. Stainless Steel 304 Surface Characterization After Immersion in HB1 + Cl Cement Extract
3.3. Carbon Steel A36 Surface Characterization After Immersion in HB1 + Cl Cement Extract
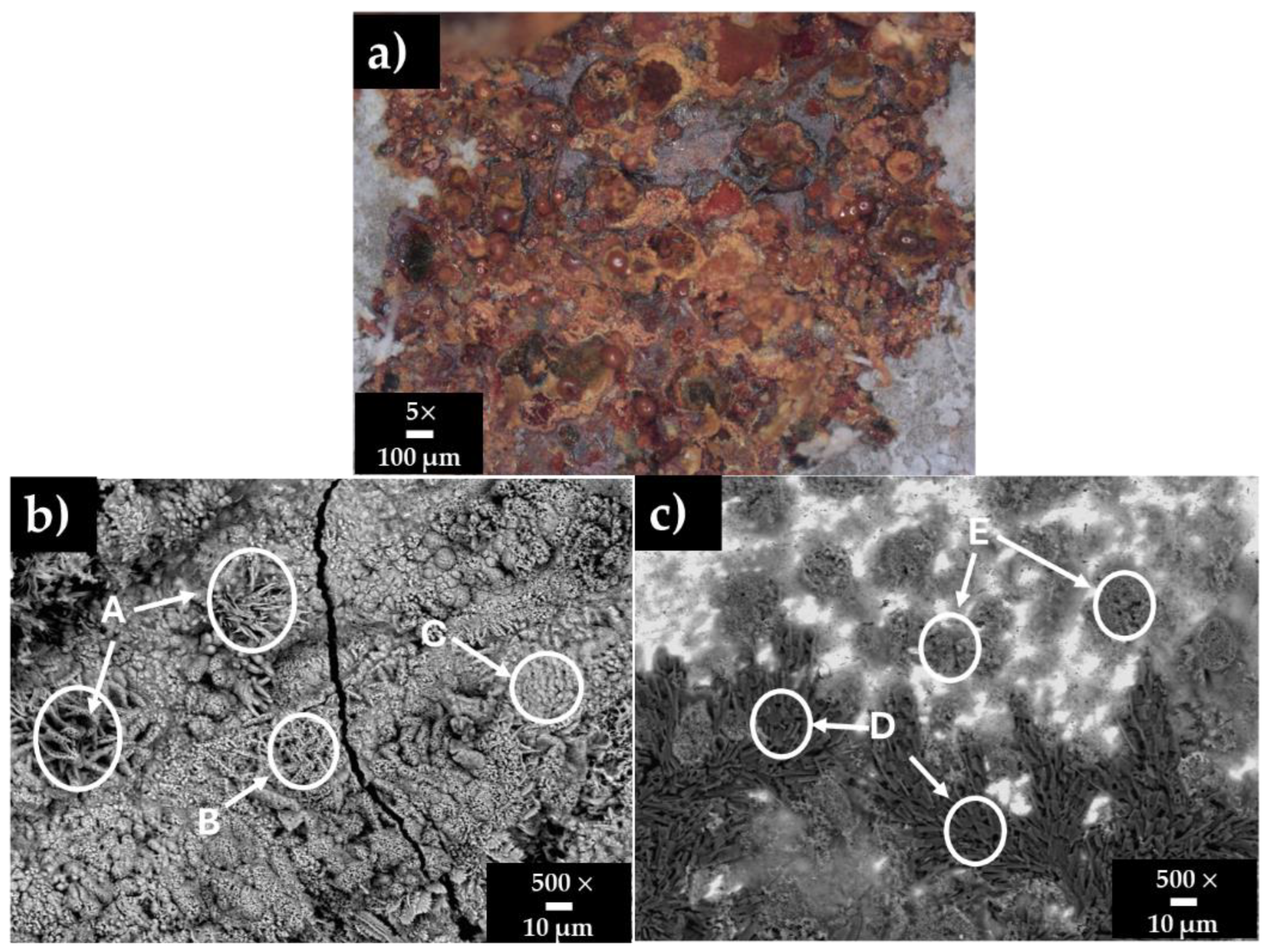
3.4. Steel Surface Damage After Immersion Tests in HB1 + Cl Cement Extract
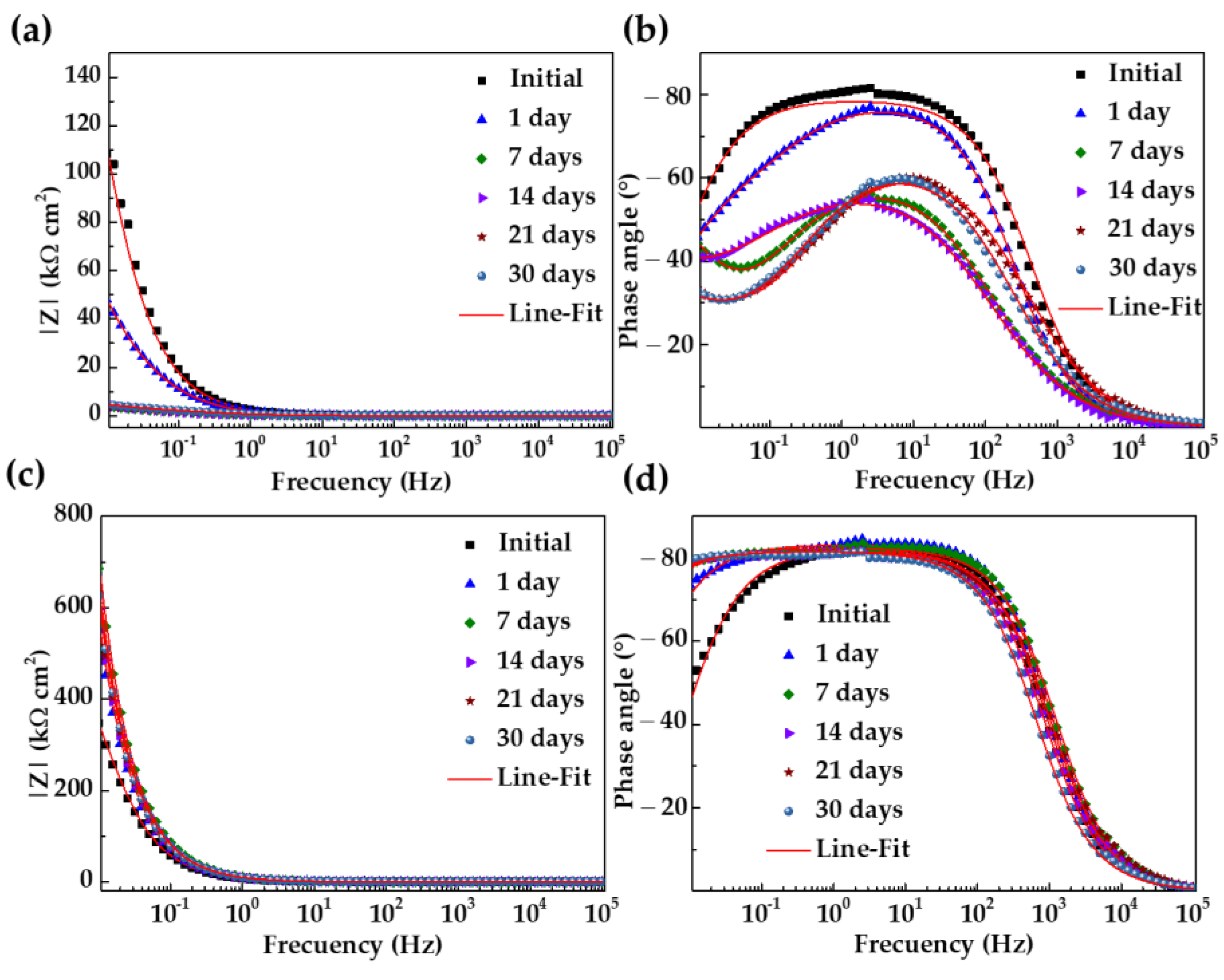
3.5. Activity at the Steel–Cement Extract Interface: Nyquist and Bode EIS Diagrams
4. Conclusions
- The ionic composition of the HB1 + Cl cement extract solution presented elevated contents of , , and (2866 mg L−1), and its pH ≈ 12.99 was attributed to the formation of NaOH and KOH. The pH tended to decrease due to the air dissolution and stabilized, reaching ≈ 9.5 after 14 days due to the carbonate/bicarbonate buffer capacity.
- Meanwhile, the corrosion potential (OCP) value of the SS304 increased to more positive/noble values (≈+72 mV), tending to form a passive film on the surface. However, the OCP of A36 carbon steel fell to very negative/active values (≈−363 mV), indicating a severe corrosion risk.
