
High-Pressure Structures and Superconductivity of Barium Iodide


Abstract
:1. Introduction
2. Computational Details
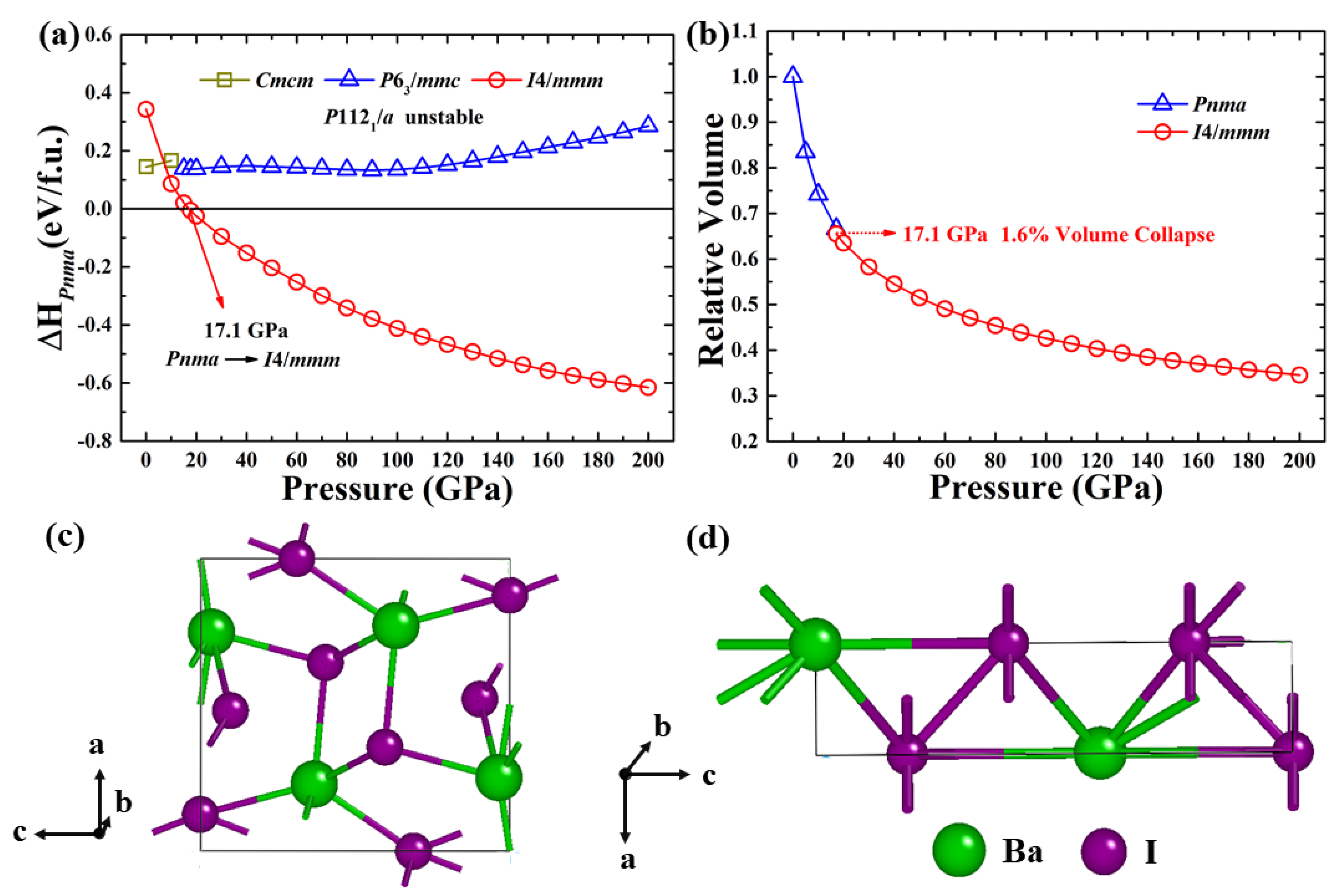
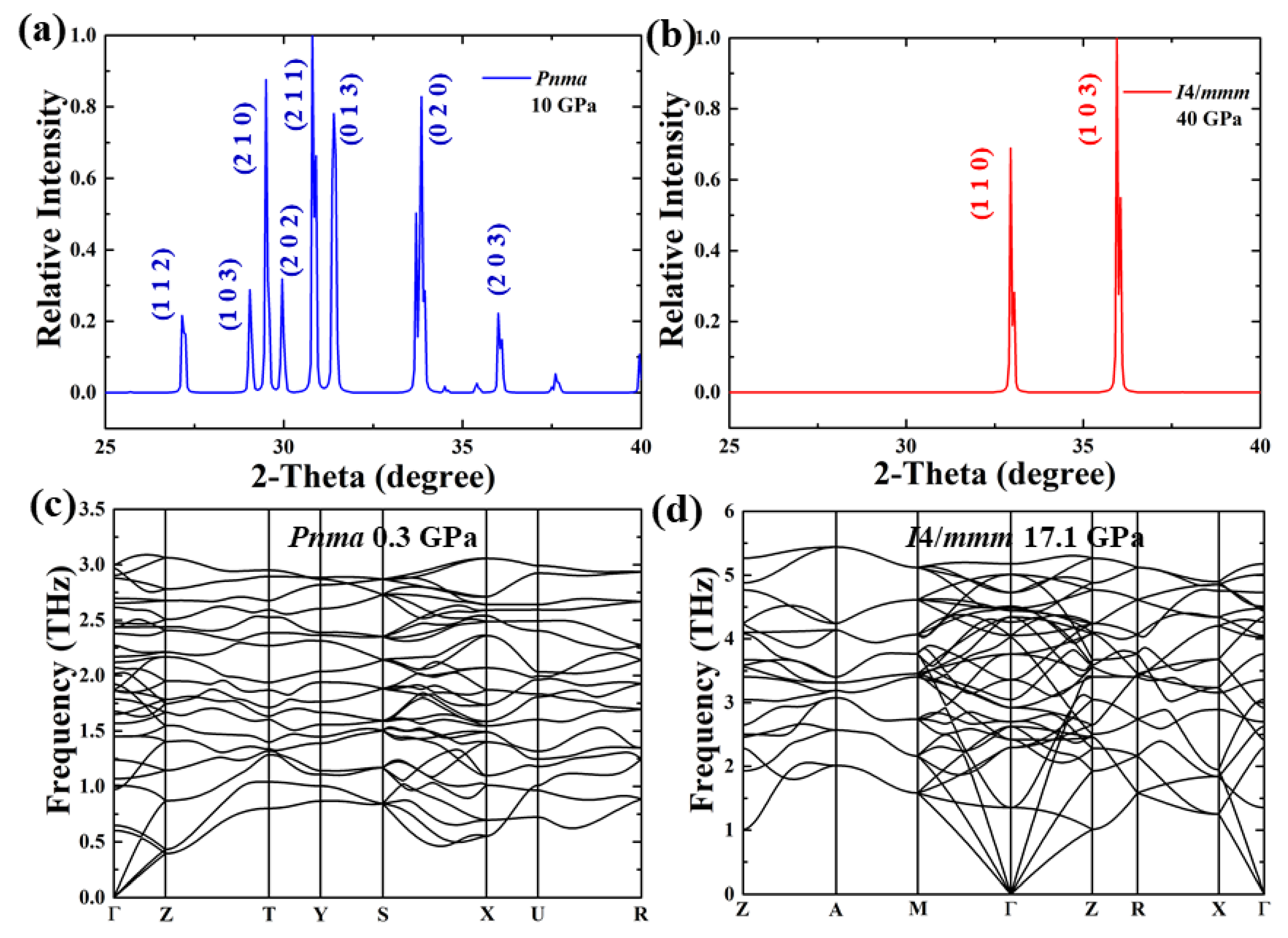
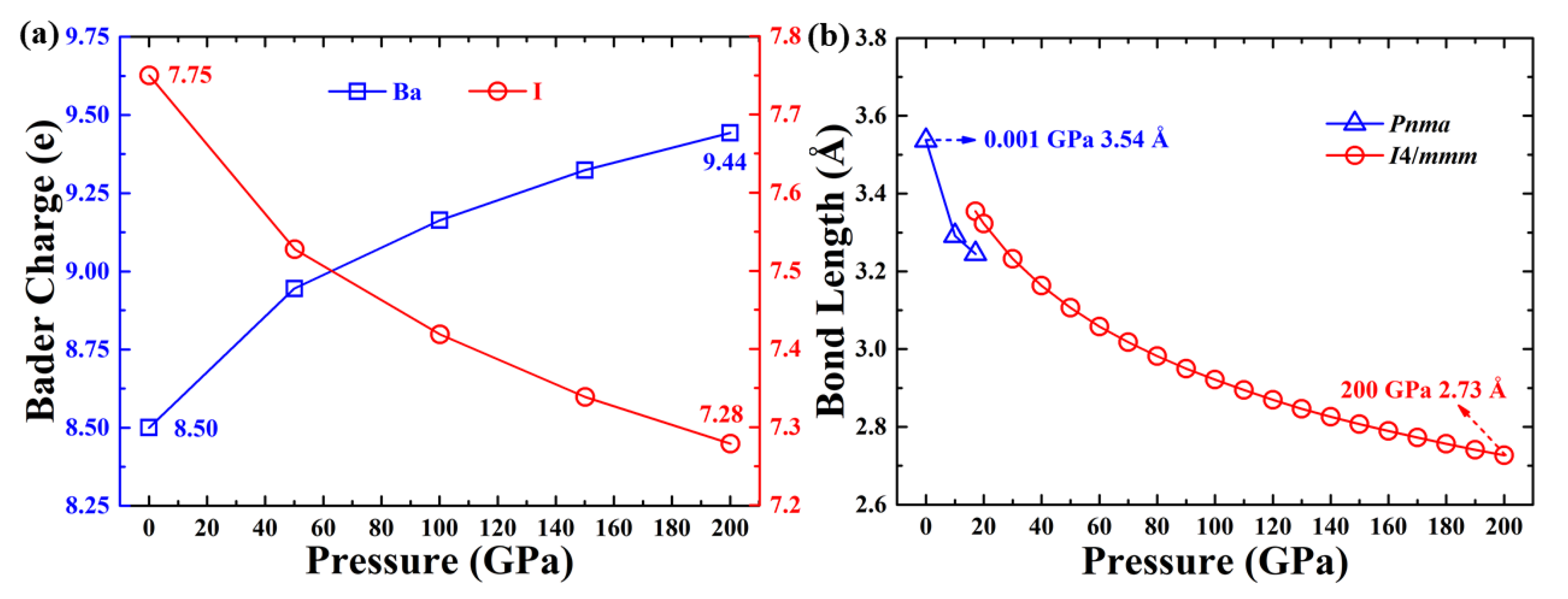
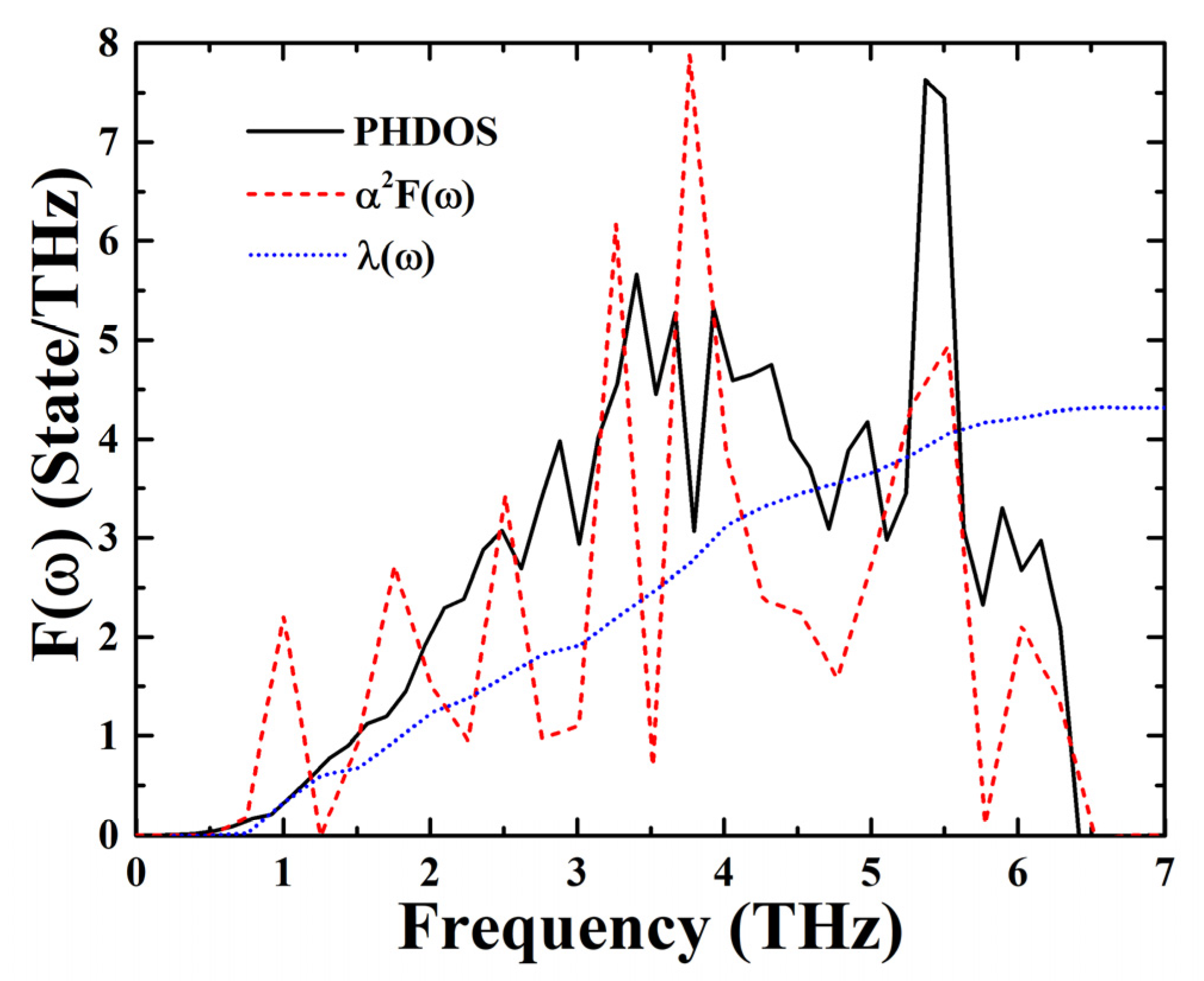
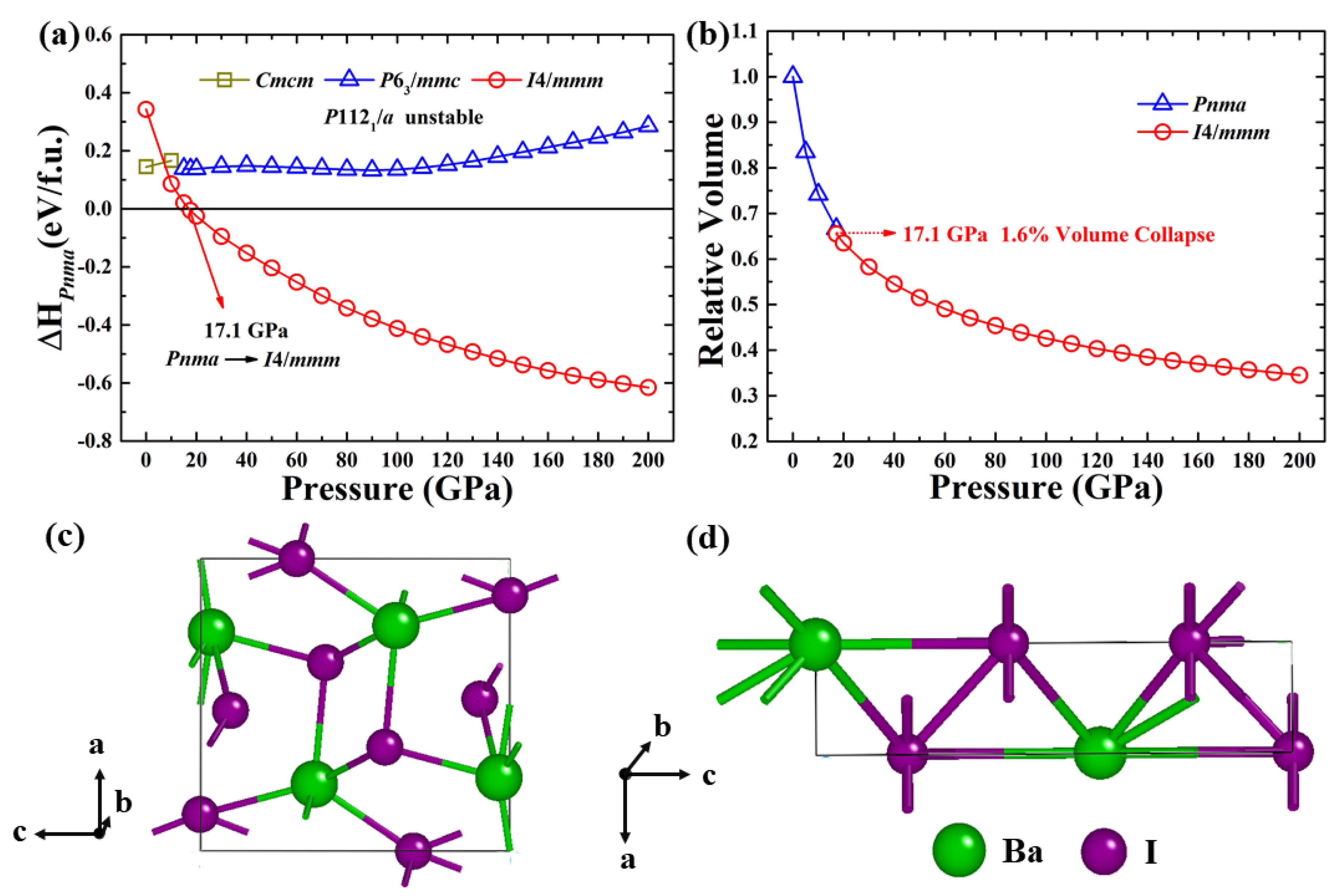
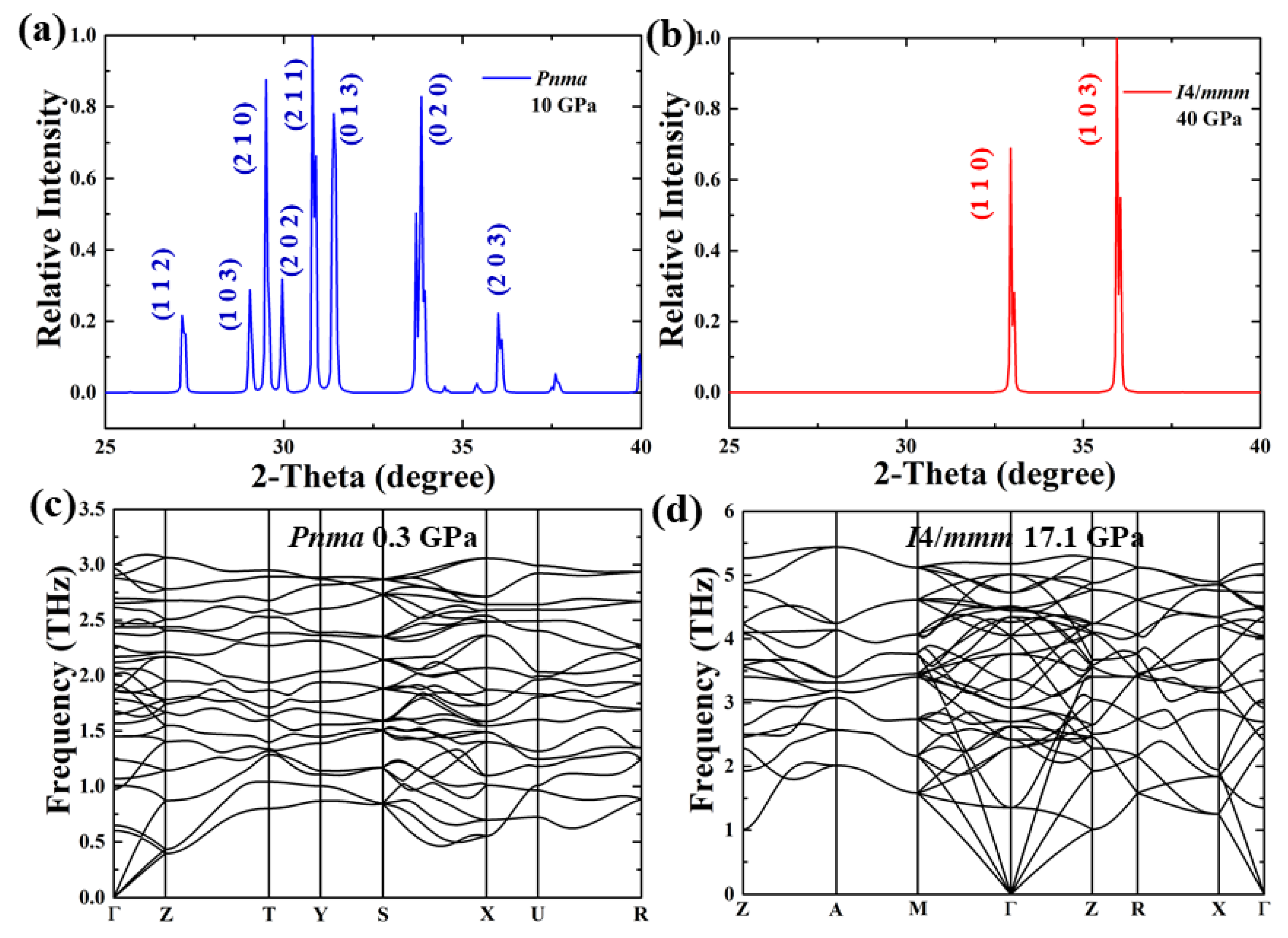
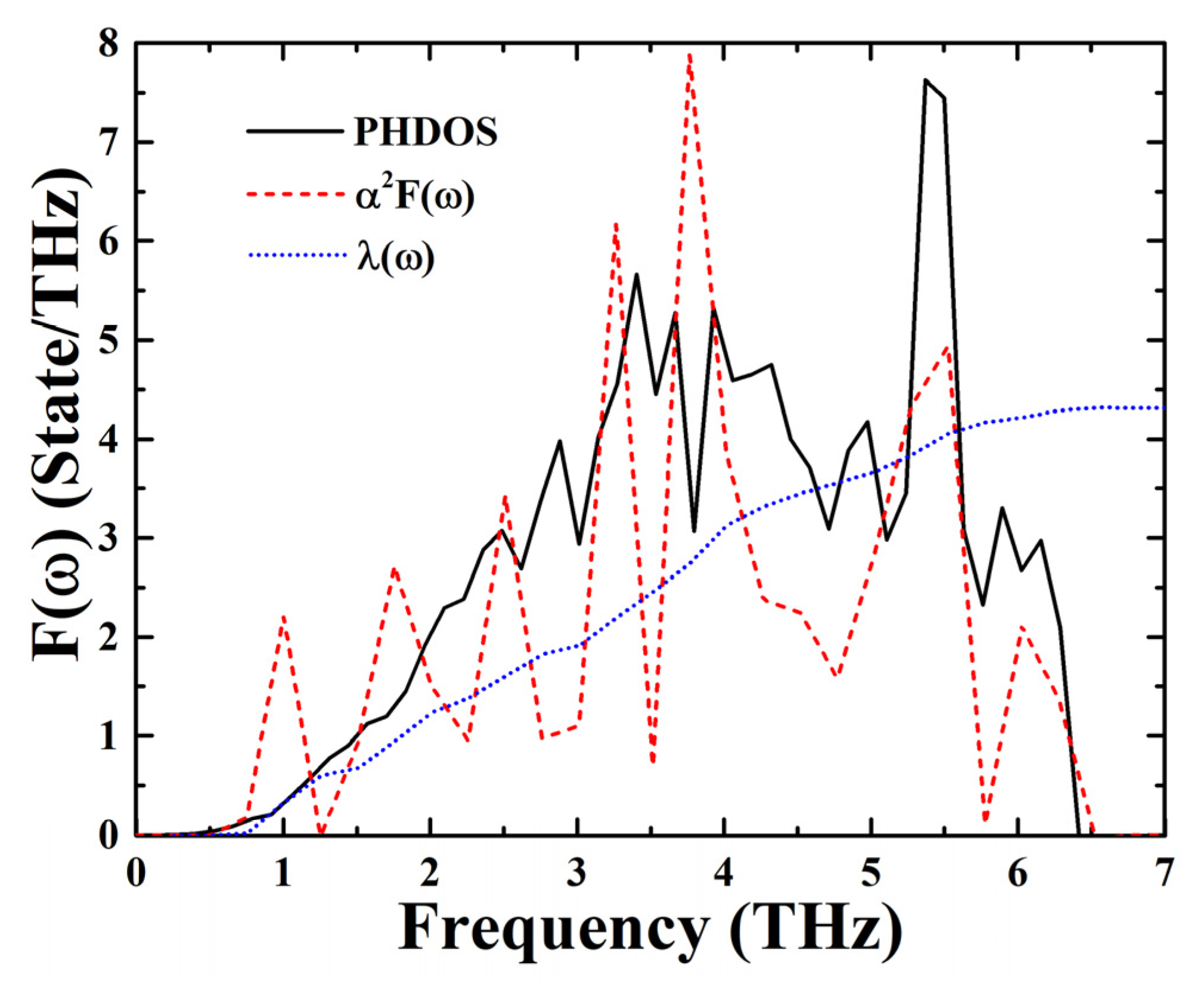
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Philip, H.Y.; Charles, J.D. Some optical properties of potassium iodide-thallium phosphors. J. Chem. Phys. 1953, 21, 892–898. [Google Scholar]
- Bradley, J.N.; Greene, P.D. Potassium iodide +silver iodide phase diagram high ionic conductivity of KAg4I5. Trans. Faraday Soc. 1966, 62, 2069–2075. [Google Scholar] [CrossRef]
