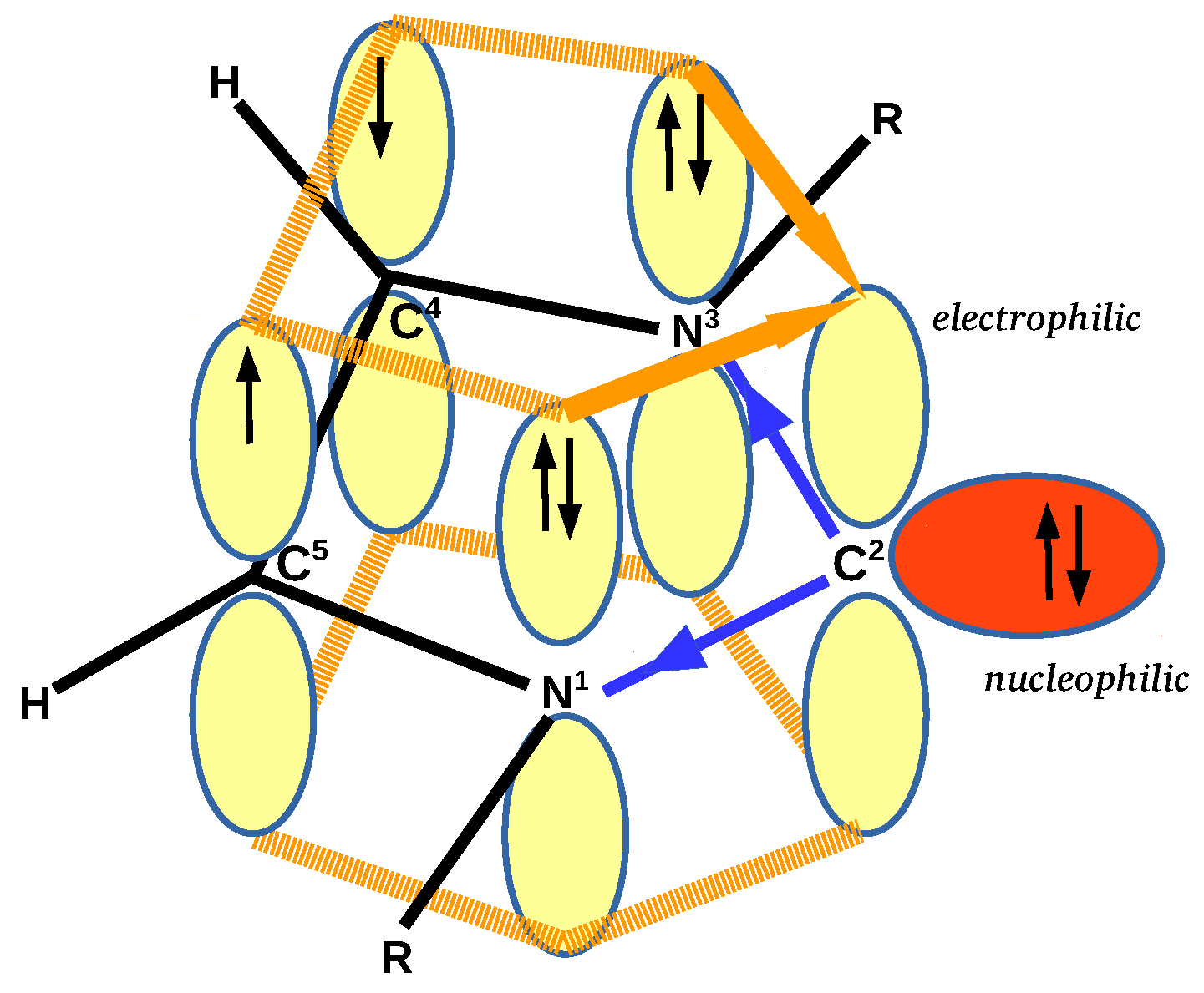
3.2. Geometric and Energetic Characteristics of the C⋯Zn Interaction
The most important parameters characterizing a given interaction are its length and its strength, where the strength can be described in various ways, e.g., by the dissociation energy. However, when it comes to the dissociation energy (or interaction energy), it actually concerns the entire dimer, i.e., it is a global quantity, and only when the described interaction is dominant can the determined dissociation energy describe this interaction [
83]. The distances C⋯Zn and the dissociation energies determined for the discussed complexes are presented in
Table 1 and
Table 2, respectively.
It is somewhat surprising that despite such diverse R substituents and different X groups, the range of C⋯Zn distances is very narrow, from ca. 2.12 Å (IMes–ZnH) to 2.20 Å (IPh–ZnMe). On the contrary, the range of the dissociation energy values is quite wide (ca. 9 kcal/mol). The strongest C⋯Zn interaction (27.4 kcal/mol) is in agreement with the shortest C⋯Zn contact in IMes–ZnH, and, conversely, the longest contact in IPh–ZnMe is characterized by the lowest dissociation energy (18.5 kcal/mol). However, the overall relationship between the length of C⋯Zn and the dissociation energy is weak if all the complexes are considered together.
It is worth mentioning that ZnH
differs substantially from both ZnMe
and ZnEt
with regard to the properties of the hydrogen atoms. Namely, taking into account electronegativities of Zn (1.7), C (2.5) and H (2.2), only in ZnH
do the hydrogen atoms have a partial negative charge (−0.172 au). For this reason, ZnH
may tend to form weak dihydrogen bonds with some hydrogen atoms of the R substituents. On the other hand, positively charged hydrogen atoms from Me (0.012 au) and Et (0.008–0.022 au) groups may readily interact with the
-electron system of the R substituents containing the benzene ring. It can also be expected that in some cases, the hydrogen atom of the R substituent can interact with the carbon atom of the X group (especially Me) to form a weak H⋯C interaction. The electrophilic and nucleophilic regions of the ZnX
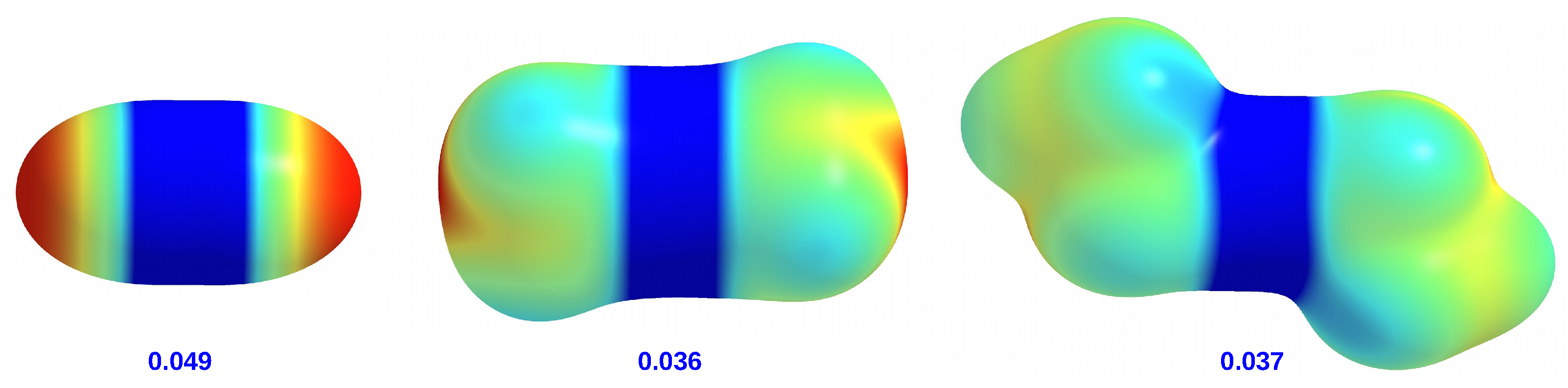
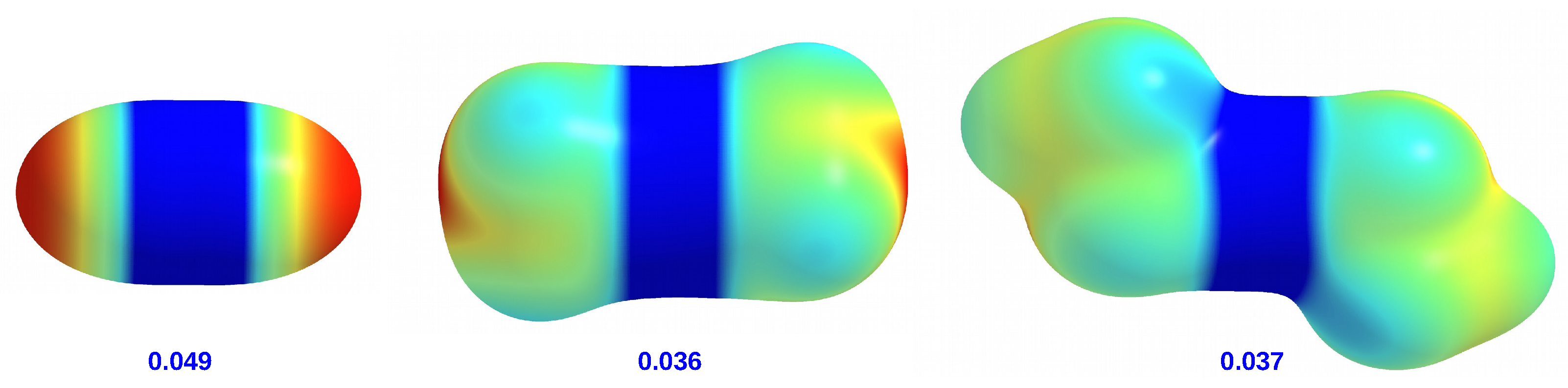
molecules can be visually represented by maps of the total electrostatic potential (ESP) projected onto the electron density isosurface (see
Figure 4).
