
Electronic, Structural, and Optical Properties of Mono-Doped and Co-Doped (210) TiO2 Brookite Surfaces for Application in Dye-Sensitized Solar Cells—A First Principles Study

 and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Bulk TiO2 Brookite
3.1.1. Structural Properties
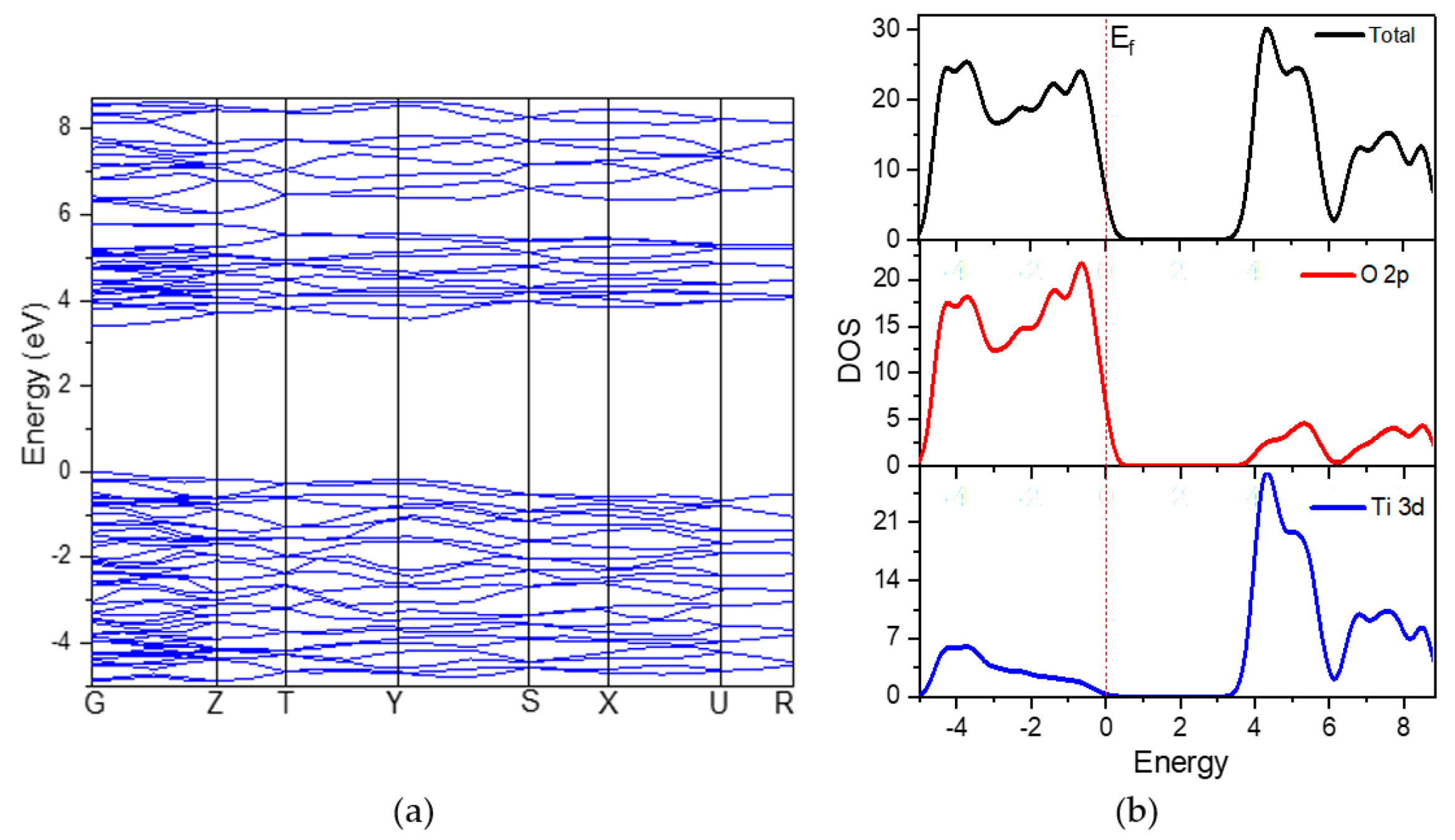
3.1.2. Electronic Properties
3.2. (210). TiO2 Surface
3.2.1. Structural Properties
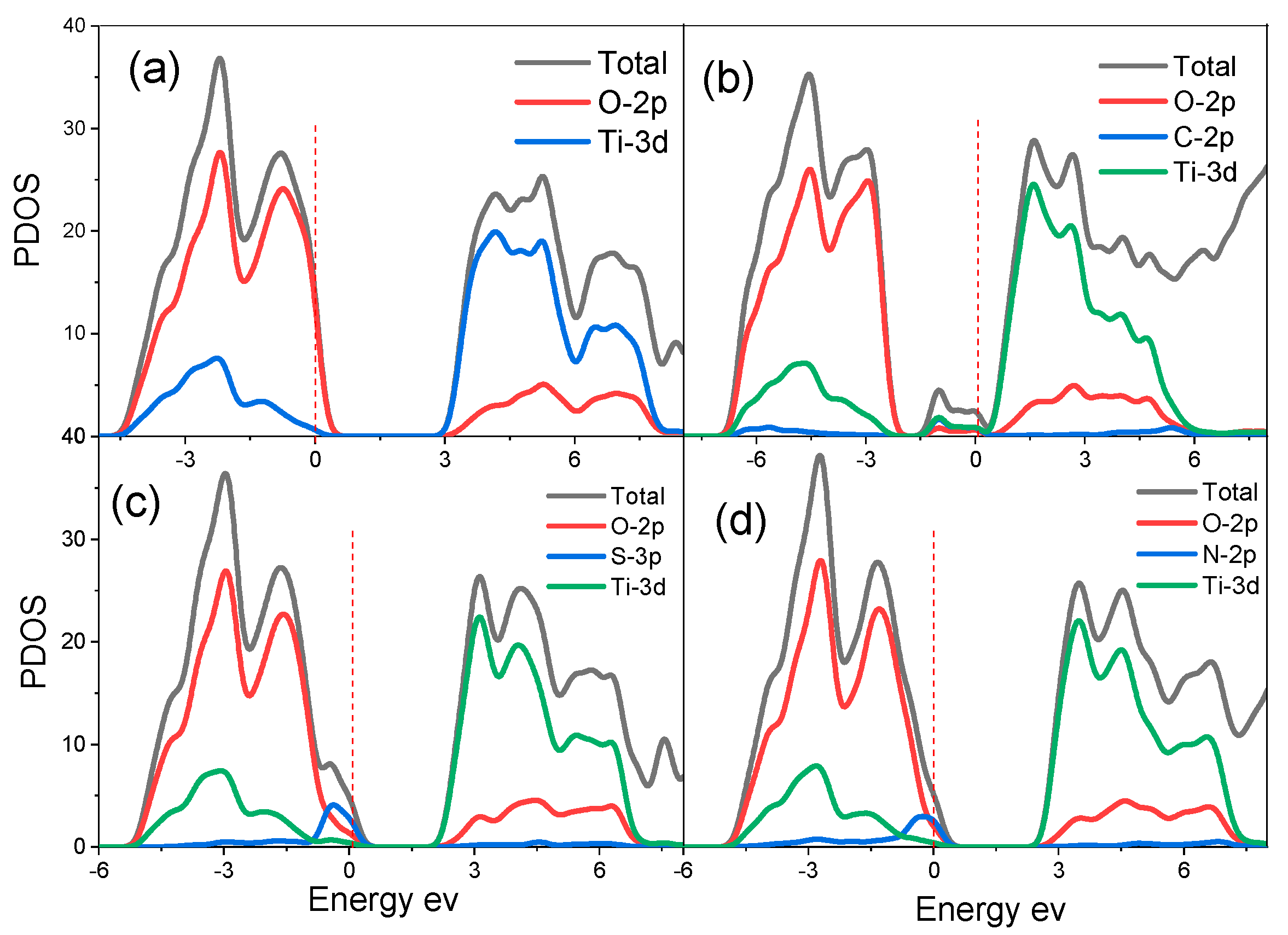
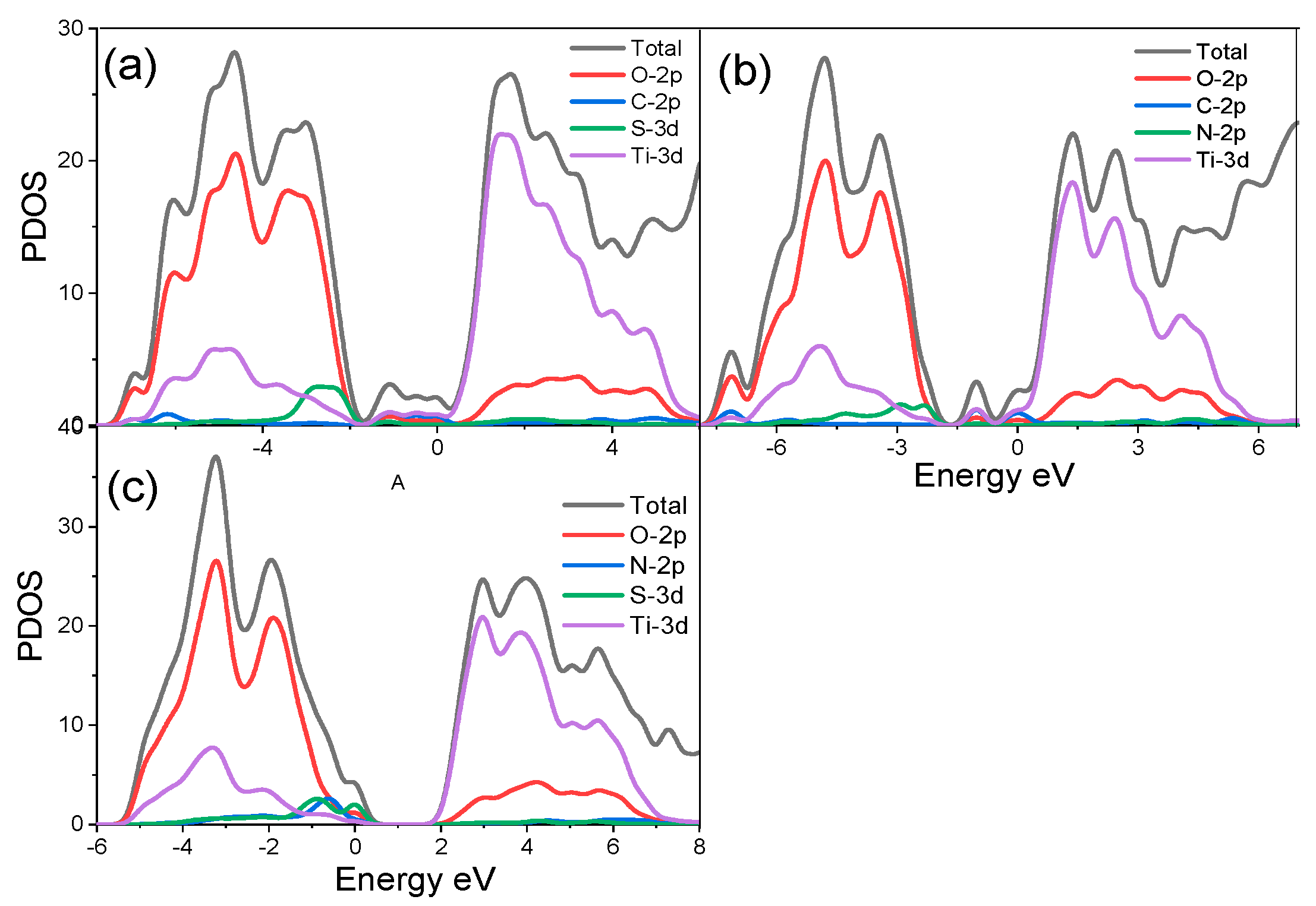
3.2.2. Electronic Properties
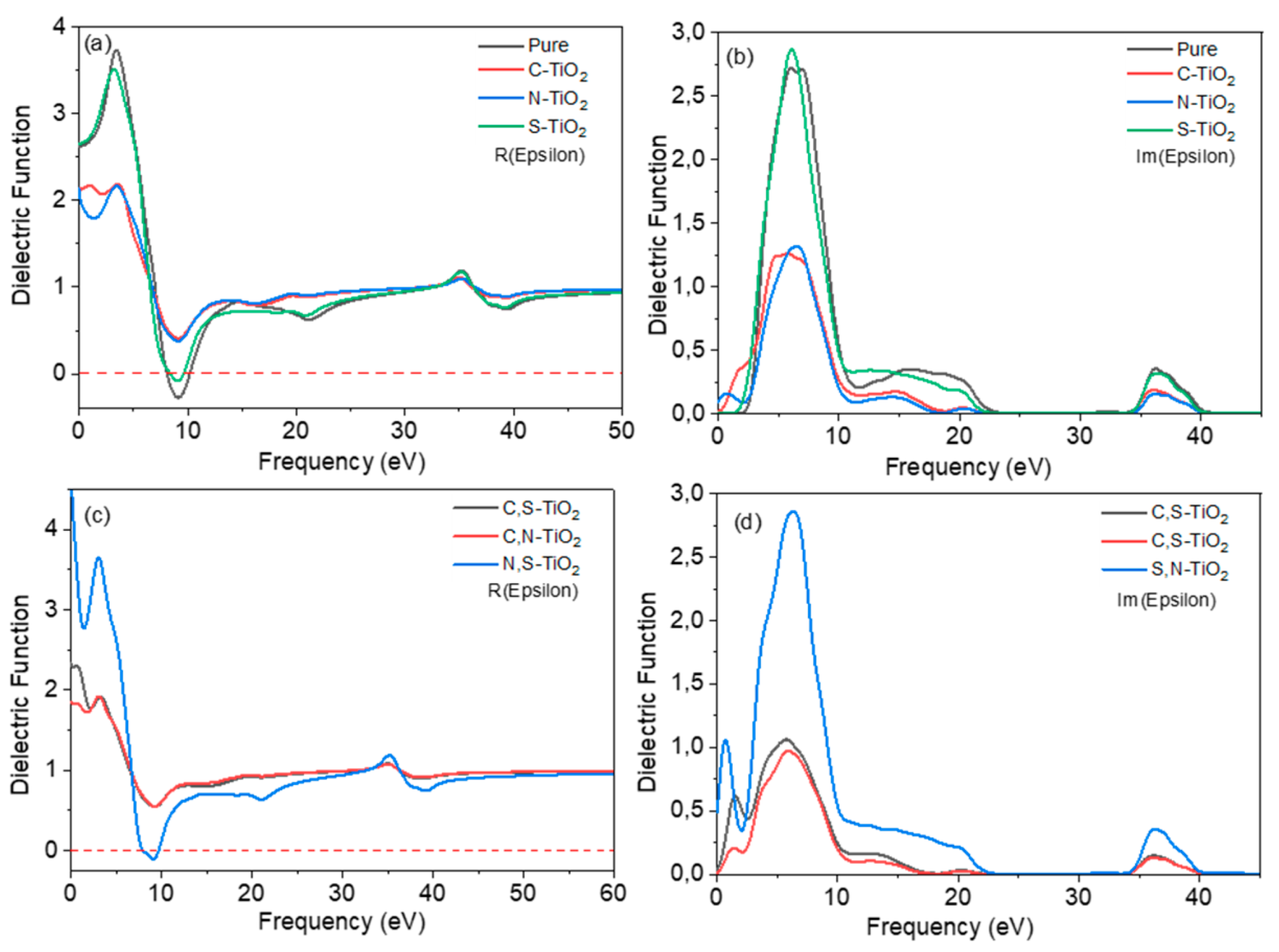
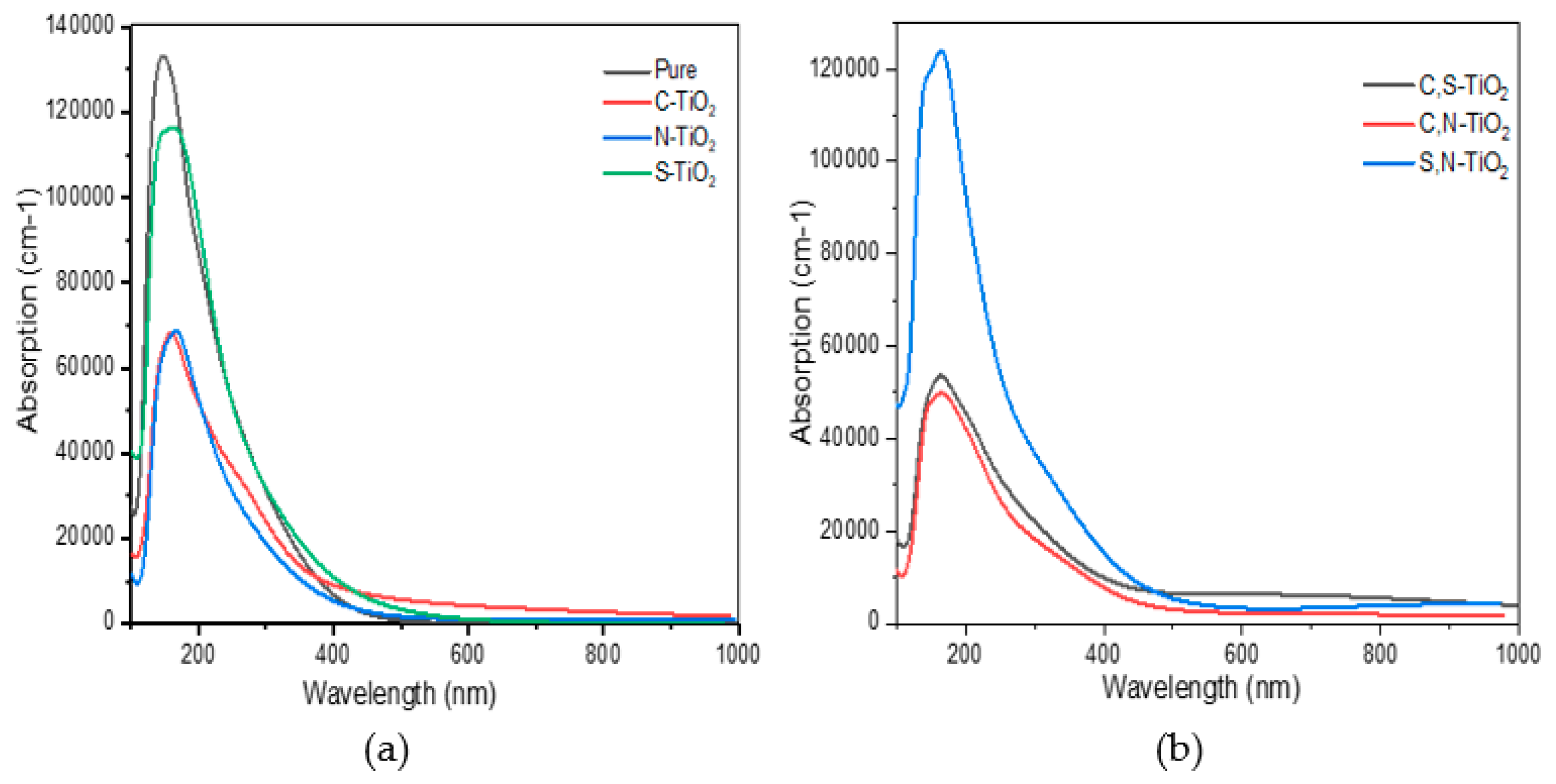
3.3. Optical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magne, C.; Dufour, F.; Labat, F.; Lancel, G.; Durupthy, O.; Cassaignon, S.; Pauporté, T. Effects of TiO2 nanoparticle polymorphism on dye-sensitized solar cell photovoltaic properties. J. Photochem. Photobiol. A 2012, 232, 22–31. [Google Scholar] [CrossRef]
- Serrano, L.A.; Park, K.; Ahn, S.; Wiles, A.A.; Hong, J.; Cooke, G. Coplanar Donor-π-acceptor dyes featuring a furylethynyl spacer for dye-sensitized solar cells. Materials 2019, 12, 839. [Google Scholar] [CrossRef] [Green Version]