- Liang, C.C. Conduction characteristics of the lithium iodide-aluminum oxide solid electrolytes. J. Electrochem. Soc. 1973, 120, 1289. [Google Scholar] [CrossRef]
- Dipak, P.; Jagir, S.S. Cadmium iodide as a new catalyst for knoevenagel condensations. J. Chem. Soc. Perkin Trans. 1993, 1, 739–740. [Google Scholar]
- Smanik, P.A.; Liu, Q.; Furminger, T.L.; Ryu, K.; Xing, S.; Mazzaferri, E.L.; Jhiang, S.M. Cloning of the human sodium lodide symporter. Biophys. Res. Commun. 1996, 226, 339–345. [Google Scholar] [CrossRef]
- Xie, W.; Li, H. Alumina-supported potassium iodide as a heterogeneous catalyst for biodiesel production from soybean oil. J. Mol. Catal. A Chem. 2006, 255, 1–9. [Google Scholar] [CrossRef]
- Xu, Y.; John, S.T.; Artem, R.O.; Cui, T.; Wang, H.; Ma, Y.; Zou, G. Superconducting high-pressure phase of cesium iodide. Phys. Rev. B 2009, 79, 144110. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Gale, J.D.; Uberuaga, B.P.; Stanek, C.R.; Marks, N.A. Importance of dispersion in density functional calculations of cesium chloride and its related halides. Phys. Rev. B 2013, 88, 054112. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Wang, J.; Deng, S.; Zhang, S.; Li, Q. Hypervalent iodine with linear chain at high pressure. Sci. Rep. 2015, 5, 14393. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, Q.; Li, H.; Yue, L.; Zhao, D.; Tian, F.; Dong, Q.; Zhang, X.; Jin, X.; Zhang, L.; et al. Pressure-tailored band engineering for significant enhancements in the photoelectric performance of CsI3 in the optical communication waveband. Adv. Funct. Mater. 2021, 2108636. [Google Scholar] [CrossRef]