- In accordance with the surface analysis (SEM-EDS, and XPS), the passive state of the SS304 was attributed to the formation of a layer of , , and , while the carbon steel A36 presented phases as its main corrosion products.
- An extended area of a uniform corrosion attacks was observed on the A36’s surface, due to the less protective Fe-corrosion layer allowing the ingress of and the diffusion of towards the steel matrix. Meanwhile, the SS304’s surface presented only small pits of ≈1 µm.
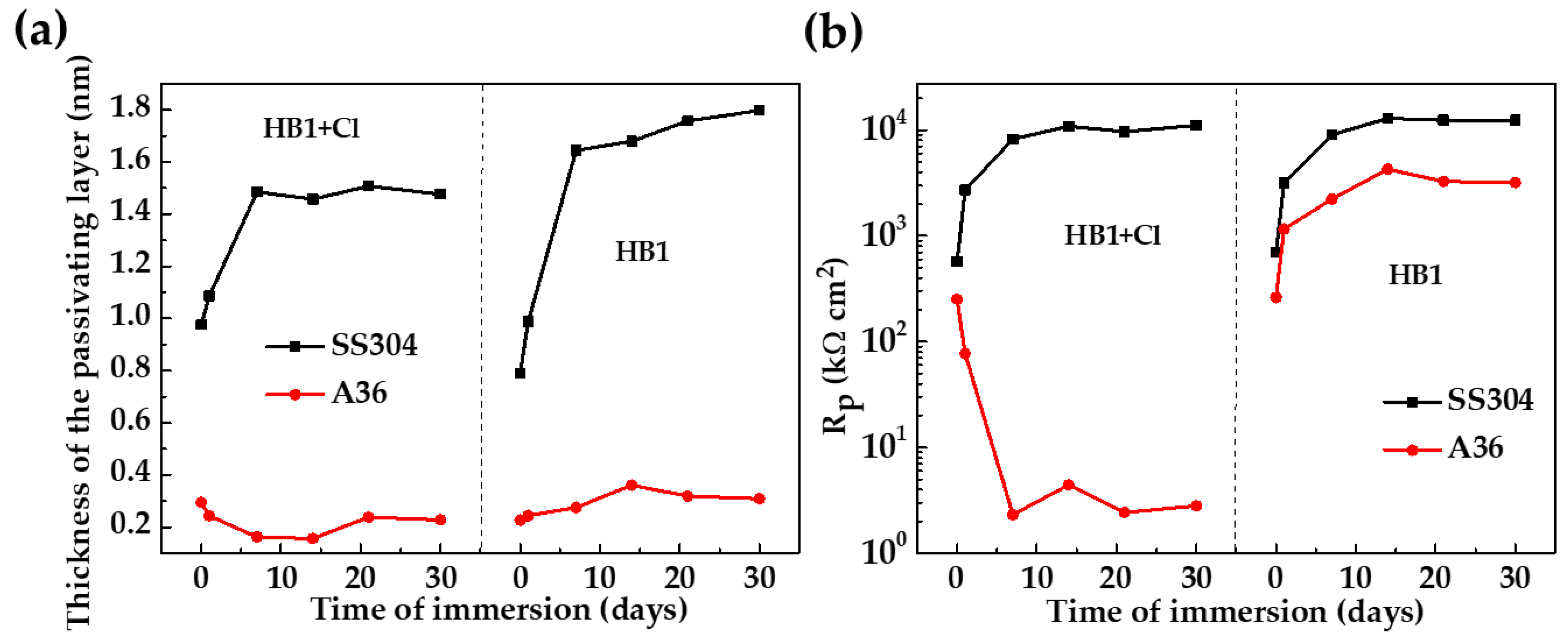
- The quantitative parameters, based on the EIS diagrams, indicated that the SS304 polarization resistance (Rp) increased by two orders of magnitude up to ≈11,080 and the thickness of the passive layer was ≈1.5 nm after 30 days of immersion; meanwhile, these parameters for the A36 were ≈2.5 and ≈0.2 nm, respectively, corroborating the active corrosion state of A36.