It is clearly visible that ZnH is distinguished by areas of strongly negative potential located on both hydrogen atoms. In the case of ZnMe and ZnEt, the ESP distributions are similar to each other, although in the former molecule there is a small negative potential region near the center of the side surface of the methyl groups. Importantly, the zinc atom in ZnH has a higher potential value (0.049 au) than in ZnMe and ZnEt (0.036 au and 0.037 au, respectively). This is in line with weak +I effects of alkyl groups.
Despite all these possibilities, however, some general conclusions can be found. Namely, it can be seen that the complexes involving ZnH
are characterized by a shorter C⋯Zn bond (2.118–2.149 Å) than those involving ZnMe
(2.170–2.198 Å). The complexes with ZnEt
, on the other hand, are in the middle, and their range (2.137–2.169 Å) overlaps with that for ZnH
. Thus, in terms of the C⋯Zn length, the complexes with ZnEt
are much more similar to those with ZnH
than with ZnMe
. This result can be explained as follows. First, a small ZnH
molecule can approach the carbene closer than ZnMe
or ZnEt
, having larger groups and therefore sterically interacting with more bulky R substituents (e.g., Ad, Dipp, Mes). Secondly, as already mentioned, the hydridic hydrogen atom of ZnH
can form weak dihydrogen bonds, as in the complex with I, i.e., imidazol-2-ylidene (see at the top of
Figure 2). In turn, the longer aliphatic chain in ZnEt
(compared to ZnMe
) allows for more favorable attractive interactions (e.g., of the
H-C type) with large R groups of the carbene unit.
It is worth emphasizing that the characteristic effect associated with the formation of the zinc bond is a significant bend of the ZnX
molecule (more precisely, the E-Zn-E angle where E is the atom directly bonded to Zn). This effect has previously been described extensively [
64,
84] and is analogous to the bending effect of the BeX
[
64,
85,
86,
87,
88,
89,
90] or MgX
[
64,
91,
92] molecule during beryllium and magnesium bond formation, respectively. The respective values of the
angles are shown in
Table 3.