- Landmann, M.; Rauls, E.; Shmidt, E.G. The electronic structure and optical response of rutile, anatase and brookite TiO2. J. Condens. Matter. Phys. 2012, 24, 195503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Lee, C.; Jung, K.W.; Jung, D. Structure related photocatalytic properties of TiO2. Bull. Korean Chem. Soc. 2009, 30, 402–404. [Google Scholar]
- Samat, M.H.; Yahya, M.Z.A.; Taib, M.F.M.; Samsi, N.S.; Hussin, N.H.; Ali, A.M.M.; Yaakob, M.K.; Aziz, S.S.S.A. First-principles study on structural, electronic, and optical properties of TiO2 for dye-sensitized solar cells photoanode. Mater. Sci. Forum 2016, 846, 719–725. [Google Scholar] [CrossRef]
- Dima, R.S.; Maluta, N.E.; Maphanga, R.R.; Sankaran, V. Computational study of TiO2 brookite (100), (010) and (210) surface doped with ruthenium for application in dye sensitised solar cells. J. Phys. Conf. Ser. 2017, 905, 012012. [Google Scholar] [CrossRef]
- Koelsch, M.; Cassaignon, S.; Guillemoles, J.F.; Jolivet, J.P. Comparison of optical and electrochemical properties of anatase and brookite TiO2 synthesized by the sol–gel method. Thin Solid Films 2002, 403, 312–319. [Google Scholar] [CrossRef]
