
Marine Atmospheric Corrosion of Carbon Steel: A Review




Abstract
:1. Introduction
2. Basic Concepts
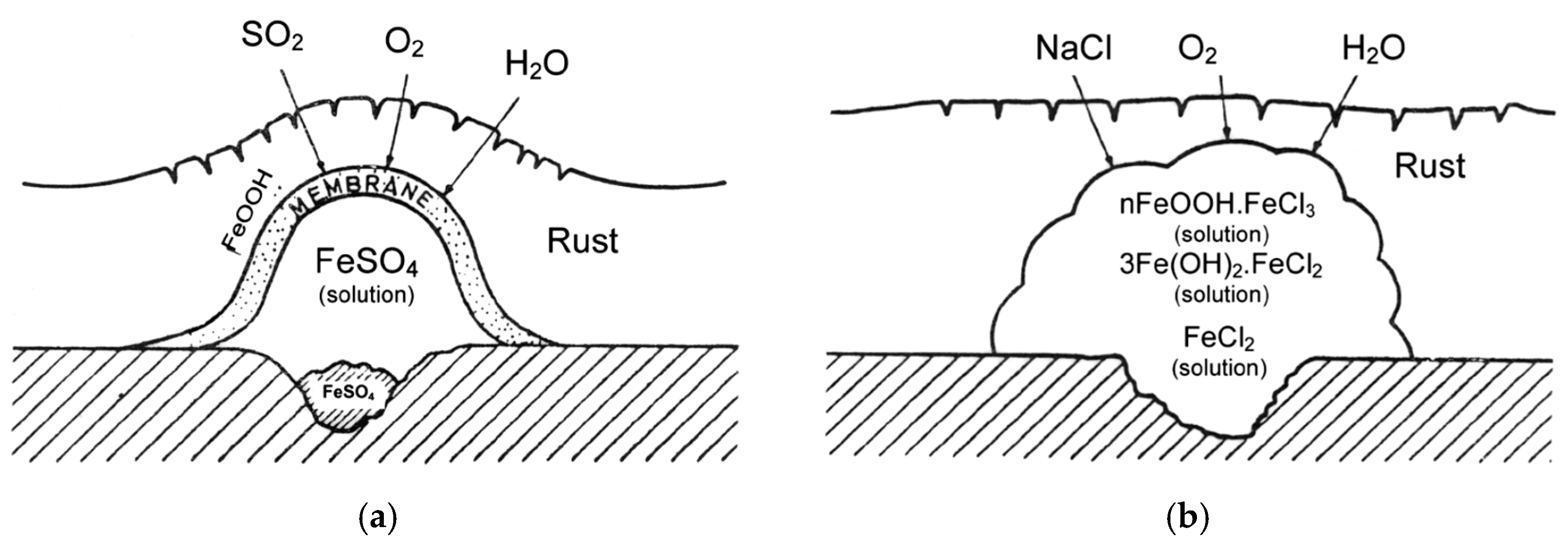