- The effect of the high concentration of chlorides (2866 mg ) on the corrosion activities of the studied steels could be considered a consequence of the competitive adsorption process between and the high concentration of in the HB1 cement system.
- The reported results suggest that chloride-containing hybrid pumice–Portland cement extract, which simulates the marine environment, is highly corrosive toward carbon steel A36, while the passivating properties of SS304 remain unaffected.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neville, A.M. Properties of Concrete, 4th ed.; Prentice/Pearson Hill: Harlow, UK, 2009; pp. 1–103. [Google Scholar]
- Pourbaix, M. Thermodynamics and corrosion. Corros. Sci. 1990, 30, 963–988. [Google Scholar] [CrossRef]
- Freire, L.; Carmezim, M.J.; Ferreira, M.G.S.; Montemor, M.F. The passive behaviour of AISI 316 in alkaline media and the effect of pH: A combined electrochemical and analytical study. Electrochim. Acta 2010, 21, 6174–6181. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, X.; Cao, Y.; Yang, H. Natural passivation behavior and its influence on chloride-induced corrosion resistance of stainless steel in simulated concrete pore solution. J. Mater. Res. Technol. 2020, 9, 12378–12390. [Google Scholar] [CrossRef]
- Szklarska-Smialowska, Z. Mechanism of pit nucleation by electrical breakdown of the passive film. Corros. Sci. 2002, 44, 1143–1149. [Google Scholar] [CrossRef]
- Evans, U.R. The passivity of metals. Part I. The isolation of the protective film. J. Chem. Soc. 1927, 1020–1040. [Google Scholar] [CrossRef]
- Strehblow, H.H. Corrosion Mechanism in Theory and Practice; Marcus, P., Oudar, J., Eds.; Marcel Dekker: New York, NY, USA, 1995; p. 201. [Google Scholar]
- Burstein, G.T.; Pistorius, P.C.; Mattin, S.P. The nucleation and growth of corrosion pits on stainless steel. Corros. Sci. 1993, 35, 57–62. [Google Scholar] [CrossRef]
- Rodriguez, R.; Gaboreau, S.; Gance, J.; Ignatiadis, I.; Betelu, S. Reinforced concrete structures: A review of corrosion mechanisms and advances in electrical methods for corrosion monitoring. Constr. Build. Mater 2021, 269, 121240. [Google Scholar] [CrossRef]
- ISO 9223_2012; Corrosion of metals and alloys–Corrosivity of atmospheres–Classification, Determination and Estimation. ISO: Geneva, Switzerland, 2012.
- Millano-González, V.; Traconis de Rincón, O.; Torres Acosta, A.A.; Sanchez Gómez, M.A.; Castro-Borges, P.; Perez-Quiroz, J.T.; Pérez-López, T.; Vera, R.; Salta, M.; Pedrón, M. Modeling Electrochemical Performance of Reinforced Concrete in Natural Marine Airborne-Exposure Environments: DURACON Project, 10-Year Evaluation. Corrosion 2023, 79, 719–731. [Google Scholar] [CrossRef]
- Glass, G.K.; Buenfeld, N.R. The presentation of the chloride threshold level for corrosion of steel in concrete. Corros. Sci. 1997, 39, 1001–1013. [Google Scholar] [CrossRef]
- Glass, G.K.; Reddy, B.; Buenfeld, N.R. The participation of bound chloride in passive film breakdown on steel in concrete. Corros. Sci. 2000, 42, 2013–2021. [Google Scholar] [CrossRef]
- Gouda, V.K. Corrosion and corrosion inhibition of reinforcing steel: I. immersed in alkaline solutions. Br. Corros. J. 1970, 5, 198–203. [Google Scholar] [CrossRef]
- Tanash, A.O.; Muthusamy, K.; Mat Yahaya, F.; Ismail, M.A. Potential of recycled powder from clay Brick, sanitary ware, and concrete waste as a cement substitute for concrete: An overview. Constr. Build. Mater. 2023, 401, 132760. [Google Scholar] [CrossRef]
- Supriya; Chaudhury, R.; Sharma, U.; Thapliyal, P.C.; Singh, L.P. Low-CO2 emission strategies to achieve net zero target in cement sector. J. Clean. Prod. 2023, 417, 137466. [Google Scholar] [CrossRef]
- Andrew, R.M. Global CO2 Emissions from Cement Production. Earth Syst. Sci. 2018, 10, 195–217. [Google Scholar] [CrossRef]
- Aitcin, P.-C. Portland cement. In Science and Technology of Concrete Admixtures, 1st ed.; Aïtcin, P., Flatt, R.J., Eds.; Woodhead Publishing: Sawston, UK, 2016; pp. 27–51. [Google Scholar] [CrossRef]
- Juenger, M.C.G.; Winnefeld, F.; Provis, J.L.; Ideker, J.H. Advances in alternative cementitious binders. Cem. Concr. Res. 2011, 41, 1232–1243. [Google Scholar] [CrossRef]
- Ramezanianpour, A.A. Cement Replacement Materials: Properties, Durability, Sustainability, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–43. [Google Scholar] [CrossRef]