The greatest effect occurs in the complexes of IAd (by far the greatest bend of ca. 128 occurs in IAd–ZnEt). This result cannot be due solely to the large size of the adamantyl group, as in the case of much smaller methyl groups in IMe, the angle does not differ significantly, especially when X = Me (137.7 vs. 136.5 for IMe and IAd, respectively). On the other hand, the smallest bend of E-Zn-E (151.1) takes place in the IDipp–ZnEt complex. For a given X, the I-ZnX complexes also exhibit a relatively weak bending effect of E-Zn-E. It can be easily noticed that, with the exception of IMe and IDipp, for a given carbene, the E-Zn-E bending effect is weakest for X = Me and by far strongest for X = Et, which most likely results from considerable steric interactions R⋯Et. The angle does not correlate with the distance C⋯Zn or the dissociation energy.
It has been shown [
64] that another structural effect that occurs during the formation of the carbene–ZnX
complex is the opening of the N-C-N angle (
) in the carbene. The results shown in
Table 4 confirm this. However, the variation in
(the range of
amounts to 1.4
–2.7
) is not large due to significant ring stiffness of the imidazol-2-ylidene unit.
For a given carbene, ZnH most often leads to the greatest opening effect. This effect is also slightly greater for ZnEt than for ZnMe. Again, does not correlate well with either the C⋯Zn distance or the dissociation energy. It therefore seems that small changes occurring in the ring of the imidazol-2-ylidene unit are due to various subtle effects.
As mentioned earlier, the angle between the plane of the ZnX
molecule (more precisely its E-Zn-E fragment) and the plane of the carbene (its imidazol-2-ylidene ring) depends largely on the size of the R substituent in the carbene molecule. This issue will now be discussed in more detail. The relevant values of the torsional angle are given in
Table 5.
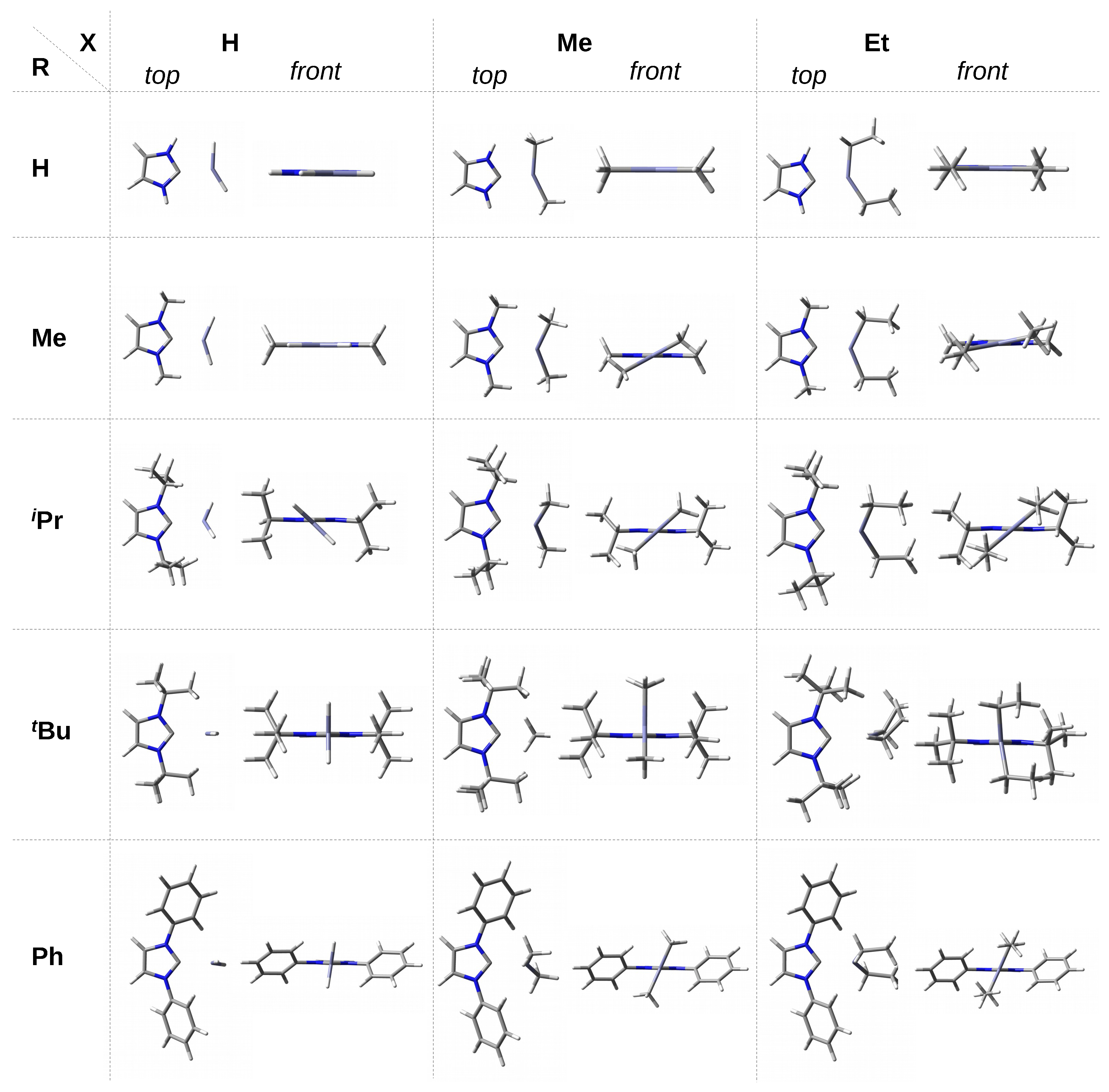
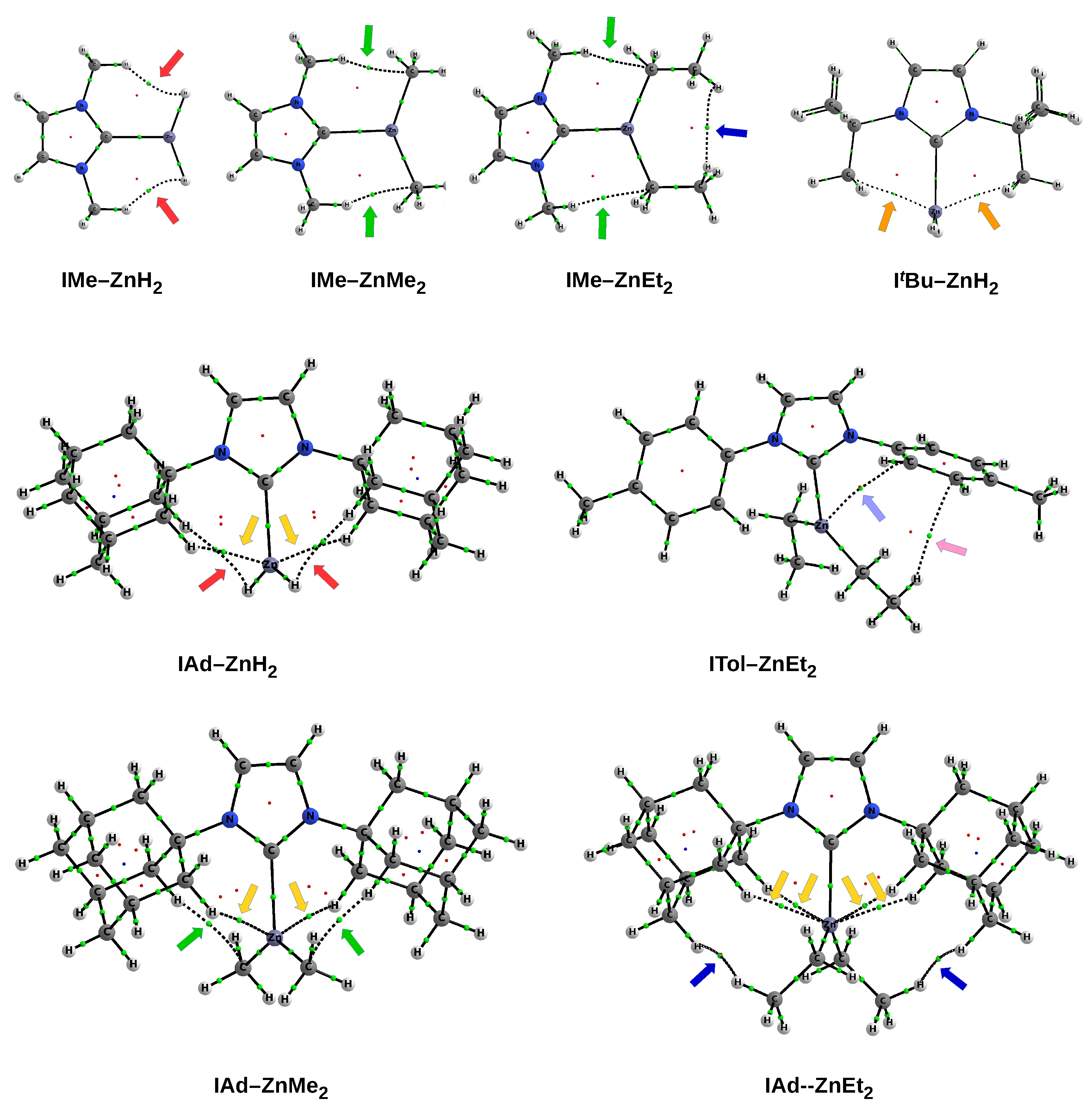
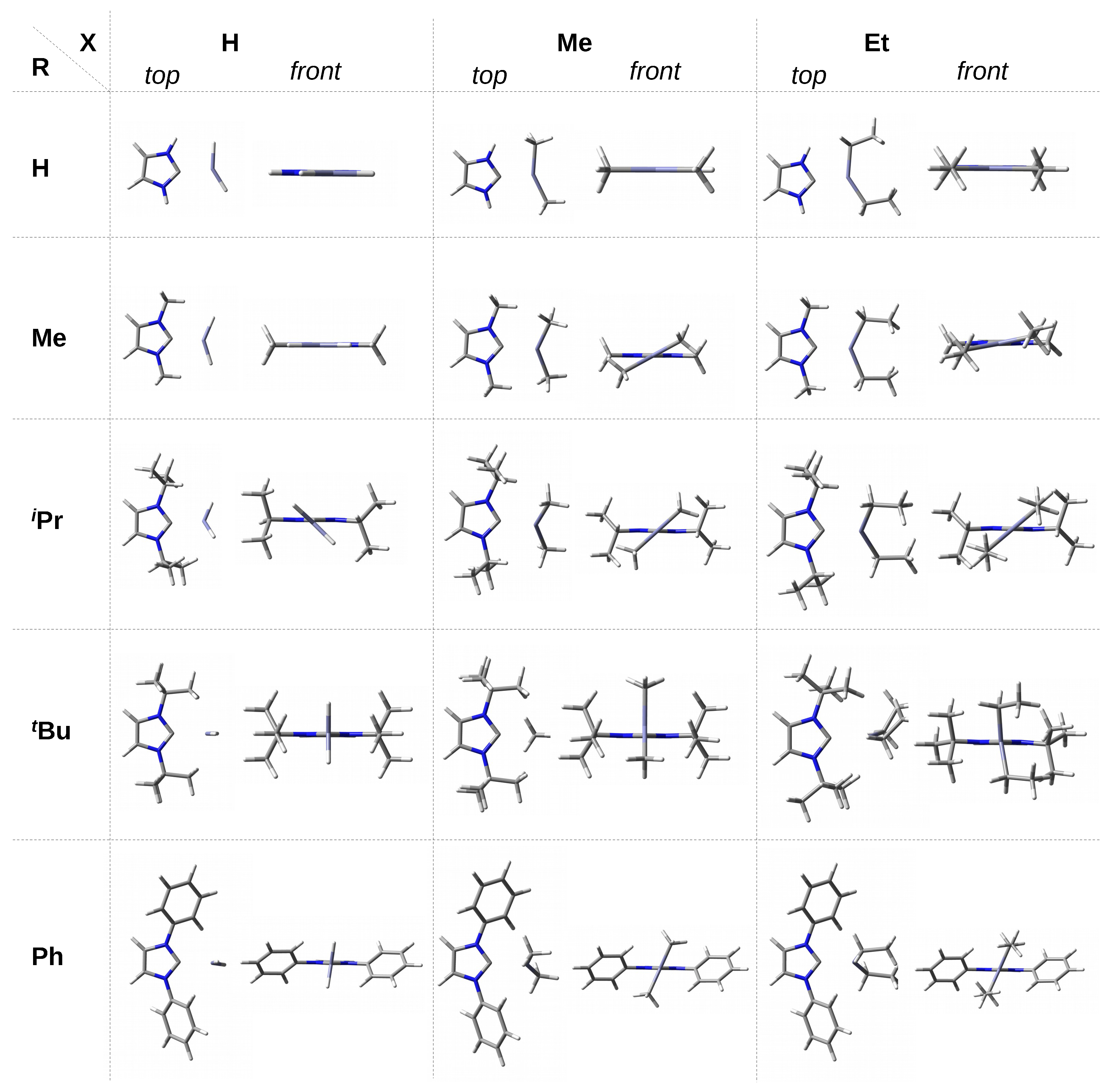
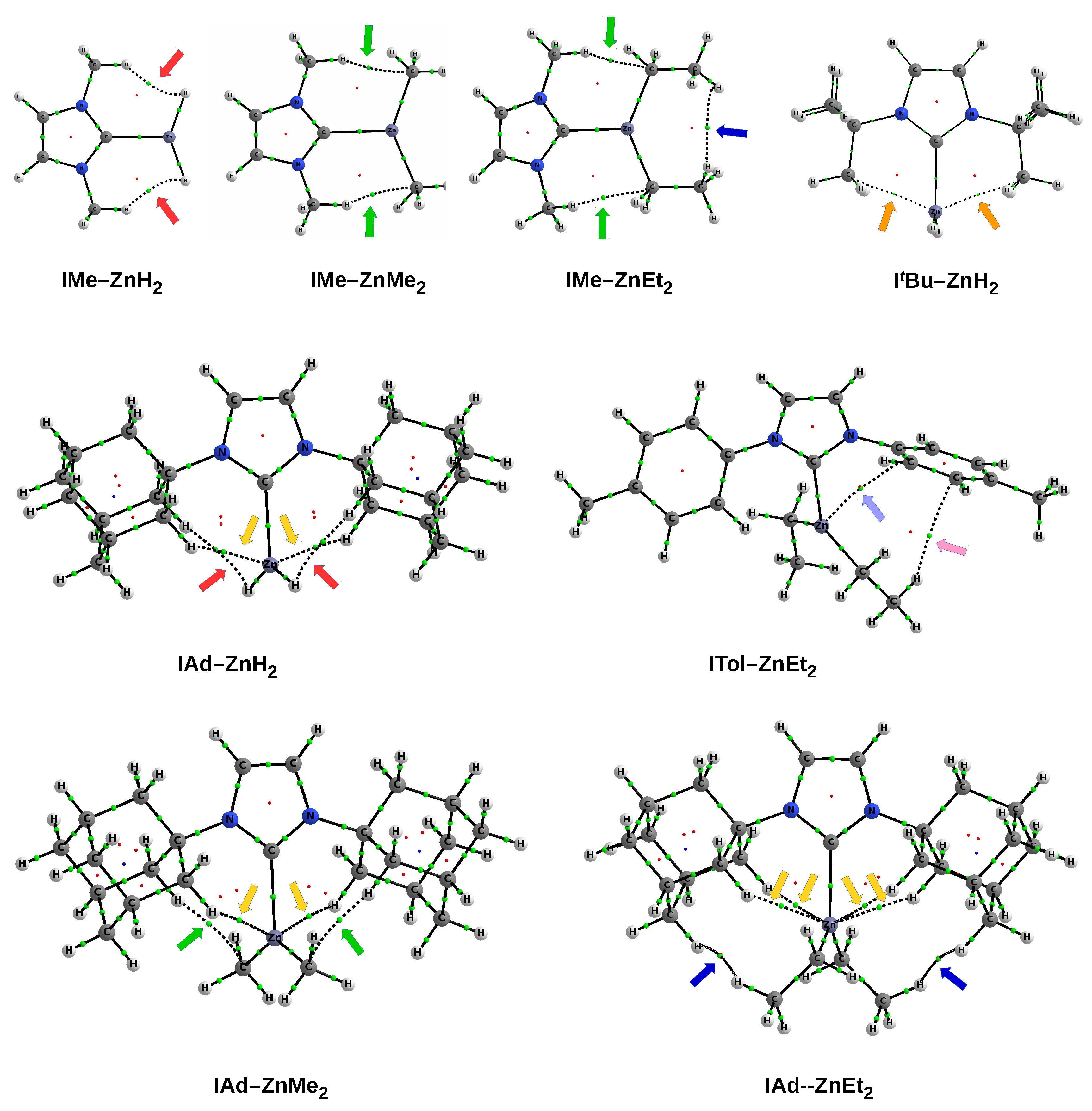
In the case of the smallest imidazol-2-ylidene, the I–ZnX
complex is flat (see also the front view in the top row of