2.1. Sulfur Dioxide
2.2. Saltwater Aerosols
2.3. Hydrogen Chloride Vapours
3. Experimentation on Marine Atmospheric Corrosion
4. The Marine Atmosphere
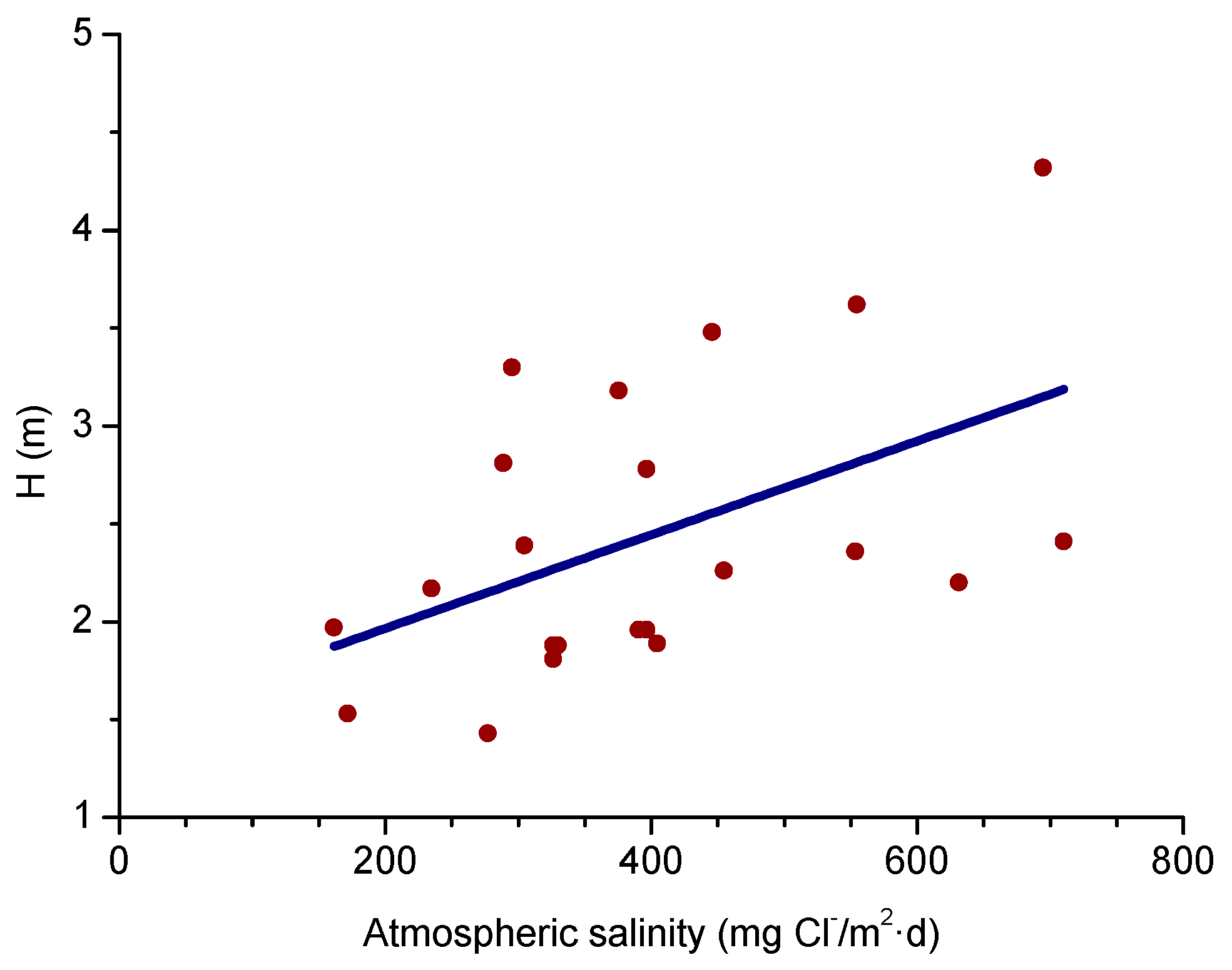
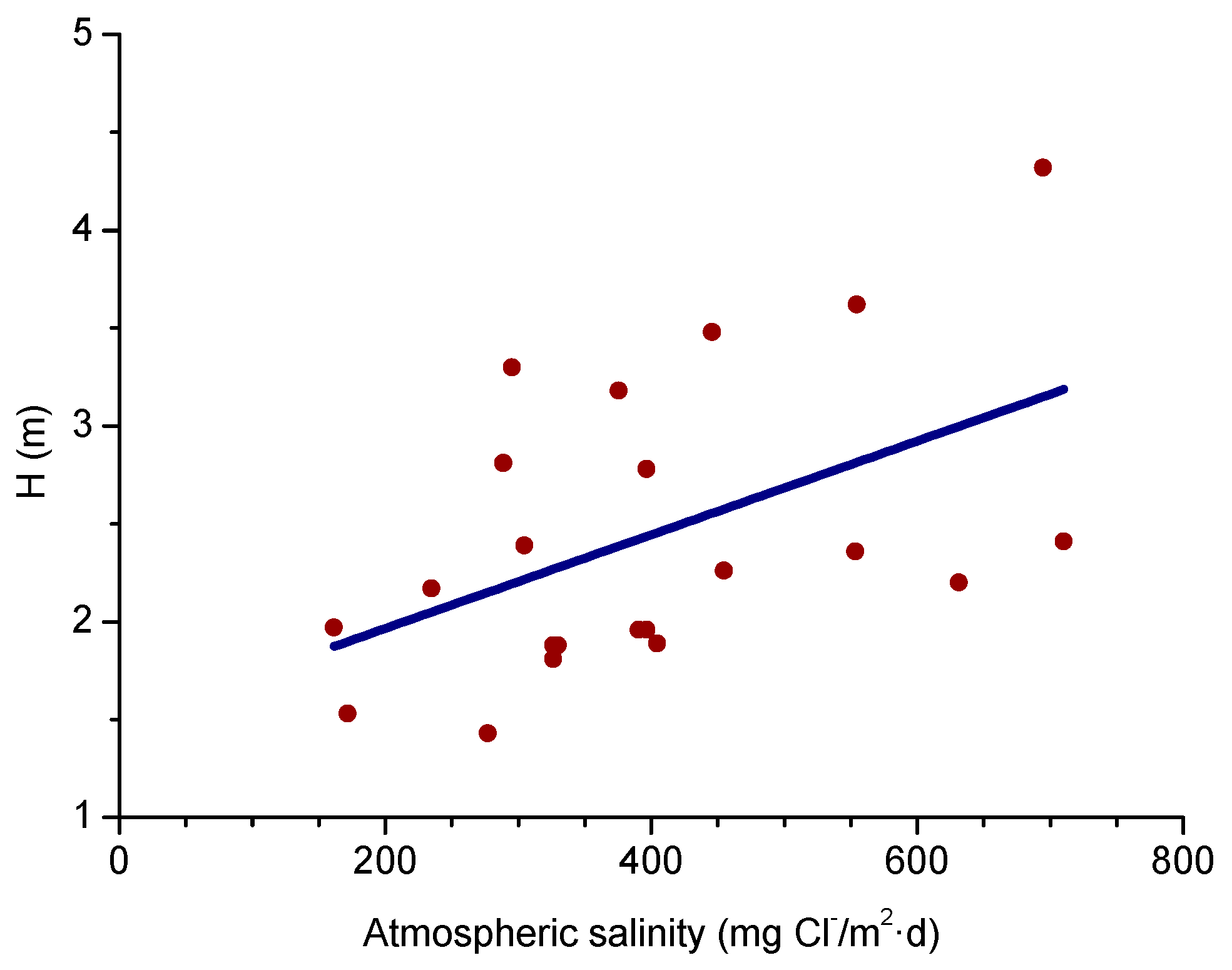
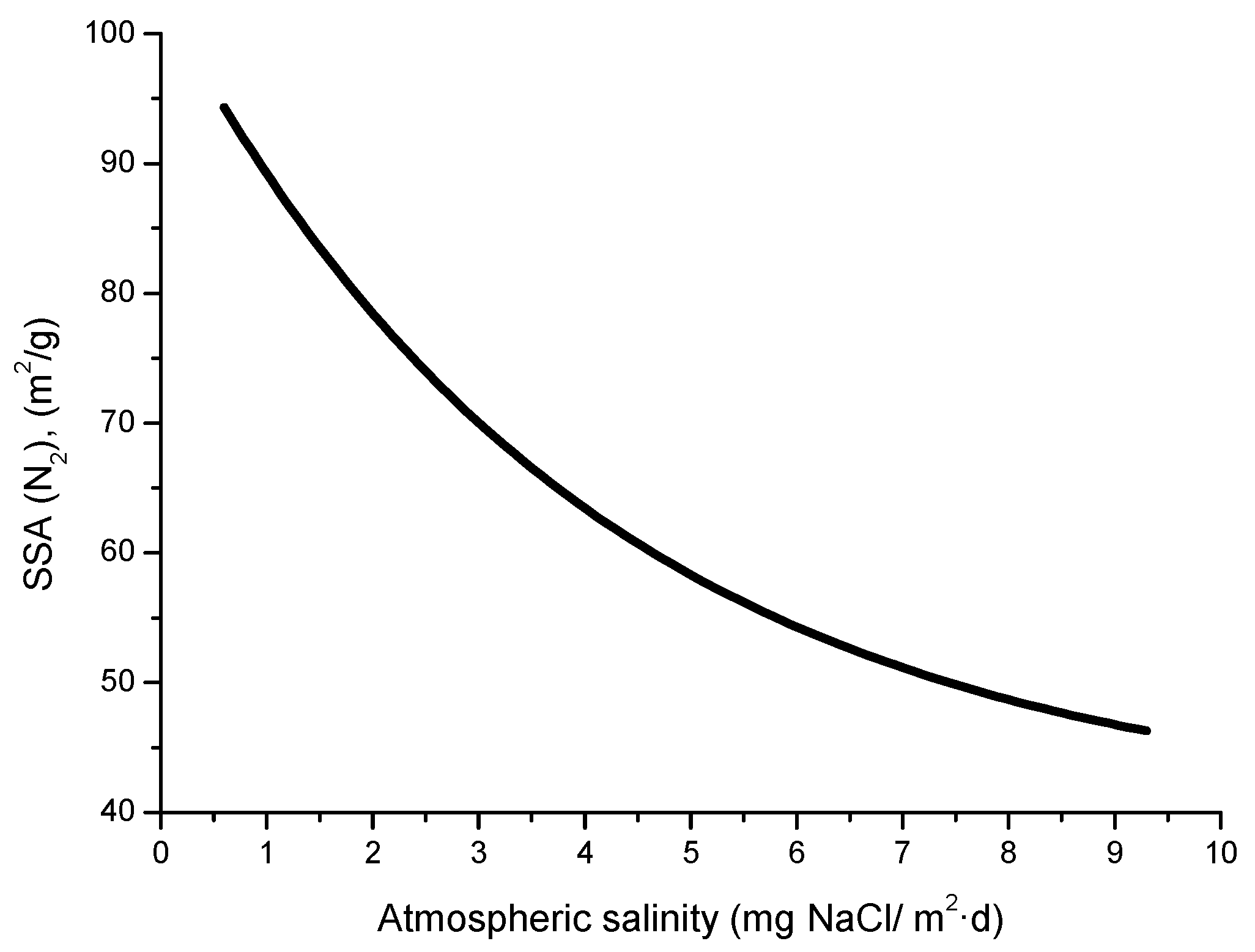