- Lothenbach, B.; Scrivener, K.; Hooton, R.D. Supplementary cementitious material. Cem. Concr. Res. 2011, 41, 1244–1256. [Google Scholar] [CrossRef]
- Juenger, M.C.G.; Snellings, R.; Bernal, S. Supplementary cementitious materials: New sources, characterization and performance insights. Cem. Concr. Res. 2019, 122, 257–273. [Google Scholar] [CrossRef]
- Játiva, A.; Etxeberria, M. Exploring the Utilization of Activated Volcanic Ash as a Substitute for Portland Cement in Mortar Formulation: A Thorough Experimental Investigation. Materials 2024, 17, 1123. [Google Scholar] [CrossRef]
- Wang, Y.; Shui, Z.; Gao, X.; Yu, R.; Huang, Y.; Cheng, S. Understanding the chloride binding and diffusion behaviors of marine concrete based on Portland limestone cement-alumina enriched pozzolans. Constr. Build. Mater. 2019, 198, 207–217. [Google Scholar] [CrossRef]
- Al-Amoudi, Q.S.B. Durability of plain and blended cements in marine environments. Adv. Cem. Res. 2002, 14, 89–100. [Google Scholar] [CrossRef]
- Juenger, M.C.G.; Siddique, R. Recent advances in understanding the role of supplementary cementitious materials in concrete. Cem. Concr. Res. 2015, 78, 71–80. [Google Scholar] [CrossRef]
- Canham, I.; Page, C.L.; Nixon, P.J. Aspects of the pore solution chemistry of blended cements related to the control of alkali silica reaction. Cem. Concr. Res. 1987, 17, 839–844. [Google Scholar] [CrossRef]
- Vollpratcht, A.; Lothenbach, B.; Snellings, R.; Haufe, J. The pore solution of blended cements: A review. Mater. Struct. 2016, 49, 3341–3367. [Google Scholar] [CrossRef]
- Aperador, W.; Mejía de Gutiérrez, R.; Bastidas, D.M. Steel corrosion behaviour in carbonated alkali-activated slag concrete. Corros. Sci. 2009, 51, 2027–2033. [Google Scholar] [CrossRef]
- Puertas, F.; Palacios, M.; Vázquez, T. Carbonation process of alkali-activated slag mortars. J. Mater. Sci. 2006, 41, 3071–3082. [Google Scholar] [CrossRef]
- Poursaee, A. Corrosion of steel bars in saturated Ca(OH)2 and concrete pore solution. Concr. Res. Lett. 2010, 1, 90–97. [Google Scholar]
- Jiang, H.; Jin, Z.; Zhang, X.; Qian, L.; Zhou, Z. The Effect of Temperatures on the Passivation Behavior of Q235 Steel in the Simulated Concrete Pore Solution. Materials 2023, 16, 588. [Google Scholar] [CrossRef]
- Li, L.; Sagüés, A.A. Chloride corrosion threshold of reinforcing steel in alkaline solutions-open circuit immersion tests. Corrosion 2001, 57, 19–28. [Google Scholar] [CrossRef]
- Zhang, F.; Jinshan, P.; Changjian, L. Localized corrosion behaviour of reinforcement steel in simulated concrete pore solution. Corros. Sci. 2009, 51, 2130–2138. [Google Scholar] [CrossRef]
- Behera, P.K.; Misra, S.; Mondal, K. Corrosion Behavior of Strained Rebar in Simulated Concrete Pore Solution. J. Mater. Eng. Perform. 2020, 29, 1939–1954. [Google Scholar] [CrossRef]
- Pokorný, P.; Vacek, V.; Prodanovic, N.; Zabloudil, A.; Fojt, J.; Johánekm, V. The Influence of Graded Amount of Potassium Permanganate on Corrosion of Hot-Dip Galvanized Steel in Simulated Concrete Pore Solutions. Materials 2022, 15, 7864. [Google Scholar] [CrossRef]
- Zakroczymski, T.; Fan, C.J.; Szklarska-Smialowska, Z. Kinetics of passive film formation on iron in 0.05M NaOH. J. Electrochem. Soc. 1985, 132, 2282–2287. [Google Scholar] [CrossRef]
- Montemor, M.F.; Simoes, A.M.; Ferreira, M.G. Analytical characterization of the passive film formed on steel in solutions simulating the concrete interstitial electrolyte. Corrosion 1998, 54, 347–353. [Google Scholar] [CrossRef]
- Veleva, L.; Alpuche-Aviles, M.A.; Graves-Brook, M.K.; Wipf, D.O. Comparative cyclic voltammetry and surface analysis of passive films grown on stainless steel 316 in concrete pore model solutions. J. Electroanal. Chem. 2002, 537, 85–93. [Google Scholar] [CrossRef]
- Veleva, L.; Alpuche-Aviles, M.A.; Graves-Brook, M.K.; Wipf, D.O. Voltammetry and surface analysis of AISI 316 stainless steel in chloride-containing simulated concrete pore environment. J. Electroanal. Chem. 2005, 578, 45–53. [Google Scholar] [CrossRef]
- Lambert, P.; Page, C.L.; Vassie, P.R.W. Investigations of reinforcement corrosion. 2. Electrochemical monitoring of steel in chloride-contaminated concrete. Mater. Struct. 1991, 24, 351–358. [Google Scholar] [CrossRef]