- Mo, S.; Ching, W. Electronic and optical properties of three phases of titanium dioxide: Rutile, anatase, and brookite. Phys. Rev. B Condens. Matter. 1995, 51, 13023–13032. [Google Scholar] [CrossRef]
- Meagher, E.P.; Lager, G.A. Polyhedral thermal expansion in the TiO2, polymorphs: Refinement of the crystal structures of rutile and brookite at high temperature. Canad. Mineral. 1979, 17, 77–85. [Google Scholar]
- Beltrán, A.; Gracia, L.; Andrés, J. Density functional theory study of the brookite surfaces and phase transitions between natural titania polymorphs. J. Phys. Chem. B 2006, 110, 23417–23423. [Google Scholar] [CrossRef]
- Gao, P.; Yang, L.; Songtao, X.; Wang, L.; Guo, W.; Lu, J. Effect of Ru, Rh, Mo, and Pd adsorption on the electronic and optical properties of anatase TiO2(101): A DFT investigation. Materials 2019, 12, 814. [Google Scholar] [CrossRef] [Green Version]
- Dima, R.S.; Maluta, N.E.; Maphanga, R.R. First-principles investigation of Ru-and Pt-doped TiO2 brookite surfaces. Int. J. Electrochem. Sci. 2020, 15, 1757–1770. [Google Scholar] [CrossRef]