4.1. Atmospheric Salinity
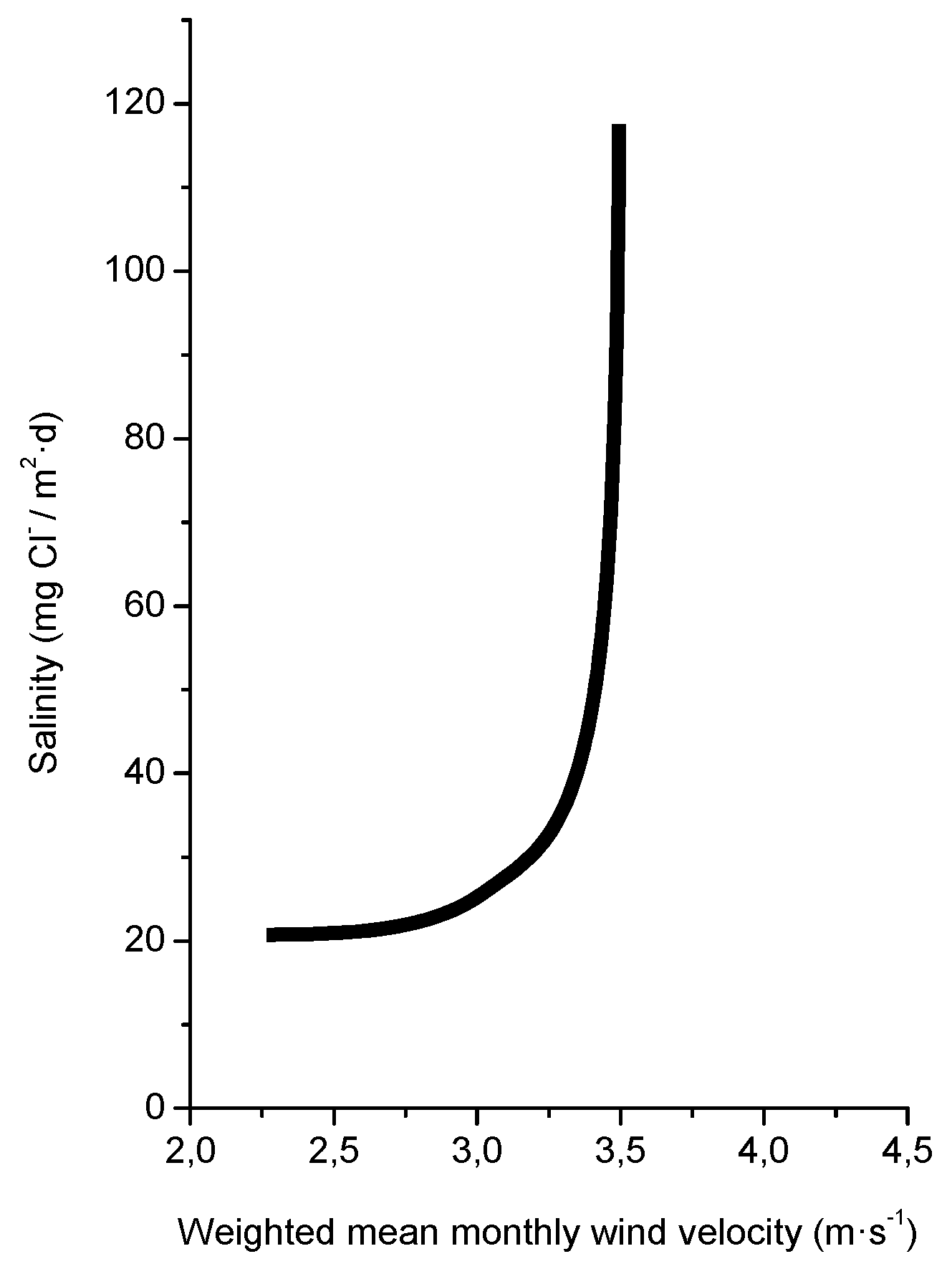
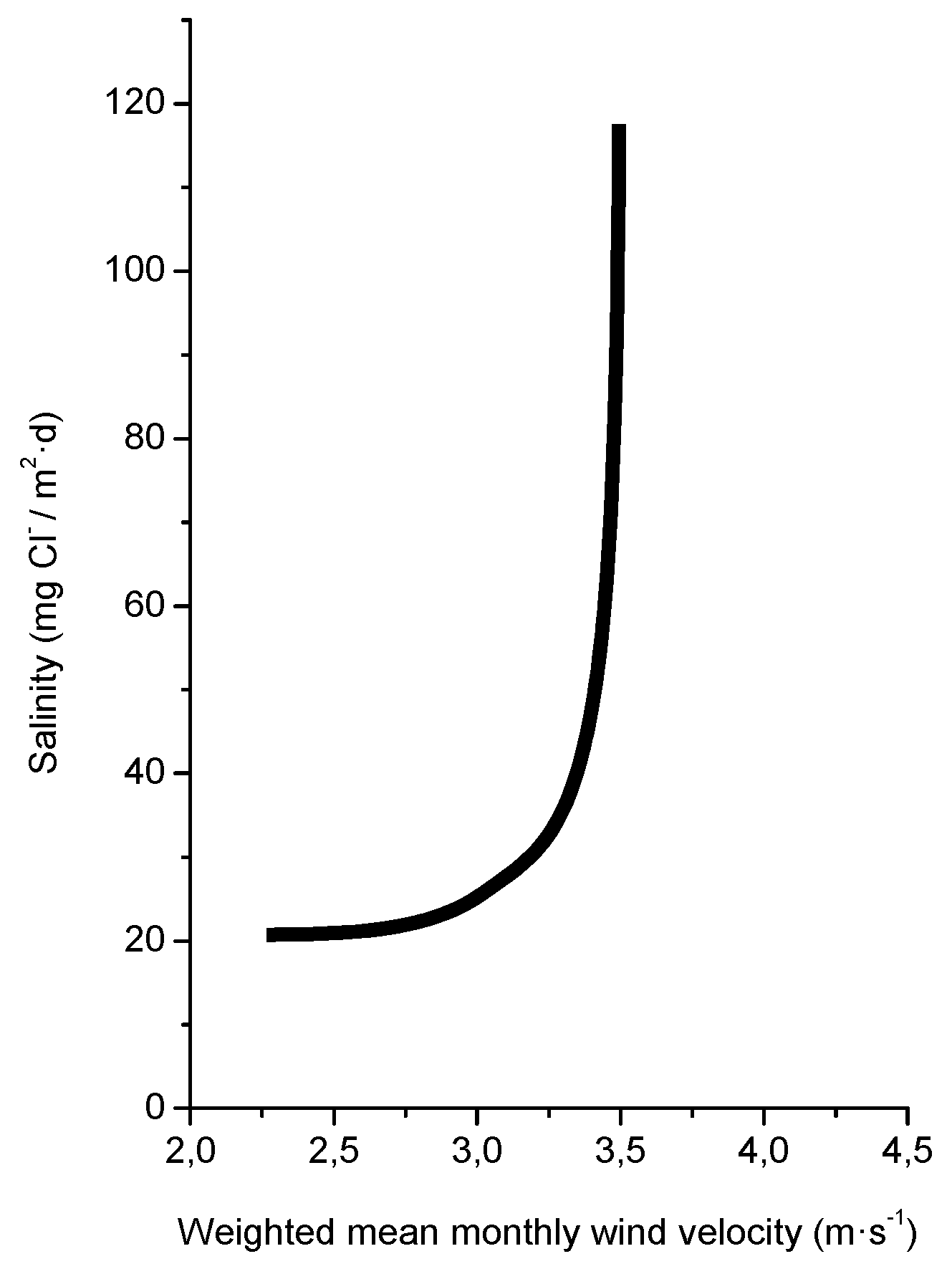
4.2. Production of Marine Aerosol