- Mangat, P.S.; Khatib, M.; Mollay, B.T. Microstructure, chloride diffusion and reinforcement corrosion in blended cement paste and concrete. Cem. Concr. Compos. 1994, 16, 73–91. [Google Scholar] [CrossRef]
- Oh, B.H.; Jang, S.Y.; Shin, Y.S. Experimental investigation of the threshold chloride concentration for corrosion initiation in reinforced concrete structures. Mag. Concr. Res. 2003, 55, 117–124. [Google Scholar] [CrossRef]
- Glass, G.K.; Buenfeld, N.R. The influence of chloride binding on the chloride induced corrosion risk in reinforced concrete. Corrosion Sci. 2000, 42, 329–344. [Google Scholar] [CrossRef]
- Tomas, M.D.A.; Hooton, R.D.; Scott, A.; Zibara, H. The efect of supplementary cementitious materials on chloride binding in hardened cement paste. Cem. Concr. Res. 2012, 42, 1–7. [Google Scholar] [CrossRef]
- Dousti, A.; Beaudoin, J.J.; Shekarchi, M. Chloride binding in hydrated MK, SF and natural zeolite-lime mixtures. Constr. Build. Mater. 2017, 154, 1035–1047. [Google Scholar] [CrossRef]
- Shi, Z.; Geiker, M.R.; De Weerdt, K.; Østnor, T.A.; Lothenbach, B.; Winnefeld, F.; Skibsted, J. Role of calcium on chloride binding in hydrated Portland cement–metakaolin–limestone blends. Cem. Concr. Res. 2017, 95, 205–216. [Google Scholar] [CrossRef]
- Chaparro, W.A.; Ruiz, J.H.; Gomez, R.D. Corrosion of reinforcing bars embedded in alkali-activated slag concrete subjected to chloride attack. Mat. Res. 2012, 15, 57–62. [Google Scholar] [CrossRef]
- Rasheeduzzafar; Ehtesham Hussain, S.; Al-Gahtani, A.S. Pore solution composition and reinforcement corrosion characteristics of microsilica blended cement concrete. Cem. Concr. Res. 1991, 21, 1035–1048. [Google Scholar] [CrossRef]
- Wang, W.; Chen, H.; Li, H.; Zhu, Z. Corrosion behavior of steel bars immersed in simulated pore solution of alkali-activated slag mortar. Constr. Build. Mater. 2017, 143, 289–297. [Google Scholar] [CrossRef]
- Wang, L.; Zhan, S.; Tang, X.; Xu, Q.; Qian, K. Pore solution chemistry of calcium sulfoaluminate cement and its effects on steel passivation. Appl. Sci. 2019, 9, 1092. [Google Scholar] [CrossRef]
- Wang, L.; Jian, Q.; Zhan, S.; Song, Y.; Ruan, S. Chloride-induced corrosion patters of reinforcements in simulated pore solution of calcium sulfoaluminate cement concrete: An analytical study. J. Build. Eng. 2024, 82, 108189. [Google Scholar] [CrossRef]
- Abd El Haleem, S.M.; Abd El Wanees, S.; Bahgat, A. Environmental Factors Affecting the Corrosion Behaviour of Reinforcing Steel. V. Role of Chloride and Sulphate Ions in the Corrosion of Reinforcing Steel in Saturated Ca(OH)2 solutions. Corros. Sci. 2014, 87, 321–333. [Google Scholar] [CrossRef]
- Liu, G.; Li, J.; Zhang, Y. Corrosion of carbon steels subjected to chloride and sulfate in simulated concrete pore solutions with different pH. Constr. Build. Mater. 2024, 440, 137445. [Google Scholar] [CrossRef]
- Bonfil, D.; Veleva, L.; Feliu, S., Jr.; Escalante-García, J.I. Corrosion Activity of Stainless Steel SS430 and Carbon Steel B450C in a Sodium Silicate Modified Limestone-Portland Cement Extract. Materials 2023, 16, 5066. [Google Scholar] [CrossRef]
- Bonfil, D.; Veleva, L.; Feliu, S., Jr.; Escalante-García, J.I. Corrosion Activity of Carbon Steel B450C and Stainless Steel SS430 Exposed to Extract Solution of a Supersulfated Cement. Materials 2022, 15, 8782. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Salas, J.; Escalante-Garcia, J.I. Hybrid binders based on volcanic pumice: Effect of the chemical composition on strength and microstructures. Cem. Concr. Res. 2024, 176, 107393. [Google Scholar] [CrossRef]
- Bonfil, D.; Veleva, L.; Escalante-Garcia, J.I. Effect of a hybrid pumice-portland cement extract on corrosion activity of stainless steel SS304 and carbon mild steel A36. Materials 2024, 17, 2255. [Google Scholar] [CrossRef]
- Krivenko, P.V.; Sanytsky, M.; Kropyvnytska, T. Alkali-sulfate activated blended Portland cements. Solid State Phenom. 2018, 276, 9–14. [Google Scholar] [CrossRef]
- ASTM-NACE/ASTM G31-12a; Standard Guide for Laboratory Immersion Corrosion Testing of Metals. ASTM International: West Conshohocken, PA, USA, 2012.
- ASTM G1-03; Standard Practice for Preparing, Cleaning, and Evaluating Corrosion Test Specimens. ASTM International: West Conshohocken, PA, USA, 2017.