- Wang, T.; Shen, D.; Xu, T.; Jiang, R. Photocatalytic degradation properties of V-doped TiO2 to automobile exhaust. Sci. Total Environ. 2017, 586, 347. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, J.; Ning, X.; Chen, Y.; Shi, Q. First-principles study of Mn-S co doped anatase TiO2. MRX 2018, 5, 45005. [Google Scholar]
- Samat, M.H.; Ali, A.M.M.; Taib, M.F.M.; Hassan, O.H.; Yahya, M.Z.A. Hubbard U calculations on optical properties of 3d transition metal oxide TiO2. Results Phys. 2016, 6, 891–896. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Jin, L.; Zhou, C. Ab initio study of electronic structures and absorption properties of pure and Fe3+ doped anatase TiO2. Sol. Energy Mater. Sol. Cells. 2011, 95, 2322. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, J.; Yang, Y.; Lu, Z.; Shi, W. Understanding electronic and optical properties of La and Mn co-doped anatase TiO2. Comput. Condens. Matter. 2016, 6, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Dubey, R.S.; Jadkar, S.R.; Bhorde, A.B. Synthesis and characterization of various doped TiO2 nanocrystals for dye-sensitized solar Cells. J. Am. Chem. Soc. 2021, 6, 3470–3482. [Google Scholar]