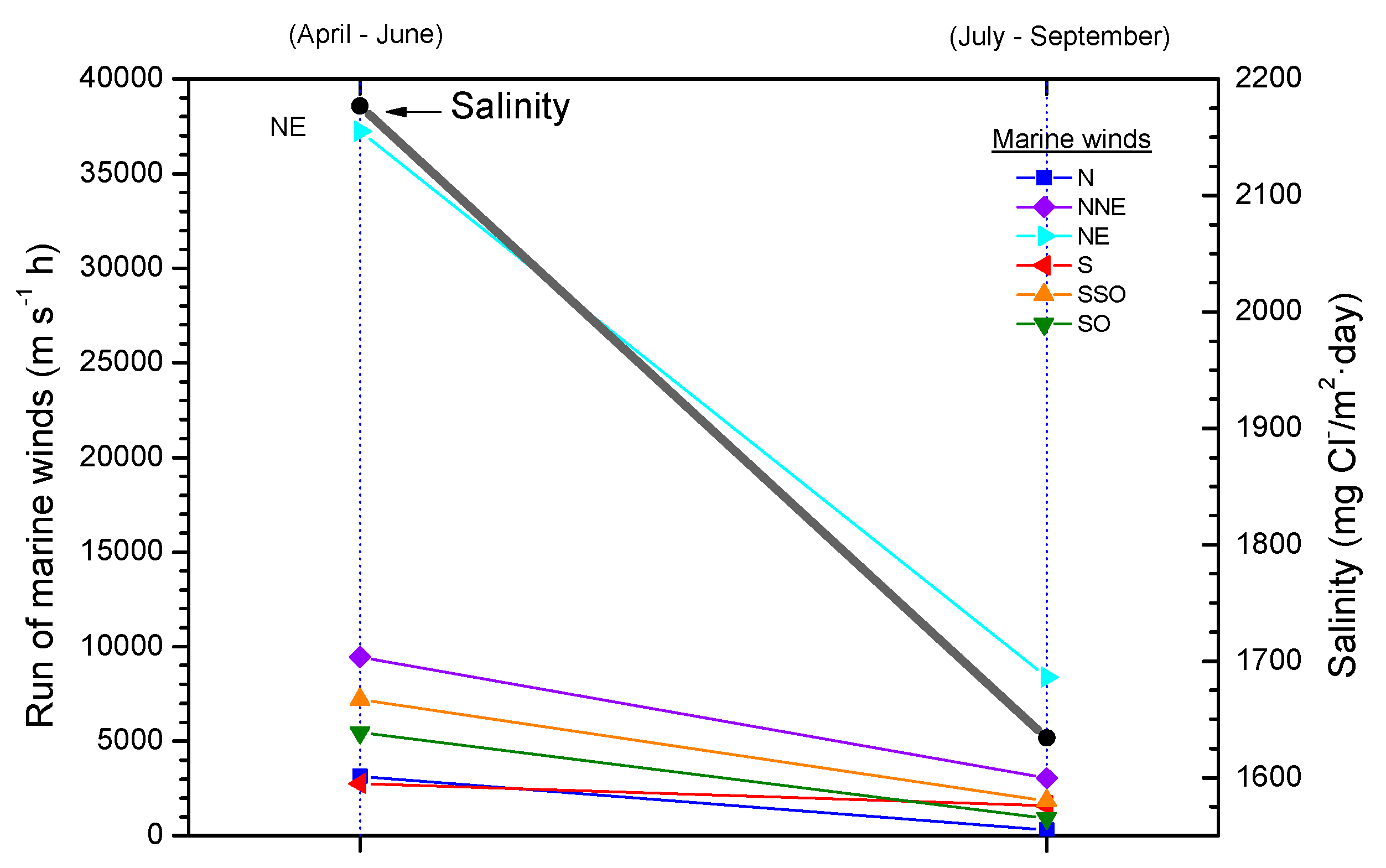
4.3. Entrainment of Marine Aerosol Inland
4.4. Effect of Salinity on Steel Corrosion
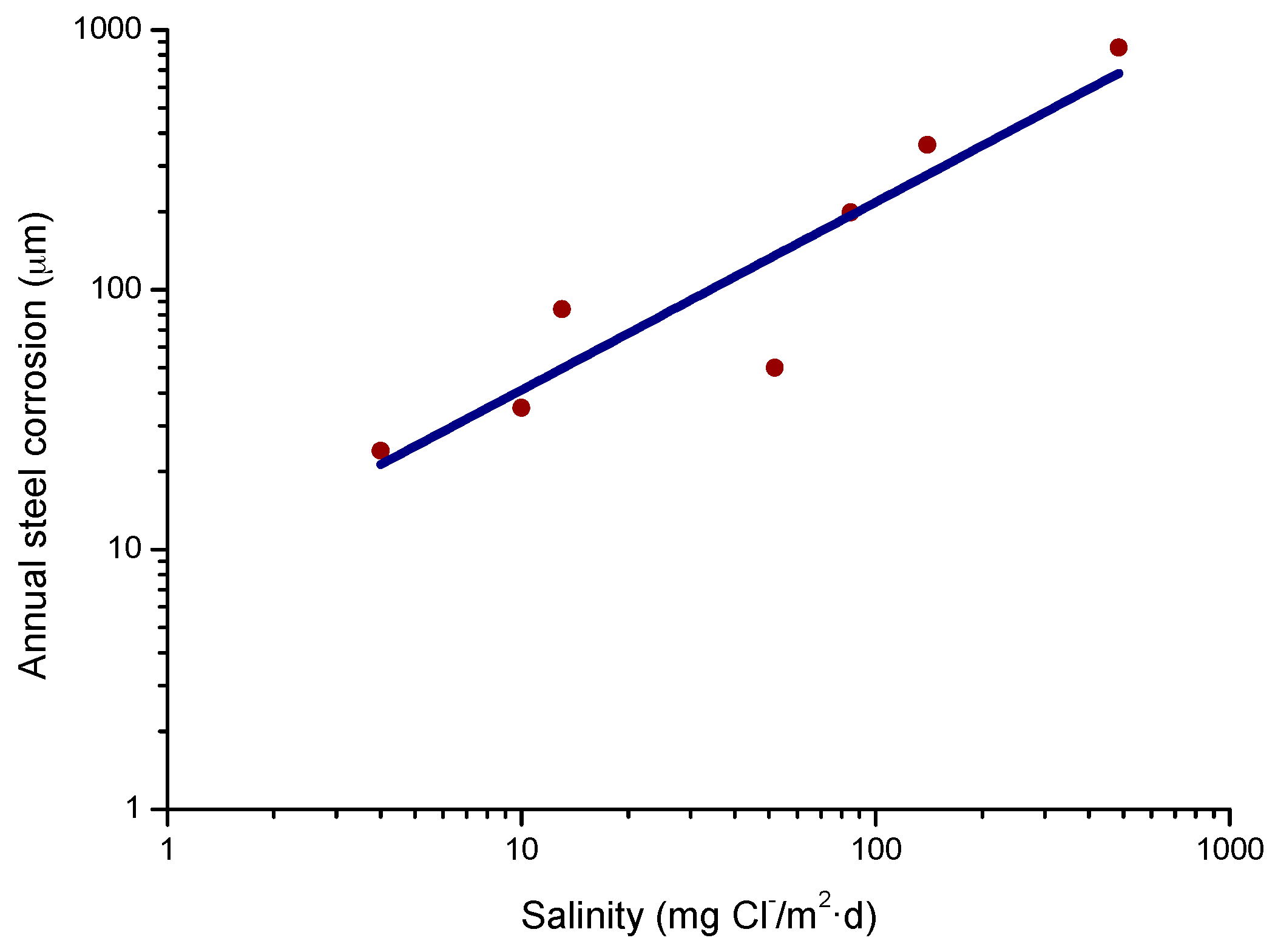