- Elizabeth, B.B.; Thomas, E.B.; Ronald, L.S. The crystal structures of barium chloride, barium bromide, and barium iodide. J. Phys. Chem. 1963, 67, 2132–2135. [Google Scholar]
- Winchell, P. Mass spectrometric investigation of barium iodide and caesium iodide vaporizations. Nature 1965, 206, 1252. [Google Scholar] [CrossRef]
- Leger, J.M.; Haines, J.; Atouf, A. The post-cotunnite phase in BaCl2, BaBr2 and BaI2 under high pressure. J. Appl. Cryst. 1995, 28, 416. [Google Scholar] [CrossRef]
- Nerine, J.C.; Giulia, H.; Alexander, D.D.; Stephen, A.P.; Edgar, L.; Cody, M.W.; Shah, K.S.; Utpal, N.R.; Arnold, B.; Lynn, A.; et al. Strontium and barium iodide high light yield scintillators. Appl. Phys. Lett. 2008, 92, 083508. [Google Scholar]
- Pradeep, K.; Agnikumar, G.V. Pressure dependence of electronic properties of BaI2. AIP Conf. Proc. 2015, 1675, 020013. [Google Scholar]
- Raman, A.; Steinfink, H. Crystal chemistry of AB2 structures. Inorg. Chem. 1967, 6, 1789. [Google Scholar] [CrossRef]
- Eremets, M.I.; Shimizu, K.T.; Kobayashi, T.C.; Amaya, K. Metallic CsI at pressures of up to 220 gigapascals. Science 1998, 281, 1333. [Google Scholar] [CrossRef] [PubMed]
- Eremets, M.I.; Shimizu, K.; Kobayashi, T.C.; Amaya, K. Metallization and superconductivity in CsI at pressures up to 220 GPa. J. Phys. Condens. Matter 1998, 10, 11519. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 094116. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Miao, M.; Lv, J.; Zhu, L.; Liu, H.; Ma, Y. An effective structure prediction method for layered materials based on 2D particle swarm optimization algorithm. J. Chem. Phys. 2012, 137, 224108. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Wang, Y.; Zhu, L.; Ma, Y. Particle-swarm structure prediction on clusters. J. Chem. Phys. 2012, 137, 084104. [Google Scholar] [CrossRef]
- Zhang, J.; Lv, J.; Li, H.; Feng, X.; Lu, C.; Redfern, S.A.T.; Liu, H.; Chen, C.; Ma, Y. Rare helium-bearing compound FeO2He stabilized at deep-earth conditions. Phys. Rev. Lett. 2018, 121, 255703. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Lv, J.; Xie, Y.; Liu, H.; Ma, Y. Route to a superconducting phase above room temperature in electron-doped hydride compounds under high pressure. Phys. Rev. Lett. 2019, 123, 097001. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Liu, H.; Lv, J.; Li, Q.; Long, C.; Wang, Y.; Chen, C.; Hemley, R.J.; Ma, Y. Stability of H3O at extreme conditions and implications for the magnetic fields of Uranus and Neptune. Proc. Natl. Acad. Sci. USA 2020, 117, 5638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Ma, Y.; Chen, C. Direct H-He chemical association in superionic FeO2H2He at Deep-Earth conditions. Natl. Sci. Rev. 2021. [Google Scholar] [CrossRef]
- Yong, X.; Liu, H.; Wu, M.; Yao, Y.; Tse, J.S.; Dia, R.; Yoo, C.S. Crystal structures and dynamical properties of dense CO2. Proc. Natl. Acad. Sci. USA 2016, 113, 11110–11115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 78, 134106. [Google Scholar] [CrossRef] [Green Version]
- Hongzhiwei Technology, Device Studio, Version 2021A, China. 2021. Available online: https://iresearch.net.cn/cloudSoftware (accessed on 7 September 2021).