- Kern, M. The hydration of carbon dioxide. J. Chem. Educ. 1960, 37, 1–14. [Google Scholar] [CrossRef]
- Huet, B.; L’Hostis, V.; Tricheux, L.; Idrissi, H. Influence of alkali, silicate and sulfate content of carbonated concrete pore solution on mild steel behavior. Mater. Corros. 2010, 61, 111–124. [Google Scholar] [CrossRef]
- Greve-Dierfeld, S.; Lothenbach, B.; Vollpracht, A.; Wu, B.; Huet, B.; Andrade, C.; Medina, C.; Thiel, C.; Gruyaert, E.; Vanoutrive, H.; et al. Understanding the carbonation of concrete with supplementary cementitous materials. A critical review by RILEM TC 281-CCC. Mater. Struct. 2020, 53, 136. [Google Scholar] [CrossRef]
- Peng, Y.; Liu, L.; Wang, S.; Lin, Y.; Sun, Y.; Xia, R. Effect of simulated pore solution on passivation characteristic of P110 steel. J. Pet. Sci. Eng. 2018, 167, 949–956. [Google Scholar] [CrossRef]
- Zhang, Q.; Meng, X.; Li, X.; Wu, L.; Suo, X.; Cao, H. Effect of anions on the anodic dissolution ofi ron: An electrochemical and density functional theory stude. Corros. Sci. 2024, 229, 111852. [Google Scholar] [CrossRef]
- Liu, G.; Li, M.; Yang, L.; Liu, C.; Zhang, Y. Electrochemical dielectic response of steel corrosion induced by chloride in simulated concrete pore solution. J. Sustain. Cem.-Based Mater. 2024, 13, 854–864. [Google Scholar] [CrossRef]
- Ghods, P.; Isgor, O.B.; McRae, G.; Miller, T. The effect of concrete pore solution composition on the quality of passive oxide films on black steel reinforcement. Cem. Concr. Compos. 2009, 31, 2–11. [Google Scholar] [CrossRef]
- Krishna Vigneshwaran, K.K.; Permeh, S.; Echeverría, M.; Lau, K.; Lasa, I. Corrosion of post-tensioned tendons with deficient grout, part 1: Electrochemical behavior of steel in alkaline sulfate solutions. Corrosion 2018, 74, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yang, L.; Wang, X.; Huang, Y. Influence of SO42− on the corrosion behavior of Q235B steel bar in simulated pore solution. Acta Metall. Sin. 2019, 55, 457–468. [Google Scholar] [CrossRef]
- Abd El Haleem, S.M.; Abd El Wanees, S.; Abd El Aal, E.E.; Diab, A. Environmental factor affecting the corrosion behavior of reinforcing steel II. Role of some anions in the initiation and inhibition of pitting corrosion of steel in Ca(OH)2 solution. Corros. Sci. 2010, 52, 292–302. [Google Scholar] [CrossRef]
- Galvele, J.R. Transport Processes and the mechanism of pitting of metals. J. Electrochem. 1976, 123, 464. [Google Scholar] [CrossRef]
- Ai, Z.; Sun, W.; Jiang, J.; Song, D.; Ma, H.; Zhang, J.; Wang, D. Passivation Characteristics of Alloy Corrosion-Resistant Steel Cr10Mo1 in Simulating Concrete Pore Solutions: Combination Effects of pH and Chloride. Materials 2016, 9, 749. [Google Scholar] [CrossRef]
- Addari, D.; Elsener, B.; Rossi, A. Electrochemistry and surface chemistry of stainless steel in alkaline media simulating con-crete pore solutions. Electrochim. Acta 2008, 53, 8078–8086. [Google Scholar] [CrossRef]
- Luo, H.; Su, H.; Dong, C.; Xiao, K.; Li, X. Electrochemical and passivation behavior investigation of ferritic stainless steel in simulated concrete pore media. Data in Brief. 2015, 96, 502–507. [Google Scholar] [CrossRef]
- Tian, Y.; Dond, C.; Wang, G.; Cheng, X.; Li, X. The effect of nickel on corrosion behavior of high-strength low alloy steel bar in simulated concrete pore solution. Constr. Build. Mater. 2020, 246, 118462. [Google Scholar] [CrossRef]
- Wahlqvist, M.; Shchukarev, A. XPS spectra and electronic structure of Group IA sulfates. J. Electron. Spectrosc. 2007, 156–158, 310–314. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Physical Electronics: Chanhassen, MN, USA, 1995; p. 81. [Google Scholar]
- Freire, L.; Catarino, M.A.; Godinho, M.I.; Ferreira, M.J.; Ferreira, M.J.; Ferreira, M.G.S.; Simoes, A.M.P.; Montemor, M.F. Electrochemical and analytical investigation of passive film formed on stainless steel in alkaline media. Cem. Concr. Comp. 2012, 34, 1075–1081. [Google Scholar] [CrossRef]
- Gardin, E.; Zanna, S.; Seyeux, A. Comparative study of the surface oxide films on lean duplex and corresponding single phase stainless steels by XPS and ToF-SIMS. Corros. Sci. 2018, 143, 403–413. [Google Scholar] [CrossRef]
- Luo, H.; Su, H.; Dong, C.; Li, X. Passivation and electrochemical behavior of 316L stainless steel in chlorinated simulated concrete pore solution. Appl. Surf. Sci. 2017, 400, 38–48. [Google Scholar] [CrossRef]
- Abreu, C.M.; Cristobal, M.J.; Losada, R.; Nóvoa, X.R.; Pena, G.; Pérez, M.C. The effect of Ni in the electrochemical properties of oxide layers grown on stainless steels. Electrochim. Acta 2006, 51, 2991–3000. [Google Scholar] [CrossRef]