Figure 2). In the case of ZnH
, such a complex structure may be partially due to the presence of a dihydrogen bond N-H⋯H-Zn, while in the case of ZnMe
and ZnEt
, it may be due to probably slightly stabilizing N-H⋯C interactions (see top view in
Figure 2). In the case of a somewhat larger IMe, the ZnX
plane starts to twist slightly when X is an ethyl group (10.7
) and, in particular, a methyl group (28.7
). The explanation should most likely be sought in the most favorable balances between supposedly destabilizing H⋯H interactions and stabilizing C-H⋯C interactions at such
angles. The torsion angle discussed is clearly greater (ca. 42
–45
) for the I
Pr–ZnX
complexes, i.e., those having much more bulky isopropyl groups. If even slightly bulkier
tert-butyl groups are present, then the ZnX
unit is positioned perpendicularly or nearly perpendicularly (ZnEt
) to the plane of the imidazol-2-ylidene ring.
The presence of the phenyl ring in the carbene substituent R introduces new possibilities of interactions. Namely, the
H-C interactions are possible, although they should be weak due to relatively long distances to the planes of the phenyl rings. Despite the large size of the phenyl groups, they give less steric hindrance than the
tert-butyl groups, which facilitates the optimization of Ph⋯ZnX
interactions by appropriately twisting the phenyl groups relative to the plane of the imidazol-2-ylidene ring. This twist is partially forced by a collision interaction between the hydrogen atoms at the positions 4 and 5 of the substituted imidazol-2-ylidene and at the position 2 of the phenyl rings. In the case of ZnMe
and ZnEt
, the torsion angle of the ZnX
plane is practically the same and amounts to ca. 67
. However, in the case of ZnH
, it is significantly greater and amounts to ca. 83
. In combination with the torsion of the phenyl rings, such arrangement of ZnMe
and ZnEt
units enables long-range
H-C intermolecular interactions (see
Figure 2). In contrast, in the case of ZnH
, the
-electron system of the phenyl groups should repel the negative hydrogens of the ZnH
molecule (
Figure 2) leading to a greater twist, which is actually the case.