- Bader, R. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893. [Google Scholar] [CrossRef]
- Baroni, S.; Giannozzi, P.; Testa, A. Green’s-function approach to linear response in solids. Phys. Rev. Lett. 1987, 58, 1861. [Google Scholar] [CrossRef]
- Giannozzi, P.; De Gironcoli, S.; Pavone, P.; Baroni, S. Ab initio calculation of phonon dispersions in semiconductors. Phys. Rev. B 1991, 43, 7231. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.J. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of material. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P. Advanced capabilities for materials modelling with QUANTUM ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Zhu, C.; Li, Q.; Zhou, Y.; Li, Q.; Ma, Y. High-pressure phase transition of cesium chloride and cesium bromide. Phys. Chem. Chem. Phys. 2014, 16, 17924. [Google Scholar] [CrossRef]
- Allen, P.B. Neutron Spectroscopy of Superconductors. Phys. Rev. B 1972, 6, 2577. [Google Scholar] [CrossRef]
- Allen, P.B.; Dynes, R.C. Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 1975, 12, 905. [Google Scholar] [CrossRef]
- Dominik, S.; Adam, Z.K.; Ewa, A.D.S.; Radosław, S. Phonon-mediated superconductivity in bismuthates by nonadiabatic pairing. Phys. Rev. B 2021, 104, 094501. [Google Scholar]
- Dominik, S.; Radosław, S. Signatures of nonadiabatic superconductivity in lithium-decorated graphene. Phys. Rev. B 2019, 99, 224512. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Lattice Parameters (Å) | Atoms | x | y | z |
|---|---|---|---|---|---|
| Pnma BaI2 | a = 9.003 | Ba1 (4c) | 0.24 | 0.75 | 0.61 |
| 0.001 GPa | b = 5.448 | I1 (4c) | 0.64 | 0.75 | 0.58 |
| c = 10.879 | I2 (4c) | 0.48 | 0.25 | 0.84 | |
| I4/mmm BaI2 | a = 4.094 | Ba (2a) | 0.00 | 0.00 | 0.00 |
| 17.1 GPa | b = 4.094 | I (4e) | 0.50 | 0.50 | 0.16 |
| c = 10.434 | |||||
| I4/mmm BaI2 | a = 3.255 | Ba (2a) | 0.00 | 0.00 | 0.00 |
| 200 GPa | b = 3.255 | I (4e) | 0.50 | 0.50 | 0.17 |
| c = 8.695 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, S.; Liu, H. High-Pressure Structures and Superconductivity of Barium Iodide. Materials 2022, 15, 522. https://doi.org/10.3390/ma15020522
Wei S, Liu H. High-Pressure Structures and Superconductivity of Barium Iodide. Materials. 2022; 15(2):522. https://doi.org/10.3390/ma15020522
Chicago/Turabian StyleWei, Shubo, and Hanyu Liu. 2022. "High-Pressure Structures and Superconductivity of Barium Iodide" Materials 15, no. 2: 522. https://doi.org/10.3390/ma15020522
APA StyleWei, S., & Liu, H. (2022). High-Pressure Structures and Superconductivity of Barium Iodide. Materials, 15(2), 522. https://doi.org/10.3390/ma15020522