- Vera, R.; Villarroel, M.; Carvajal, A.M.; Vera, E.; Ortiz, C. Corrosion Products of Reinforcement in Concrete in Marine and Industrial Environments. Mater. Chem. Phys. 2009, 114, 467–474. [Google Scholar] [CrossRef]
- Gotić, M.; Musić, S. FT-IR and FE SEM Investigation of Iron Oxides Precipitated from FeSO4 Solutions. J. Mol. Struct. 2007, 834–836, 445–453. [Google Scholar] [CrossRef]
- Mataferia, I.A. Characterization of Steel Corrosion Products in Reinforced Concrete. Master’s Thesis, University of Ottawa, Ottawa, CA, Canada, 14 May 2021. [Google Scholar] [CrossRef]
- Antunes, R.A.; Costa, I.; Araujo, D. Characterization of Corrosion Products Formed on Steels in the First Months of Atmospheric Exposure. Mater. Res. 2003, 6, 403–408. [Google Scholar] [CrossRef]
- Duffó, G.S.; Worris, W.; Raspini, I.; Saragovi, C. A study of steel rebars embedded in concrete during 65 years. Corros. Sci. 2004, 46, 2143–2157. [Google Scholar] [CrossRef]
- Rémazeilles, C.; Refait, P. On the formation of B-FeOOH (akaganeite) in chloride-containing environments. Corros. Sci. 2007, 49, 844–857. [Google Scholar] [CrossRef]
- Freire, L.; Novoa, X.R.; Montemor, M.F.; Carmezin, M.J. Study of passive films formed on mild steel in alkaline media by the application of anodic potentials. Mater. Chem. Phys. 2009, 114, 962–972. [Google Scholar] [CrossRef]
- Ghods, P.; Burkan Isgor, O.; Bensebaa, F.; Kingston, D. Angle-resolved XPS of carbon steel passivity and chloride-induced despassivation in simulated concrete pore solution. Corros. Sci. 2012, 58, 159–167. [Google Scholar] [CrossRef]
- Burak Gunay, H.; Ghods, P.; Burkan Isgor, O.; Carpenter, G.J.C.; Wu, X. Characterization of atomic structure oxide films on carbon steel in simulated concrete pore solutions using EELS. Appl. Surf. Sci. 2013, 274, 195–202. [Google Scholar] [CrossRef]
- Veleva, L. Phase transformation of iron hydroxide in the corrosion products formed in humid tropical climate. In Proceedings of the Corrosion 2003, San Diego, CA, USA, 16–20 March 2003. [Google Scholar]
- Tolulope Loto, R. Pitting corrosion evaluation and inhibition of stainless steels: A review. J. Mater. Environ. Sci. 2015, 6, 2750–2762. [Google Scholar]
- Jiang, J.-y.; Liu, Y.; Chu, H.-y.; Wang, D.; Ma, H.; Sun, W. Pitting Corrosion Behaviour of New Corrosion-Resistant Reinforcement Bars in Chloride-Containing Concrete Pore Solution. Materials 2017, 10, 903. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tan, X.; Lan, R.; Ran, G.; Li, J.; Jiang, Y. Mechanistics of inclusion-induced pitting of stainless steel: A review. J. Mater. Sci. Technol. 2024, 168, 143–156. [Google Scholar] [CrossRef]
- Freire, L.; Carmezin, M.J.; Ferreira, M.G.S.; Montemor, M.F. The electrochemical behaviour of stainless steel AISI 304 in alkaline solution with different pH in the presence of chlorides. Electrochim. Acta 2011, 56, 5280–5289. [Google Scholar] [CrossRef]
- Jiang, J.-Y.; Wang, D.; Chu, H.-Y.; Ma, H.; Liu, Y.; Gao, Y.; Shi, J.; Sun, W. The Passive Film Growth Mechanism of New Corrosion-Resistant Steel Rebar in Simulated Concrete Pore Solution: Nanometer Structure and Electrochemical Study. Materials 2017, 10, 412. [Google Scholar] [CrossRef]
- Sarango de Souza, J.; De Oliveira, L.A.; Sayeg, I.J.; Antunes, R.A. Electrochemical study of the AISI 409 ferritic stainless steel: Passive film stability and pitting nucleation and growth. Mater. Res. 2017, 20, 1669. [Google Scholar] [CrossRef]
- Orazem, M.E.; Frateur, I.; Tribollet, B.; Vivier, V.; Marcelin, S.; Pebere, N.; Bunge, A.L.; White, E.A.; Riemer, D.P.; Musiani, M. Dielectric properties of materials showing constant-phase-element (CPE) impedance response. J. Electrochem. Soc. 2013, 160, C215. [Google Scholar] [CrossRef]
- Hirschorn, B.; Orazem, M.E.; Tribollet, B.; Vivier, V.; Frateur, I.; Musiani, M. Determination of effective capacitance and film thickness from constant-phase element parameters. Electrochim. Acta 2010, 55, 6218–6227. [Google Scholar] [CrossRef]
- Ji, H.; Tian, Y.; Zhao, R.; Jin, N.; Tian, Z.; Yan, D.; Ye, H. Passivation and depassivation og HPB335 Carbon steel in simulated concrete pore solution. Int. J. Electrochem. Sci. 2020, 15, 6488–6507. [Google Scholar] [CrossRef]
- Saura, P.; Zornoza, E.; Andrade, C.; Ferrandiz-Mas, V.; Garcés, P. Composition of Corroded Reinforcing Steel Surface in Solutions Simulating the Electrolytic Environments in the Micropores of Concrete in the Propagation Period. Materials 2022, 15, 2216. [Google Scholar] [CrossRef] [PubMed]