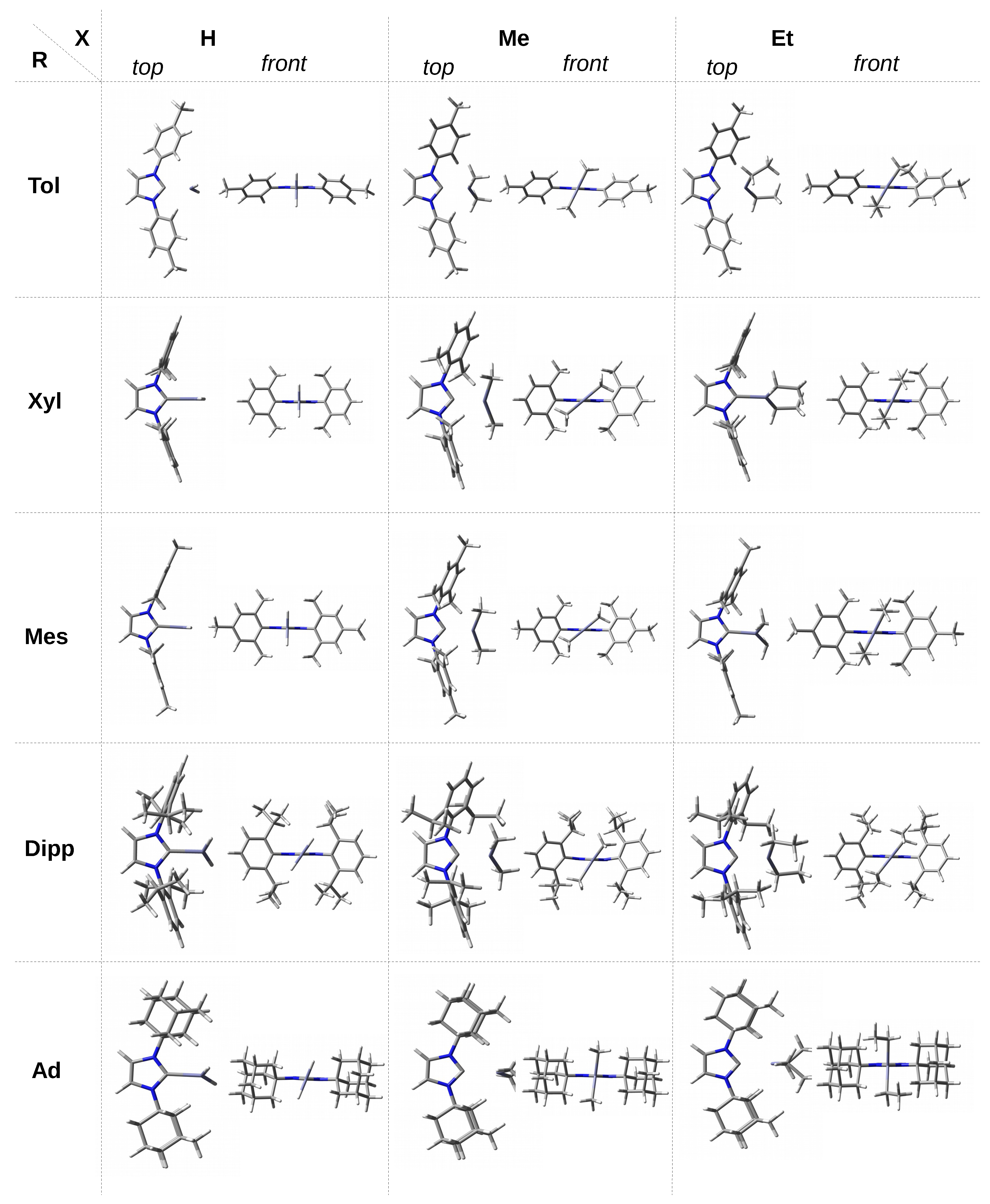
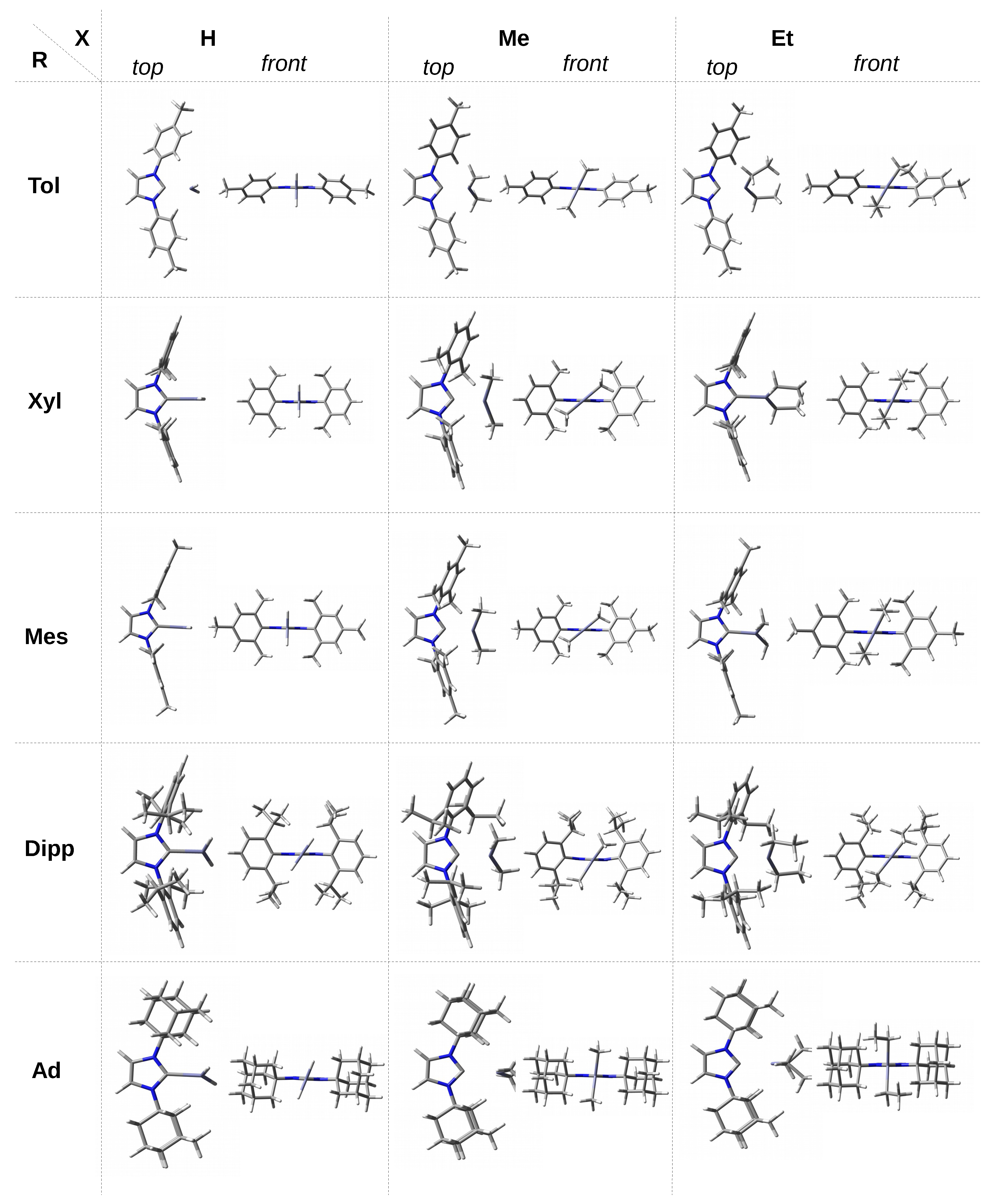
In the case of single, double, and triple methylated phenyl groups, i.e., at the transition to ITol, IXyl, and IMes, the ZnH
unit is perpendicular, which probably results from a slightly greater (as a result of the +I effect of Me) unfavorable effect of
H-Zn. On the other hand, in the case of ZnMe
and ZnEt
molecules, a considerable variation in the values of the
angle is observed. For a given X, being either Me or Et, the torsional angle for the Xyl and Mes groups is practically the same (ca. 40
and 64
, respectively). The greater angle for X = Et should most likely be attributed to the greater steric interactions than for X = Me, which are largely derived from the methyl groups at the 2 and 6 positions of the phenyl rings. In addition, note (
Figure 3) that the rings of Xyl and Mes are nearly perpendicular to the plane of the imidazol-2-ylidene unit, particularly for X = Et. In the case of ZnMe
, the change of the R substituent from Ph to Tol, i.e., the substitution of the methyl group in position 4, practically does not affect the torsional angle (change from 66.7
to 66.1
), while such a replacement has a small impact in the case of ZnEt
(change from 66.6
to 59.7
). Due to the large distance of the methyl group in Tol to X, these changes should not be ascribed to significantly altered steric effects and are rather due to the difference in subtle electronic effects.