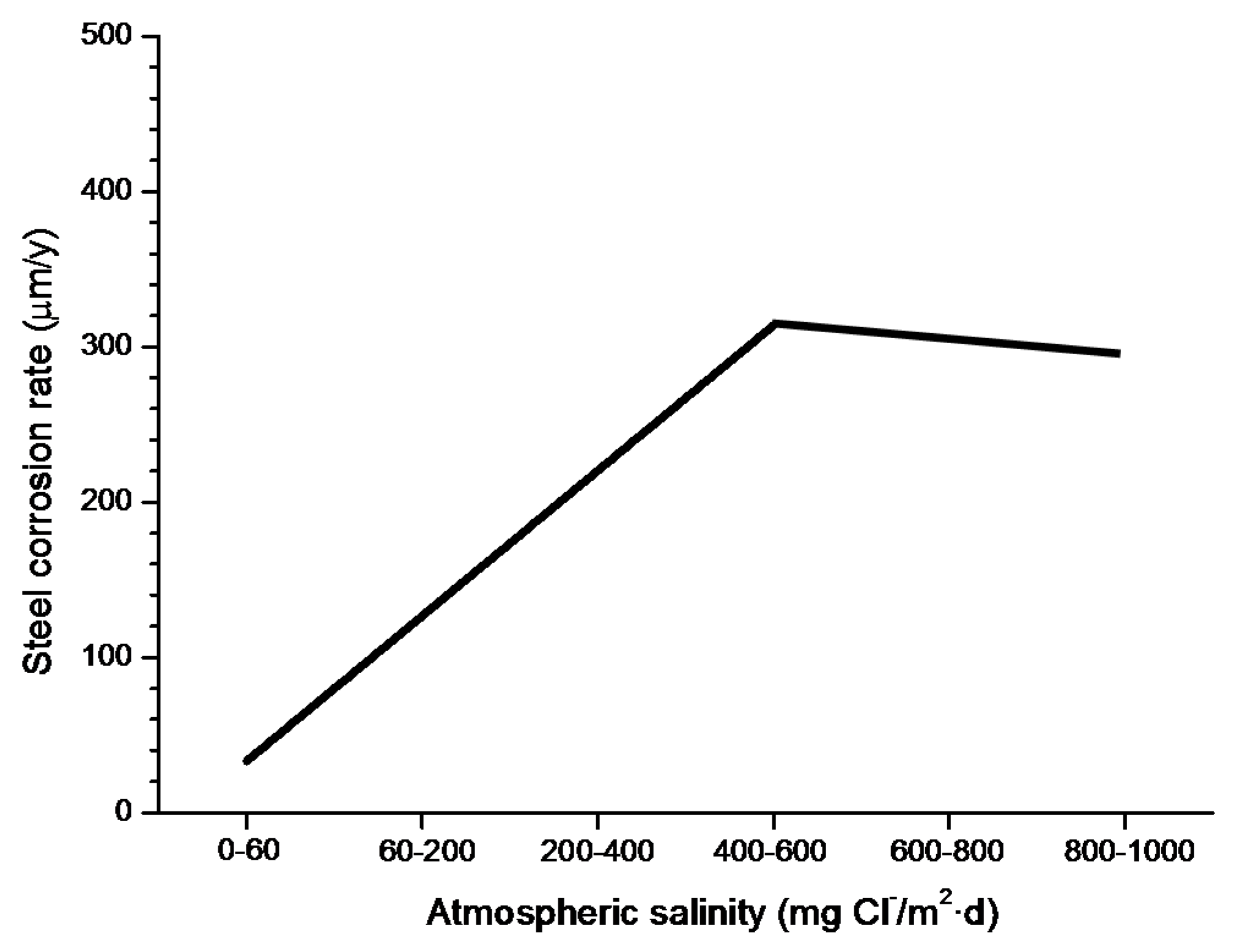
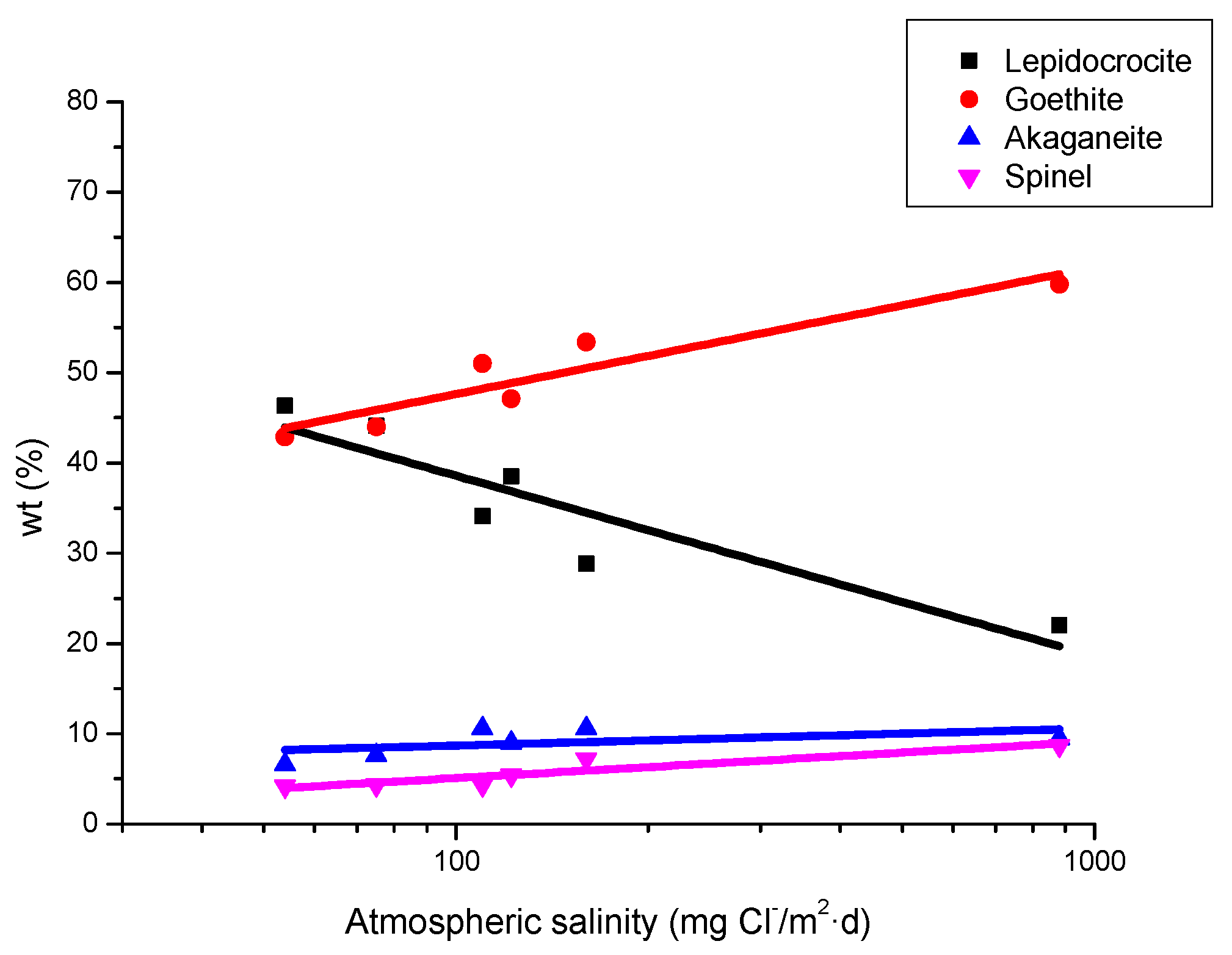
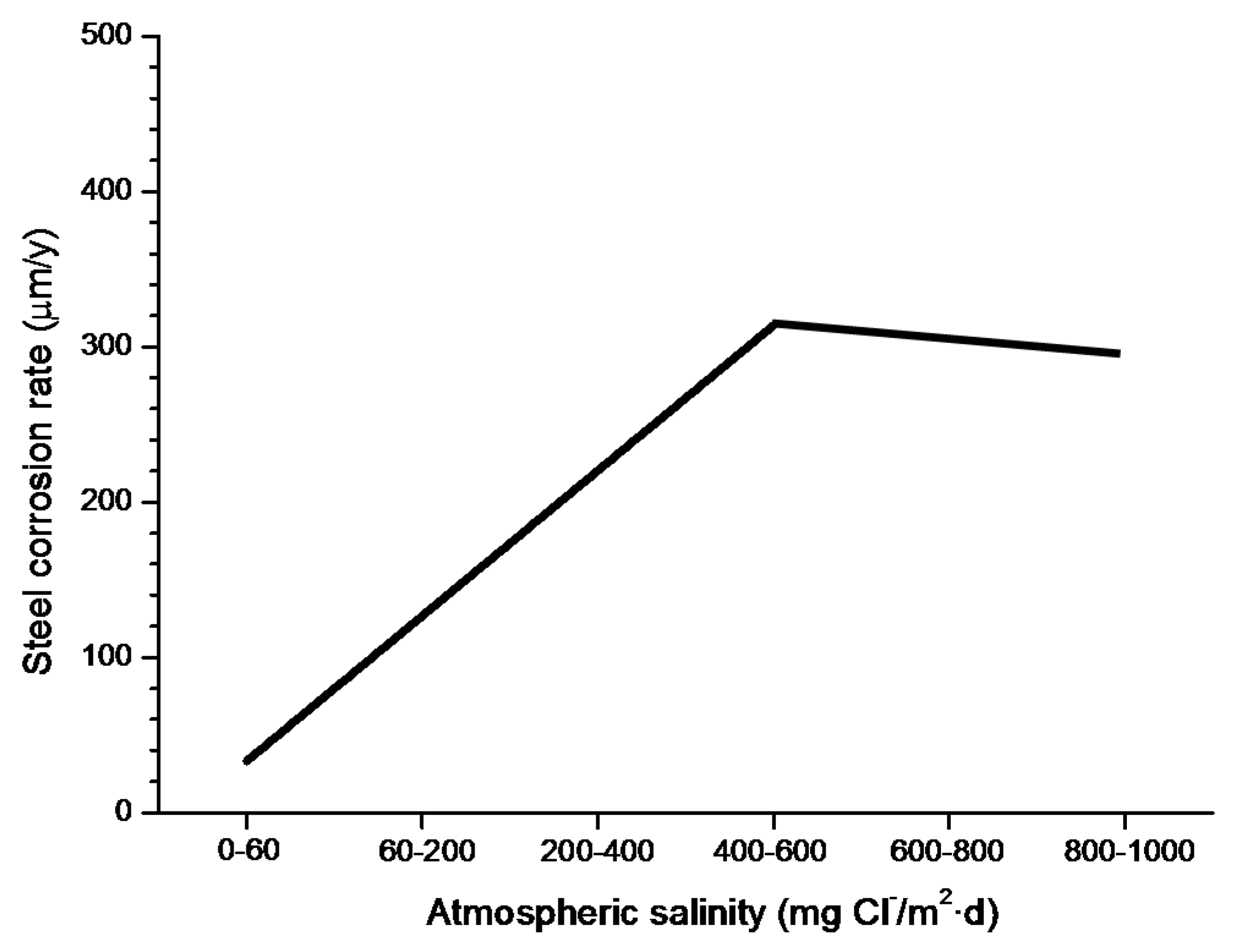