- Poursaee, A.; Hansson, C.M. Reinforcing steel passivation in mortar and pore solution. Cem. Concr. Res. 2017, 37, 1127–1133. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element (wt.%) | C | Cr | P | S | Mn | Si | Ni | Fe |
|---|---|---|---|---|---|---|---|---|
| SS304 | 0.08 | 18.00 | 0.045 | 0.03 | 2.00 | 1.00 | 8.00 | Balance |
| A36 | 0.0650 | 0.02 | 0.004 | 0.0020 | 0.41 | 0.02 | - | Balance |
| Cement | SiO2 | Al2O3 | Fe2O3 | CaO | MgO | SO3 | K2O | Na2O | Cl | LOI |
|---|---|---|---|---|---|---|---|---|---|---|
| HB1 | 43.05 | 8.85 | 2.79 | 29.78 | 0.75 | 4.87 | 2.88 | 4.35 | 0.05 | 2.63 |
| PC | 22.30 | 4.62 | 2.44 | 58.42 | 1.92 | 2.20 | 0.35 | 0.28 | - | 3.62 |
| Element ) | Li | Si | Sr | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HB1 | 1.32 | 2480.20 | 17,358.90 | 0.37 | 210.00 | 4.39 | 10,115.00 | 9.74 | 2866.20 | 1661.00 |
| PC [47] | - | 1373.58 | 420.71 | - | 256.51 | - | - | - | - | 3434.80 |
| Time (Days) | Initial | 1 | 7 | 14 | 21 | 30 | ||
|---|---|---|---|---|---|---|---|---|
| HB1 + Cl | SS304 | pH | 12.99 | 12.90 | 10.37 | 9.61 | 9.61 | 9.60 |
| OCP vs. SHE (mV) | −191.484 | −87.98 | +8.29 | +49.31 | +62.65 | +72.41 | ||
| A36 | pH | 12.99 | 12.92 | 10.50 | 9.73 | 9.62 | 9.61 | |
| OCP vs. SHE (mV) | −144.85 | −288.29 | −359.12 | −386.88 | −371.93 | −363.64 | ||
| HB1 | SS304 | pH | 12.99 | 12.92 | 10.48 | 9.61 | 9.18 | 9.07 |
| OCP vs. SHE (mV) | −204.89 | −89.81 | +128.126 | +219.75 | +245.30 | +265.08 | ||
| A36 | pH | 12.99 | 12.93 | 10.11 | 9.57 | 9.10 | 9.08 | |
| OCP vs. SHE (mV) | −89.05 | −24.84 | +48.58 | +67.456 | +79.154 | +88.18 |
| Zones/wt.% | Fe | Cr | Ni | O | Na | Ca | S | K | Si | C | Cl | Sr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | - | - | - | 43.17 | 2.88 | 17.59 | 0.93 | 1.83 | 2.87 | 16.64 | 2.31 | 13.77 |
| B | - | - | - | 37.24 | 17.20 | 9.29 | 3.98 | 5.83 | 7.40 | - | 19.06 | - |
| C | 63.16 | 16.34 | 9.45 | 4.21 | - | - | - | - | 2.70 | 2.75 | - | - |
| D | - | - | - | 51.57 | 27.44 | - | 12.46 | 0.66 | 3.85 | 2.58 | 0.98 | - |
| Zones/wt.% | Fe | O | Na | K | S | C | Si | Al | Cl |
|---|---|---|---|---|---|---|---|---|---|
| A | 47.18 | 46.34 | - | - | 1.79 | 3.16 | - | - | 1.53 |
| B | 43.00 | 49.03 | 0.90 | - | 0.88 | 3.77 | - | - | 2.41 |
| C | 48.06 | 45.53 | 1.91 | - | 1.11 | 2.43 | - | 0.96 | |
| D | 1.52 | 59.31 | 25.93 | - | - | 10.78 | 2.06 | 0.41 | - |
| E | 6.25 | 44.86 | 23.28 | 3.23 | 11.66 | 4.06 | 5.62 | 0.72 | 0.28 |
| Steel/wt.% | Fe | Cr | Ni | Mn | Si | C | |
|---|---|---|---|---|---|---|---|
| A36 | D1 | 96.91 | - | - | - | - | 3.09 |
| SS304 | D2 | 65.34 | 18.17 | 15.29 | 1.21 | - | - |
| D3 | 54.57 | 16.27 | 11.55 | 5.96 | 6.68 | 6.67 |
| Steel | Days | CPE | n | ||||
|---|---|---|---|---|---|---|---|
| SS304 | 0 | 0.01 | 570.1 | 25.66 | 0.91 | 570.1 | 6.52 |
| 1 | 0.01 | 2706.0 | 21.24 | 0.92 | 2706.0 | 7.88 | |
| 7 | 0.01 | 8203.0 | 17.51 | 0.91 | 8203.0 | 5.64 | |
| 14 | 0.02 | 10,850 | 19.95 | 0.90 | 10,850 | 1.95 | |
| 21 | 0.01 | 9651 | 19.00 | 0.90 | 9651 | 13.29 | |
| 30 | 0.02 | 11,080 | 18.56 | 0.90 | 11,080 | 9.32 | |
| A36 | 0 | 0.01 | 251.4 | 83.09 | 0.87 | 251.4 | 8.33 |
| 1 | 0.01 | 77.04 | 124.7 | 0.84 | 77.04 | 1.10 | |
| 7 | 0.01 | 2.32 | 506.0 | 0.70 | 2.32 | 4.89 | |
| 14 | 0.01 | 4.43 | 699.4 | 0.66 | 4.43 | 1.81 | |
| 21 | 0.01 | 2.43 | 329.4 | 0.70 | 2.43 | 4.01 | |
| 30 | 0.01 | 2.81 | 314.7 | 0.70 | 2.81 | 3.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonfil, D.; Veleva, L.; Escalante-Garcia, J.I. Stainless Steel 304 and Carbon Mild Steel A36 Activity in Chloride-Containing Hybrid Pumice-Portland Cement Extract Pore Environment. Materials 2025, 18, 1216. https://doi.org/10.3390/ma18061216
Bonfil D, Veleva L, Escalante-Garcia JI. Stainless Steel 304 and Carbon Mild Steel A36 Activity in Chloride-Containing Hybrid Pumice-Portland Cement Extract Pore Environment. Materials. 2025; 18(6):1216. https://doi.org/10.3390/ma18061216
Chicago/Turabian StyleBonfil, David, Lucien Veleva, and Jose Ivan Escalante-Garcia. 2025. "Stainless Steel 304 and Carbon Mild Steel A36 Activity in Chloride-Containing Hybrid Pumice-Portland Cement Extract Pore Environment" Materials 18, no. 6: 1216. https://doi.org/10.3390/ma18061216
APA StyleBonfil, D., Veleva, L., & Escalante-Garcia, J. I. (2025). Stainless Steel 304 and Carbon Mild Steel A36 Activity in Chloride-Containing Hybrid Pumice-Portland Cement Extract Pore Environment. Materials, 18(6), 1216. https://doi.org/10.3390/ma18061216