- Huang, L.H.; Sun, C.; Liu, Y.L. Pt/N-co doped TiO2 nanotubes and its photocatalytic activity under visible light. Appl. Surf. Sci. 2007, 253, 7029. [Google Scholar] [CrossRef]
- Zhao, L.; Xie, Y.; Lin, Q.; Zheng, R.; Diao, Y. Preparation of C, N and P co-doped TiO2 and its photocatalytic activity under visible light. Funct. Mater. Lett. 2019, 12, 1950045. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, J.; Cho, J.H. Electronic properties of N- and C-doped TiO2. Appl. Phys. Lett. 2005, 87, 011904. [Google Scholar] [CrossRef]
- Kuo, C.; Chen, W.; Chen, T. The electronic structure changes and the origin of the enhanced optical properties in N-doped anatase TiO2-A theoretical revisit. Int. J. Appl. Phys. 2014, 116, 093709. [Google Scholar] [CrossRef]
- Chen, H.; Li, X.; Wan, R.; Kao-Walter, S.; Lei, Y.; Leng, C. A DFT study on modification mechanism of (N,S) interstitial co-doped rutile TiO2. Chem. Phys. Lett. 2018, 695, 8–18. [Google Scholar] [CrossRef]
- Guo, Y.; Guo, T.; Chen, J.; Wei, J.; Bai, L.; Ye, X.; Ding, Z.; Xu, W.; Zhou, Z. Synthesis of C-N-S co-doped TiO2 mischcrystal with an isobandgap characteristic and its photocatalytic activity under visible light. Catal. Sci. Technol. 2018, 8, 418–4121. [Google Scholar] [CrossRef]