Compared to the Ph, Tol, Xyl, and Mes groups, the Dipp group introduces new possibilities. Namely, on the one hand, a large spatial hindrance and, on the other hand, considerable flexibility in the arrangement of the isopropyl groups allow quite large interactions with the ZnX
unit. In addition, the faces of the phenyl rings are largely obscured by these groups. As a result, the observed twist angle will be the result of subtle Dipp⋯X interactions, not only unfavorable steric, but also some favorable ones, i.e., locally stabilizing. In the case of ZnH
and ZnEt
, the angle is similar (54.7
and 53.1
respectively), while it is clearly different (58.3
) in the case of ZnMe
. This may result from both the less symmetrical arrangement of the phenyl rings of the Dipp groups (
Figure 3) and the relatively strong C-H⋯C interactions, which can be expected considering the distribution of ESP around methyl groups (
Figure 4). The adamantyl group is undoubtedly the largest spatially; therefore, it should lead to the greatest steric effects on ZnX
. Indeed, the twist angles for both ZnMe
and especially ZnEt
are considerably large (83.7
and 88.0
, respectively). What may seem surprising at first is the relatively small twist angle for IAd–ZnH
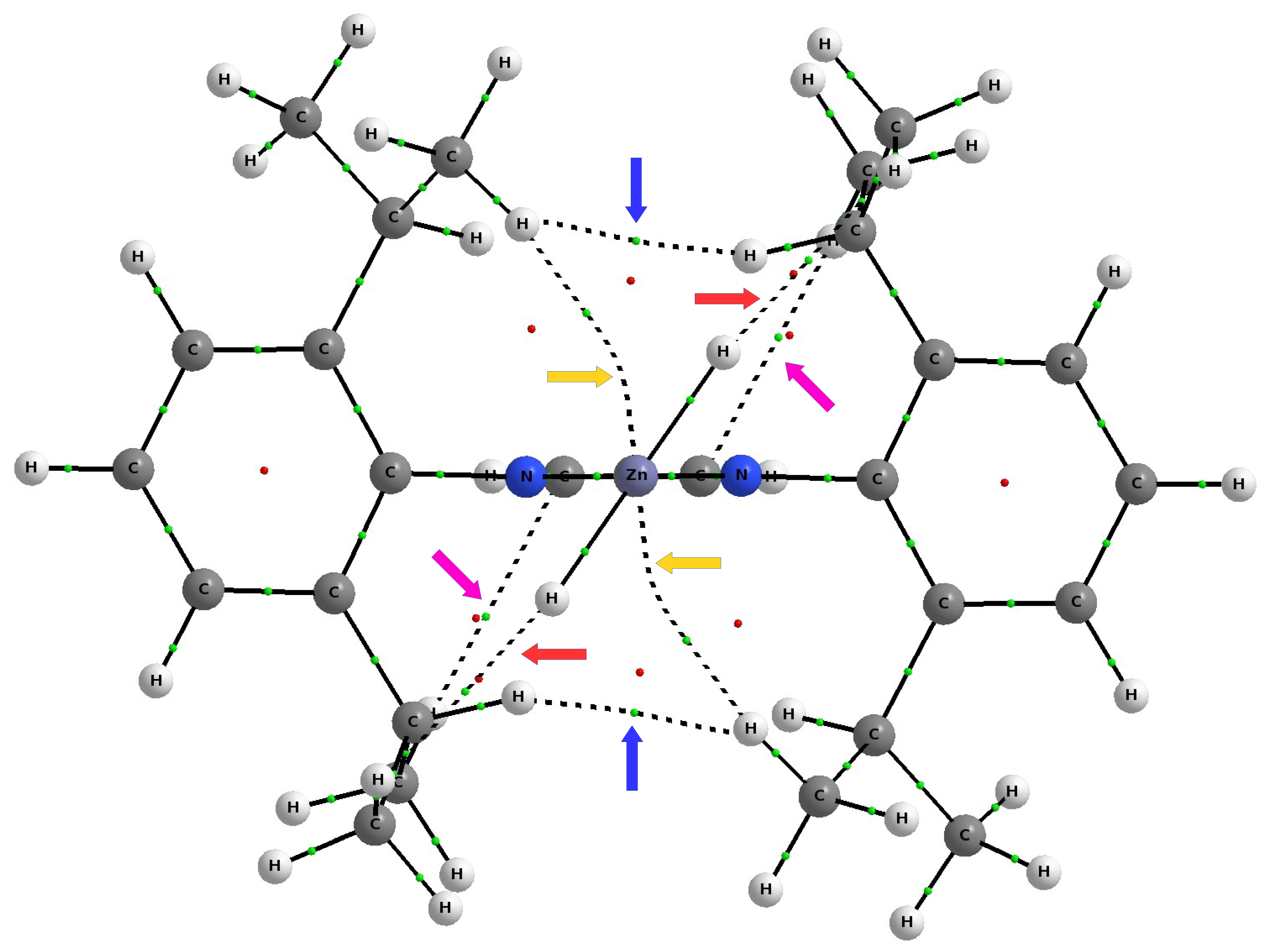
, which amounts to 68.5
only. However,
Figure 3 suggests that this may be due to the presence of two dihydrogen bonds of the C-H⋯H-Zn type, although their length is as large as 2.328 Å. Additionally, such a deviation (
= 68.5
) from the perpendicularly oriented ZnH
molecule allows for the presence of two C-H⋯Zn interactions (2.264 Å), which in principle can be called (
)-agostic bonds [
93,
94,
95,
96,
97,
98,
99,
100,
101].
These latter two examples, i.e., the complexes IDipp–ZnX and IAd–ZnX, show that the torsional angle of the ZnX unit plane with respect to the plane of the imidazol-2-ylidene ring may depend not only on steric effects, but also on the possibility of obtaining favorable long-range interactions, e.g., of the C-H⋯C, C-H⋯H-C, or C-H⋯Zn type. To my knowledge, although in the context of NHC-M systems, the steric effects derived from substituents in the 1 and 3 positions of imidazol-2-ylidene are often described, the significant influence of possible stabilizing interactions has not previously been adequately addressed.