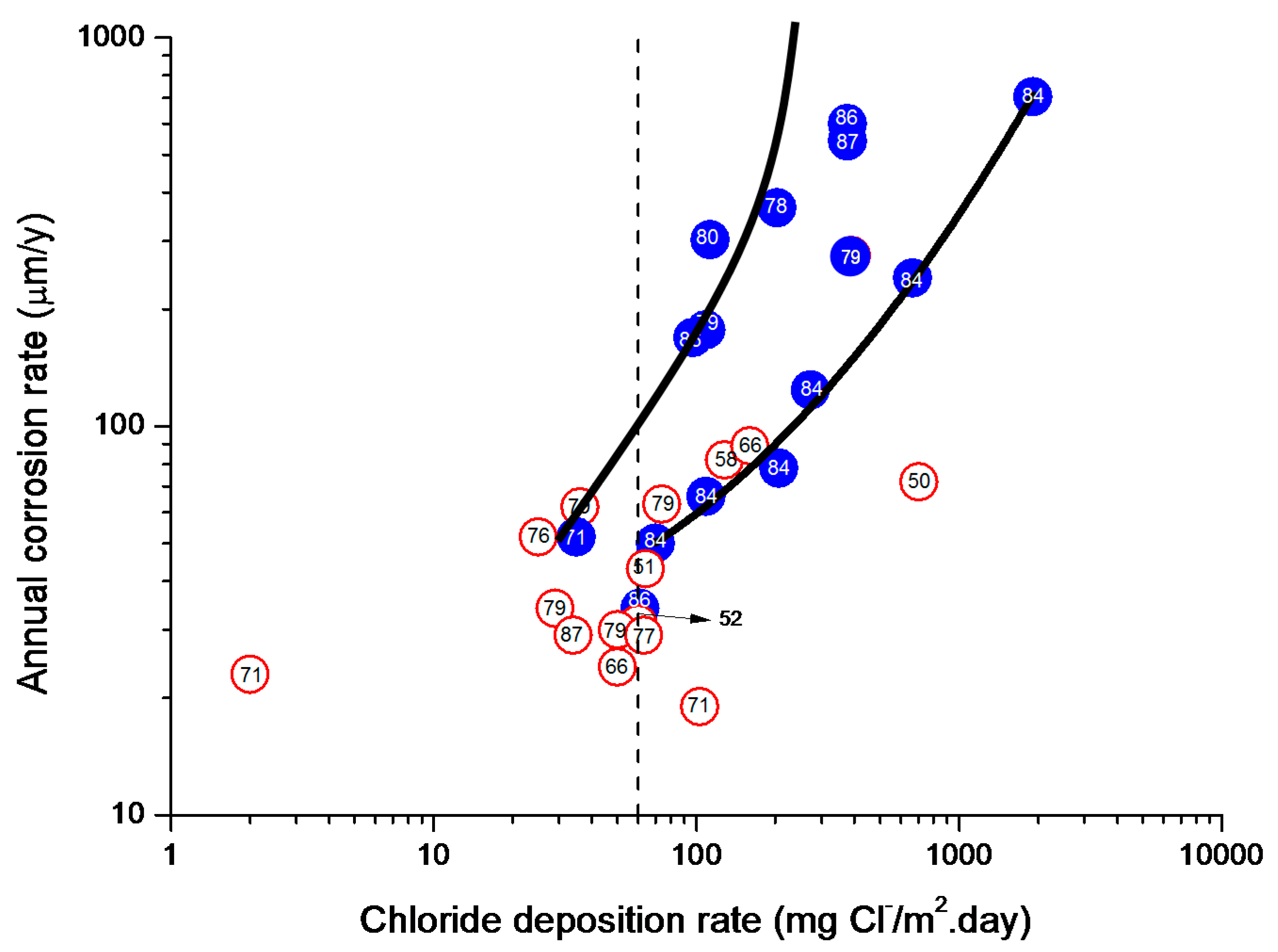
4.4.1. Steel Corrosion versus Salinity
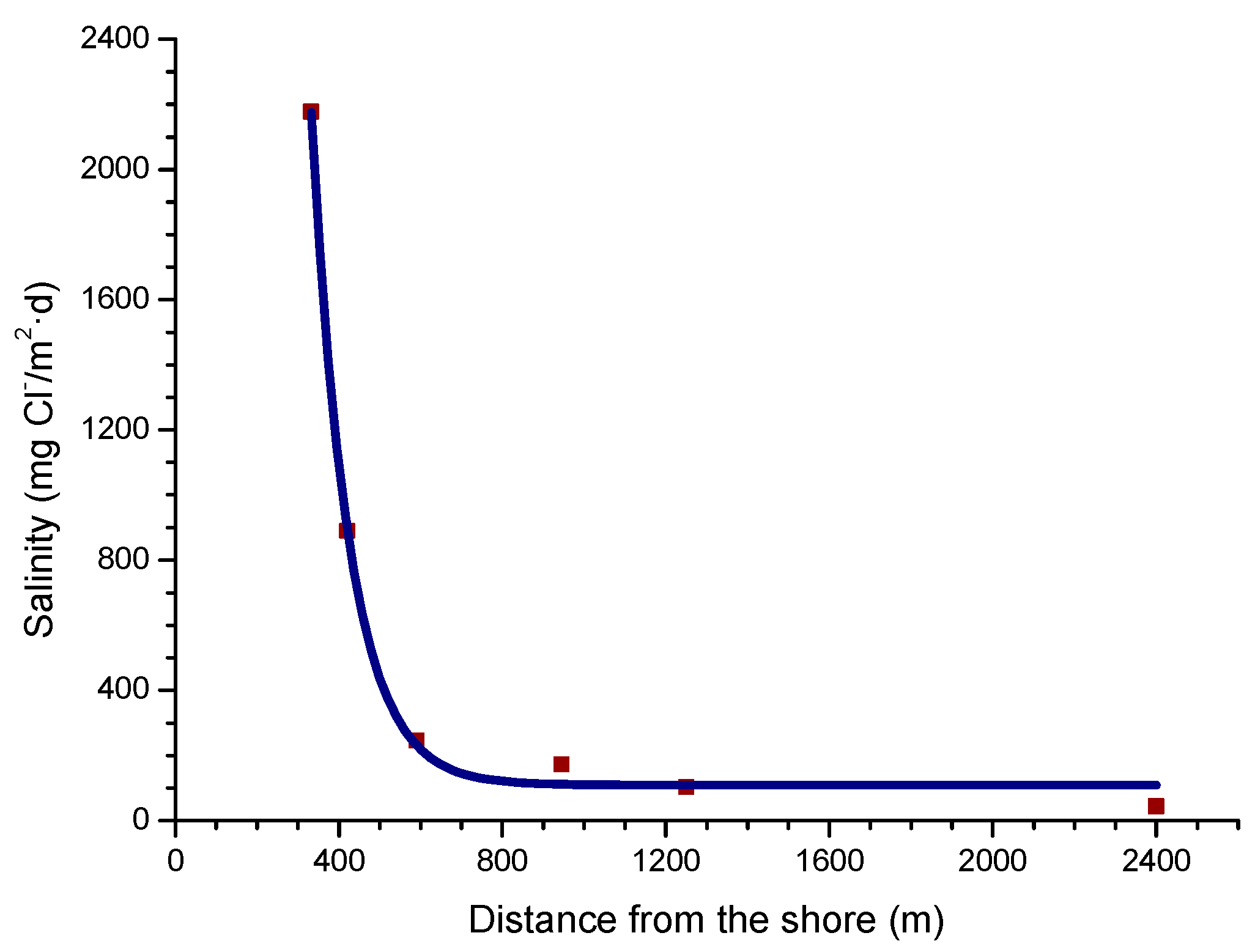
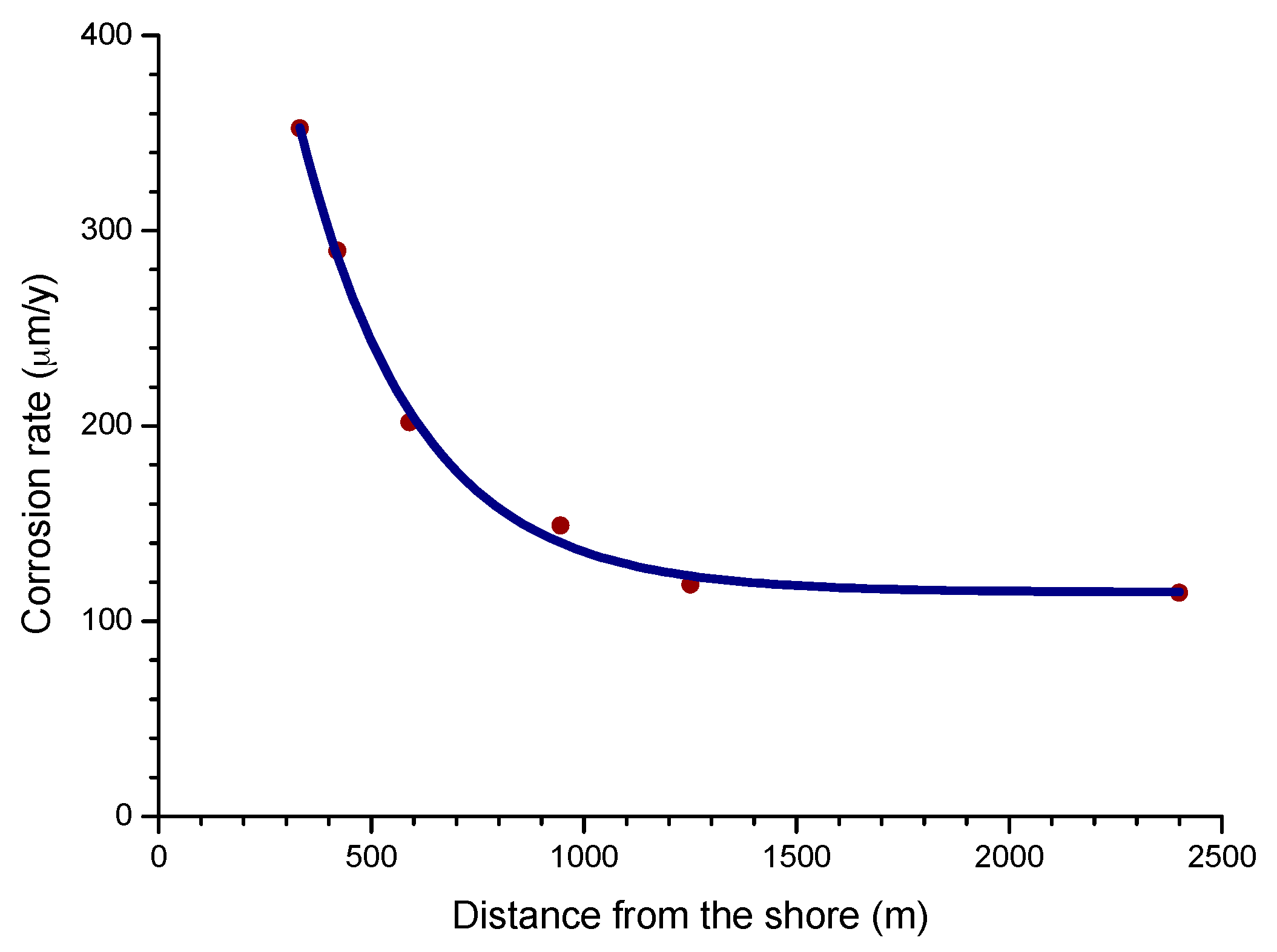