- Mamakhel, A.; Yu, J.; Søndergaard-Pedersen, F.; Hald, P.; Iversen, B.B. Facile synthesis of brookite TiO2 nano particles Electronic supplementary. Chem. Comm. 2020, 56, 1584–1587. [Google Scholar] [CrossRef]
- Li, W.; Chu, L.; Gong, X.; Lu, G. A comparative DFT study of adsorption and catalytic performance of Au nanoparticles at anatase and brookite TiO2 surfaces. Surf. Sci. 2011, 605, 1369. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, R.; Li, J.; Li, L.; Lin, S. First-principles study on transition metal-doped anatase TiO2. Nanoscale Res. Lett. 2014, 9, 46. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lattice Parameter | Literature Experimental [9] | Literature Calculations [10] | This Work Calculations |
|---|---|---|---|
| a (Å) | 9.184 | 9.157 | 9.166 |
| b (Å) | 5.447 | 5.430 | 5.436 |
| c (Å) | 5.145 | 5.122 | 5.135 |
| V (Å)3 | 25,8.40 | 27,0.37 | 25,5.86 |
| s | p | d | Total | Charge | ||
|---|---|---|---|---|---|---|
| Pure | O | 1.89 | 4.90 | 0 | 6.79 | −0.76 |
| Ti | 2.21 | 6.24 | 2.26 | 10.69 | 1.32 | |