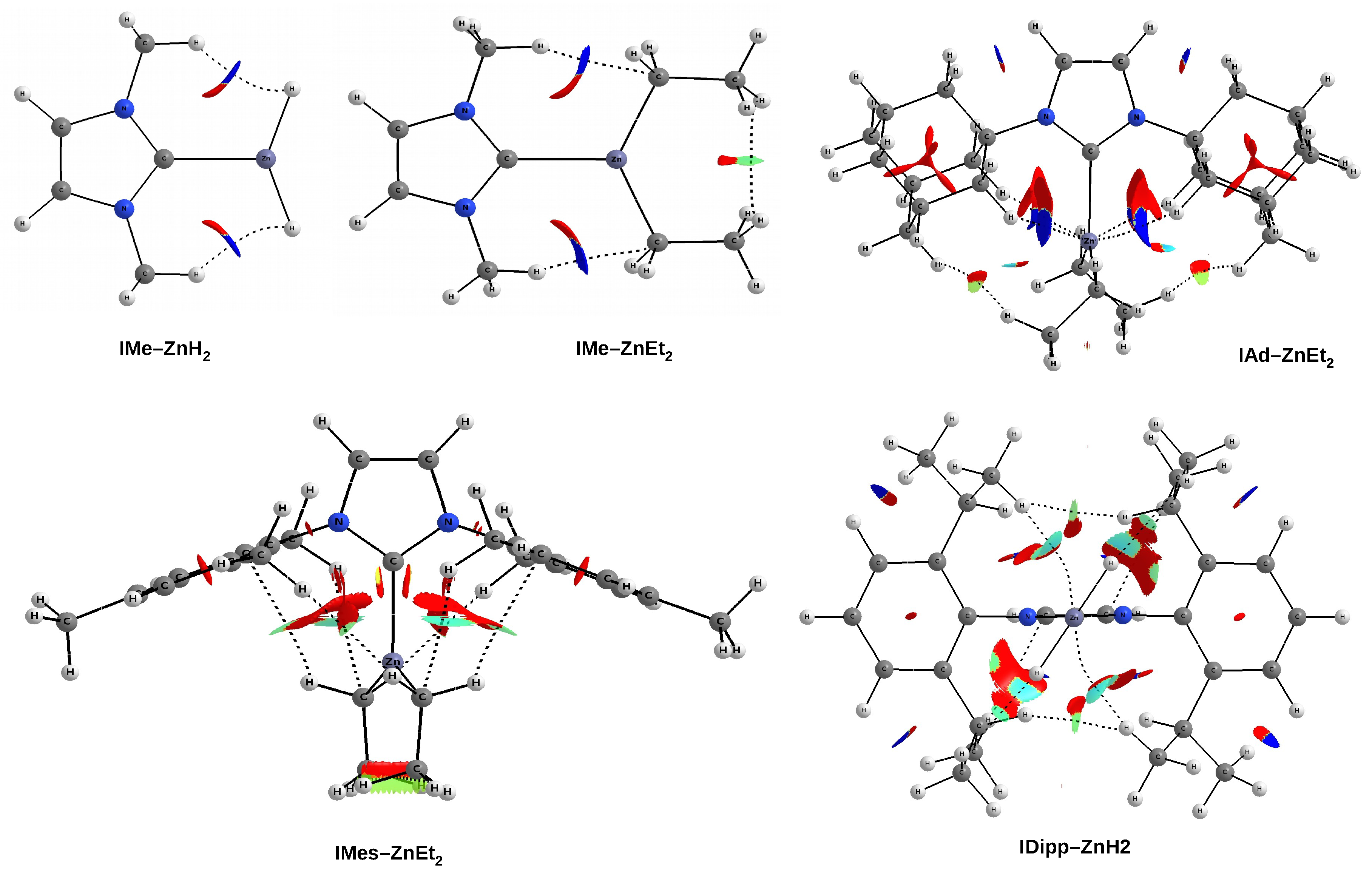
3.4. NCI-Based Analysis
Parameters based on QTAIM are often local as they relate to specific critical points on the molecular graph and are in a way blind to what is happening in the immediate vicinity of these points. One way out of this limitation is the NCI method [
75,
76], which is based on the value of the reduced electron density gradient,
. This method allows one to isolate and then illustrate weak interactions by applying appropriate cutoffs on the values of both the electron density and its reduced gradient. Then, the individual weak interactions are displayed as certain regions of real space rather than simply as a bond critical point between a pair of atoms [
75]. To further investigate the nature of the weak interactions existing between the R substituents in imidazol-2-ylidene and the ZnX
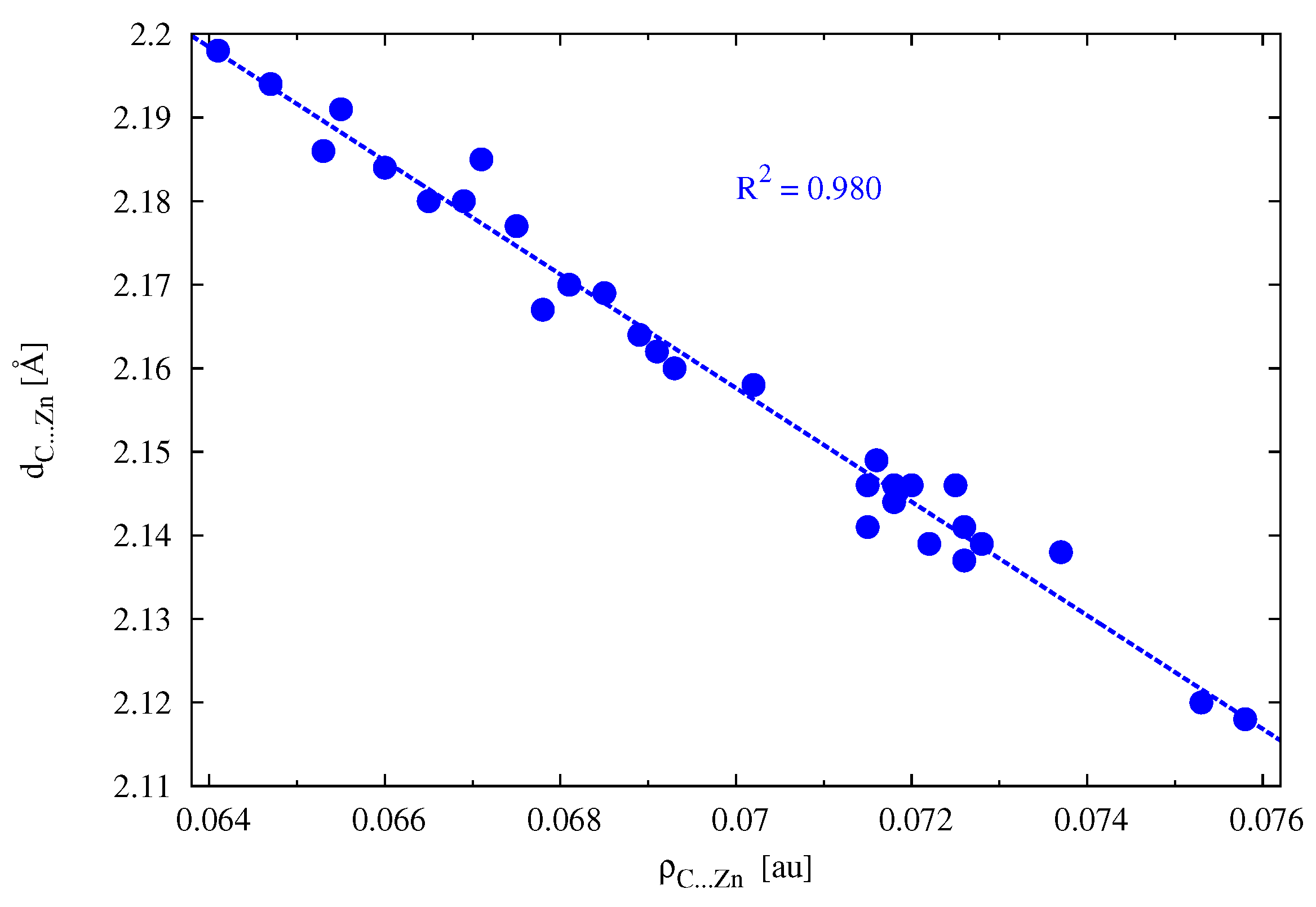
moiety, these interactions were ‘isolated’ by using a fairly low electron density cutoff of 0.030 au. (Note that the C⋯Zn bond considered earlier is characterized by much higher values in the range of 0.064–0.076 au;
Table 6.) To further increase the sensitivity of distinguishing the strength of the respective weak interactions within the NCI method [
75], a very narrow scale of sgn(
)
values was used, from −0.008 au (blue) to zero (red), so that all (weak) repulsive interactions are shown in red. On the contrary, all the other colors are concerned with weak attractive interactions. Representative examples of NCI plots are shown in
Figure 8.
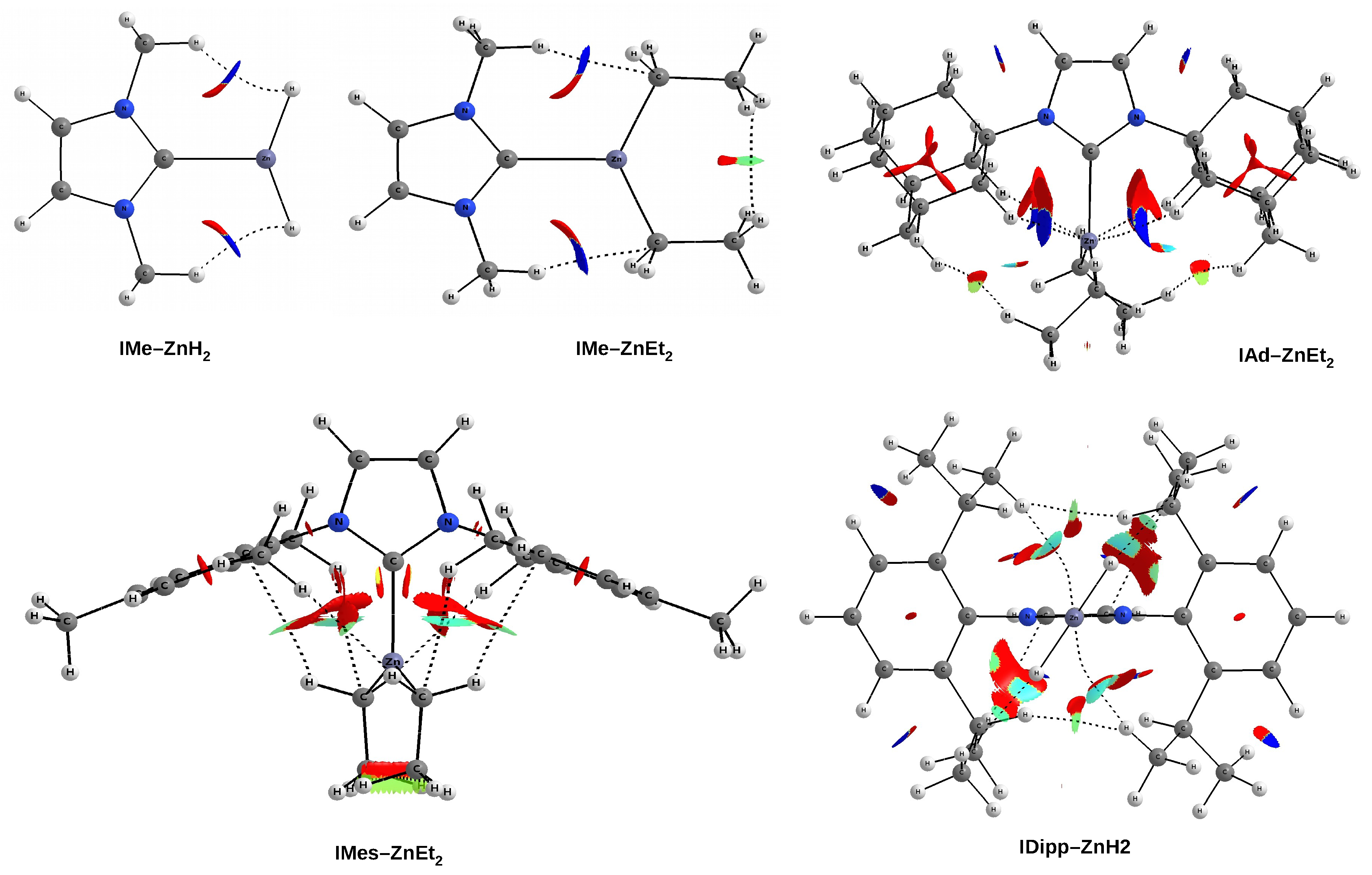
As already mentioned (
Figure 6), the IMe–ZnH
and IMe–ZnEt
complexes are characterized by the presence of a pair of bond paths for C-H⋯H-Zn dihydrogen bond and C-H⋯C tetrel bond, respectively. These paths pierce the blue portions of the
s isosurface, suggesting that the interactions are relatively strong (although they obviously belong to weak non-covalent interactions). In the case of the latter of these complexes, it is clearly visible that the C-H⋯H-C dihydrogen interaction should be much weaker, but also stabilizing (green). The case of the IAd–ZnEt
complex shows that despite the presence of distinct areas of repulsion, the agostic C-H⋯Zn bonds are attractive and stronger than the C-H⋯H-C contacts that indicate the interaction between the Ad and Et groups. A similar situation also takes place in IMes–ZnEt
, although the attractive interactions to the zinc and the C-H
hydrogen bonds should be weaker. It is worth taking a closer look at the interaction zone between the ethyl groups of ZnEt
. Namely, this zone presents two sub-areas with opposite characteristics due to the nature of the interaction. The C⋯C interaction is clearly repulsive, while the interaction between the hydrogen atoms themselves is surprisingly (very weakly) attractive (greenish-yellow).
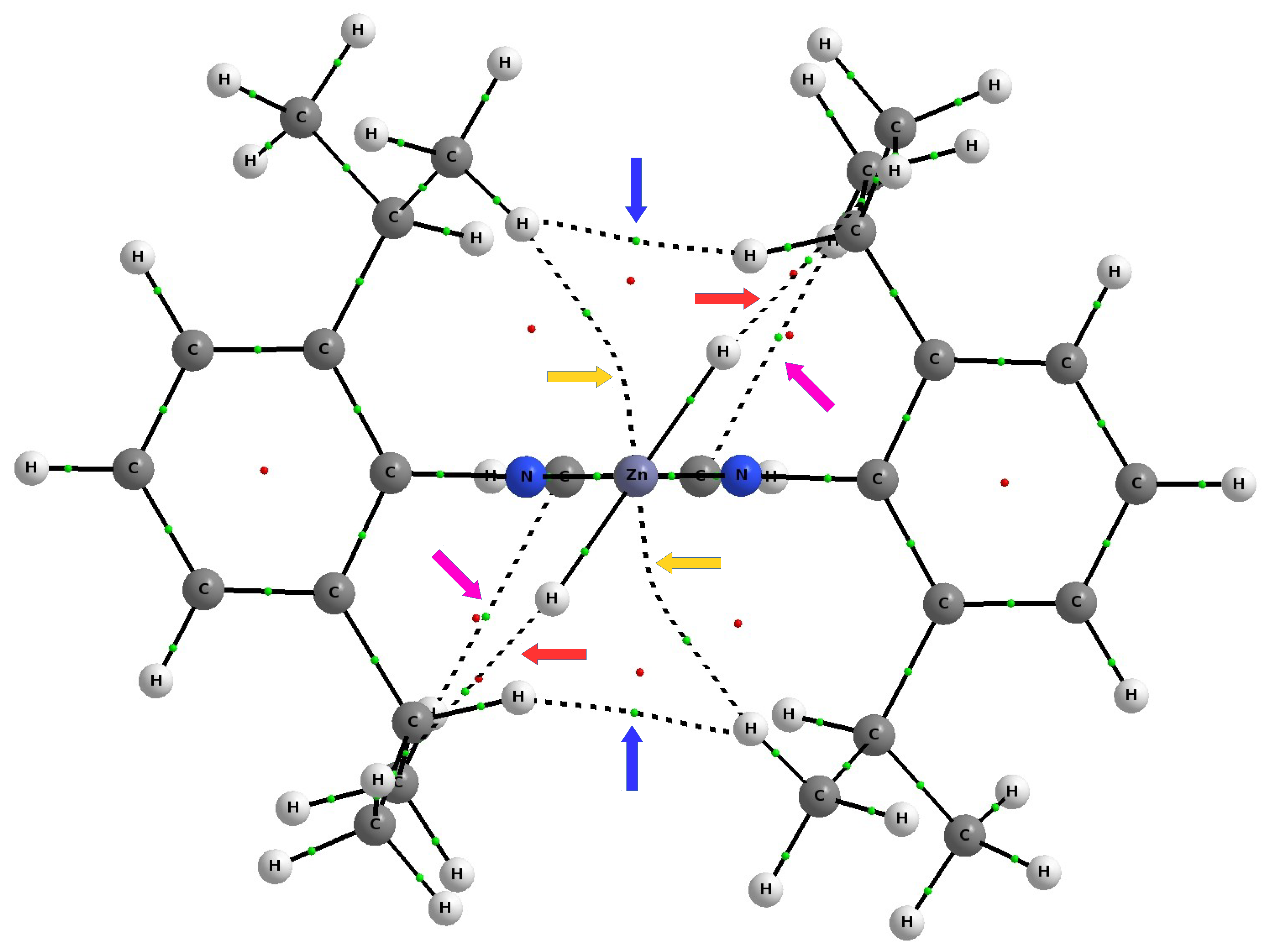
As discussed earlier (see
Figure 7), the complex IDipp–ZnH
exposes four pairs of bond paths for various types of weak non-covalent interactions (C-H
, C-H⋯H-C, C-H⋯Zn, C-H⋯H-Zn). Their presence causes, in addition to the pronounced areas of repulsion, weakly binding regions to also be present, which again shows that the structure of the IR–ZnX
(and more generally NCH–M) complexes not only depends on the steric repulsion of R⋯X, but may also to some extent depend on various binding interactions. Depending on the possibility of their formation, dihydrogen bonds, tetrel bonds, and agostic bonds should play a particular role here. Even if these individual interactions are weak, their overall binding effect may be non-negligible.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}