| C-doped | O | 1.89 | 4.90 | 0 | 6.79 | −0.45 |
| C | 1.4 | 3.06 | 0 | 4.52 | −0.52 | |
| Ti | 2.27 | 6.27 | 2.31 | 10.85 | 1.32 | |
| N-doped | O | 1.89 | 4.90 | 0 | 6.79 | −0.56 |
| N | 1.78 | 3.93 | 0 | 5.71 | −0.71 | |
| Ti | 2.23 | 6.25 | 2.29 | 10.77 | −1.33 | |
| S-doped | O | 1.89 | 4.90 | 0 | 6.79 | −0.54 |
| S | 1.87 | 4.54 | 0 | 6.42 | −0.42 | |
| Ti | 2.26 | 6.32 | 2.32 | 10.90 | 1.35 | |
| C- S-doped | O | 1.89 | 4.91 | 0 | 6.80 | −0.54 |
| C | 1.50 | 3.01 | 0 | 4.51 | −0.31 | |
| S | 1.88 | 4.44 | 0 | 6.35 | −0.32 | |
| Ti | 2.29 | 6.30 | 2.49 | 11.08 | 1.34 | |
| C-N-doped | O | 1.89 | 4.90 | 0 | 6.79 | −0.77 |
| C | 1.51 | 2.78 | 0 | 4.30 | −0.30 | |
| N | 1.74 | 4.06 | 0 | 5.08 | −0.80 | |
| Ti | 2.25 | 6.27 | 2.38 | 10.90 | −1.19 | |
| N-S-doped | O | 1.90 | 4.90 | 0 | 6.80 | −0.79 |
| N | 1.76 | 4.03 | 0 | 5.79 | −0.79 | |
| S | 1.91 | 4.38 | 0 | 6.29 | −0.29 | |
| Ti | 2.25 | 6.31 | 2.36 | 10.92 | 1.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dima, R.S.; Phuthu, L.; Maluta, N.E.; Kirui, J.K.; Maphanga, R.R. Electronic, Structural, and Optical Properties of Mono-Doped and Co-Doped (210) TiO2 Brookite Surfaces for Application in Dye-Sensitized Solar Cells—A First Principles Study. Materials 2021, 14, 3918. https://doi.org/10.3390/ma14143918
Dima RS, Phuthu L, Maluta NE, Kirui JK, Maphanga RR. Electronic, Structural, and Optical Properties of Mono-Doped and Co-Doped (210) TiO2 Brookite Surfaces for Application in Dye-Sensitized Solar Cells—A First Principles Study. Materials. 2021; 14(14):3918. https://doi.org/10.3390/ma14143918
Chicago/Turabian StyleDima, Ratshilumela S., Lutendo Phuthu, Nnditshedzeni E. Maluta, Joseph K. Kirui, and Rapela R. Maphanga. 2021. "Electronic, Structural, and Optical Properties of Mono-Doped and Co-Doped (210) TiO2 Brookite Surfaces for Application in Dye-Sensitized Solar Cells—A First Principles Study" Materials 14, no. 14: 3918. https://doi.org/10.3390/ma14143918
APA StyleDima, R. S., Phuthu, L., Maluta, N. E., Kirui, J. K., & Maphanga, R. R. (2021). Electronic, Structural, and Optical Properties of Mono-Doped and Co-Doped (210) TiO2 Brookite Surfaces for Application in Dye-Sensitized Solar Cells—A First Principles Study. Materials, 14(14), 3918. https://doi.org/10.3390/ma14143918