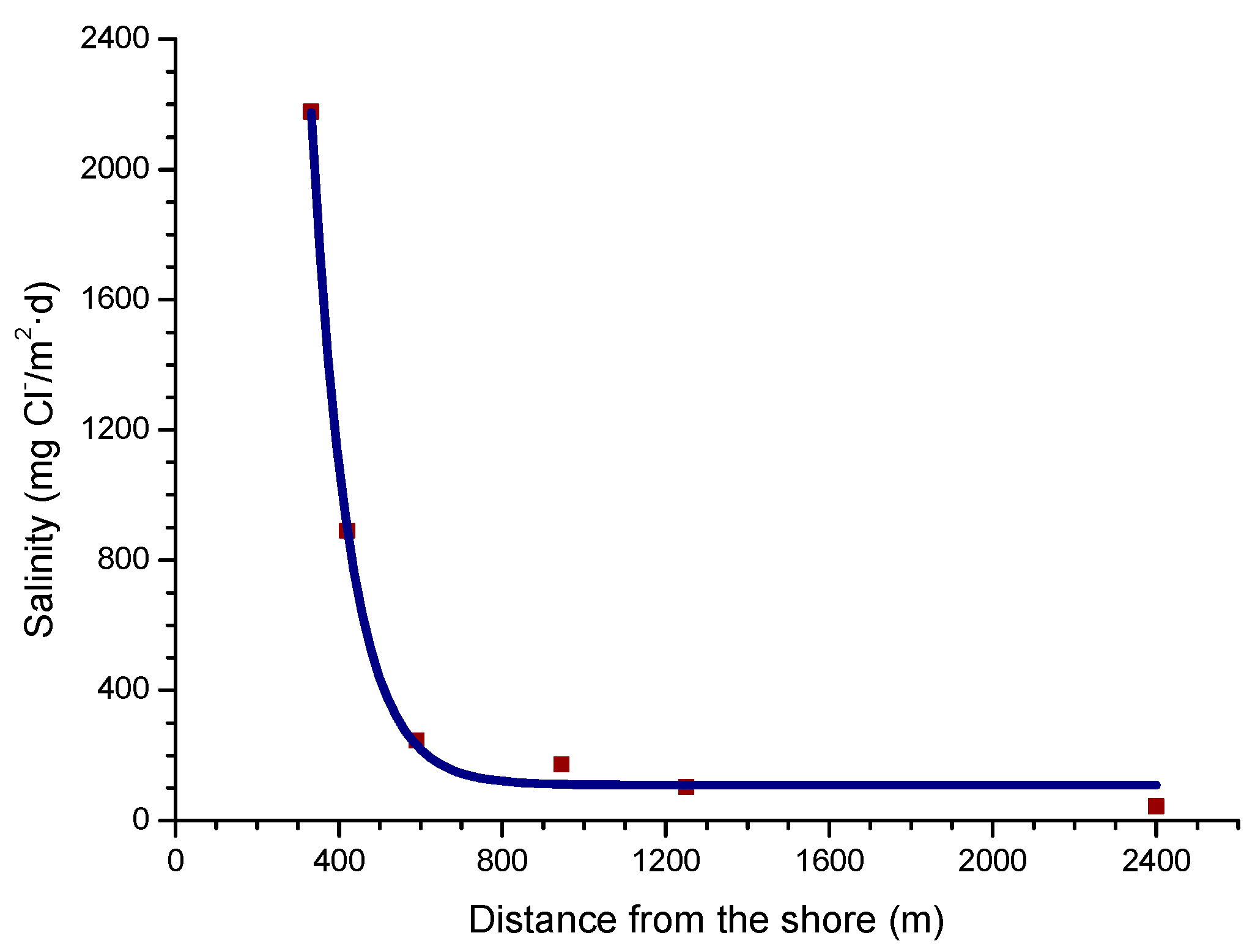
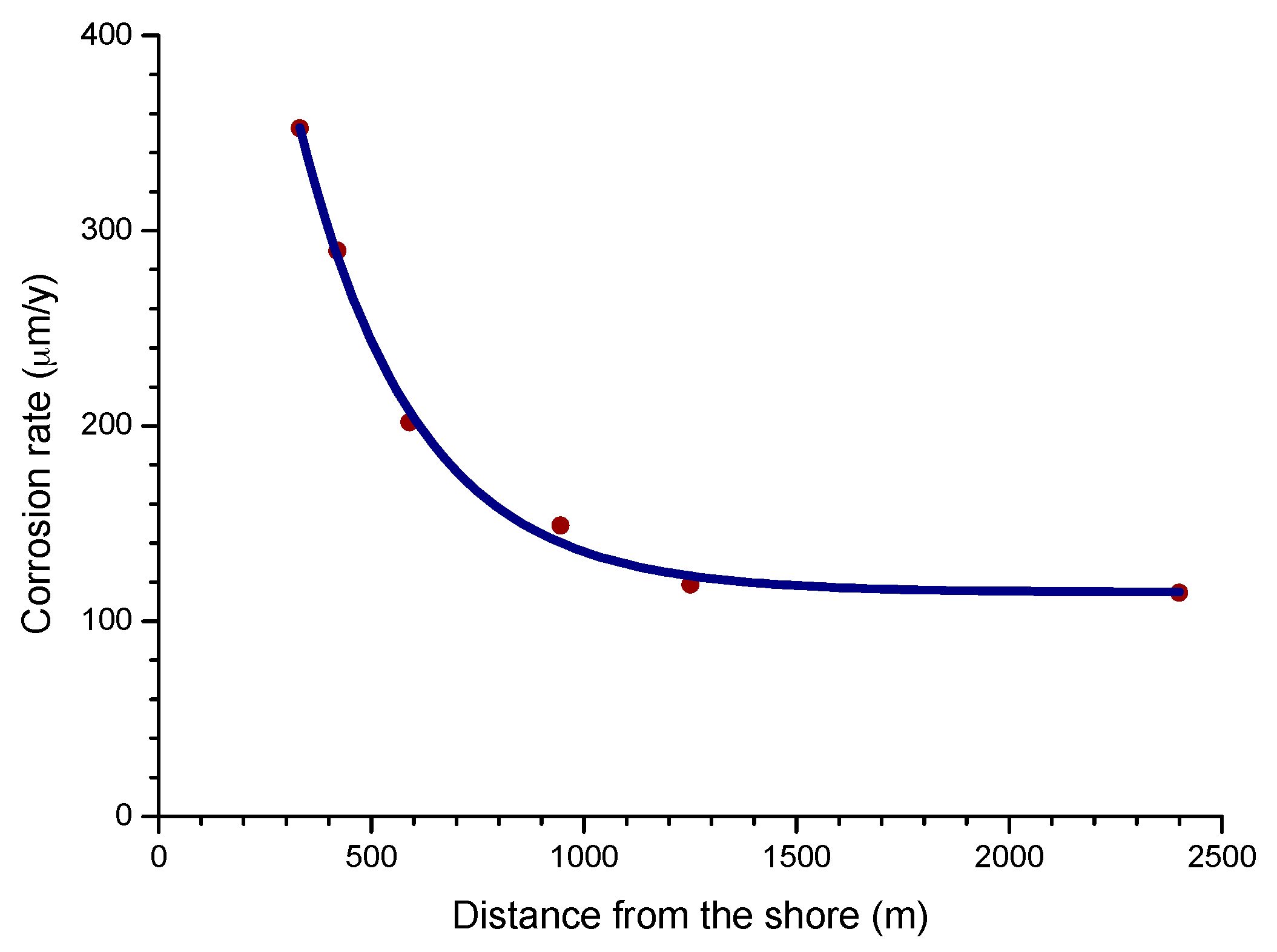
4.4.2. Steel Corrosion versus Distance from the Shore
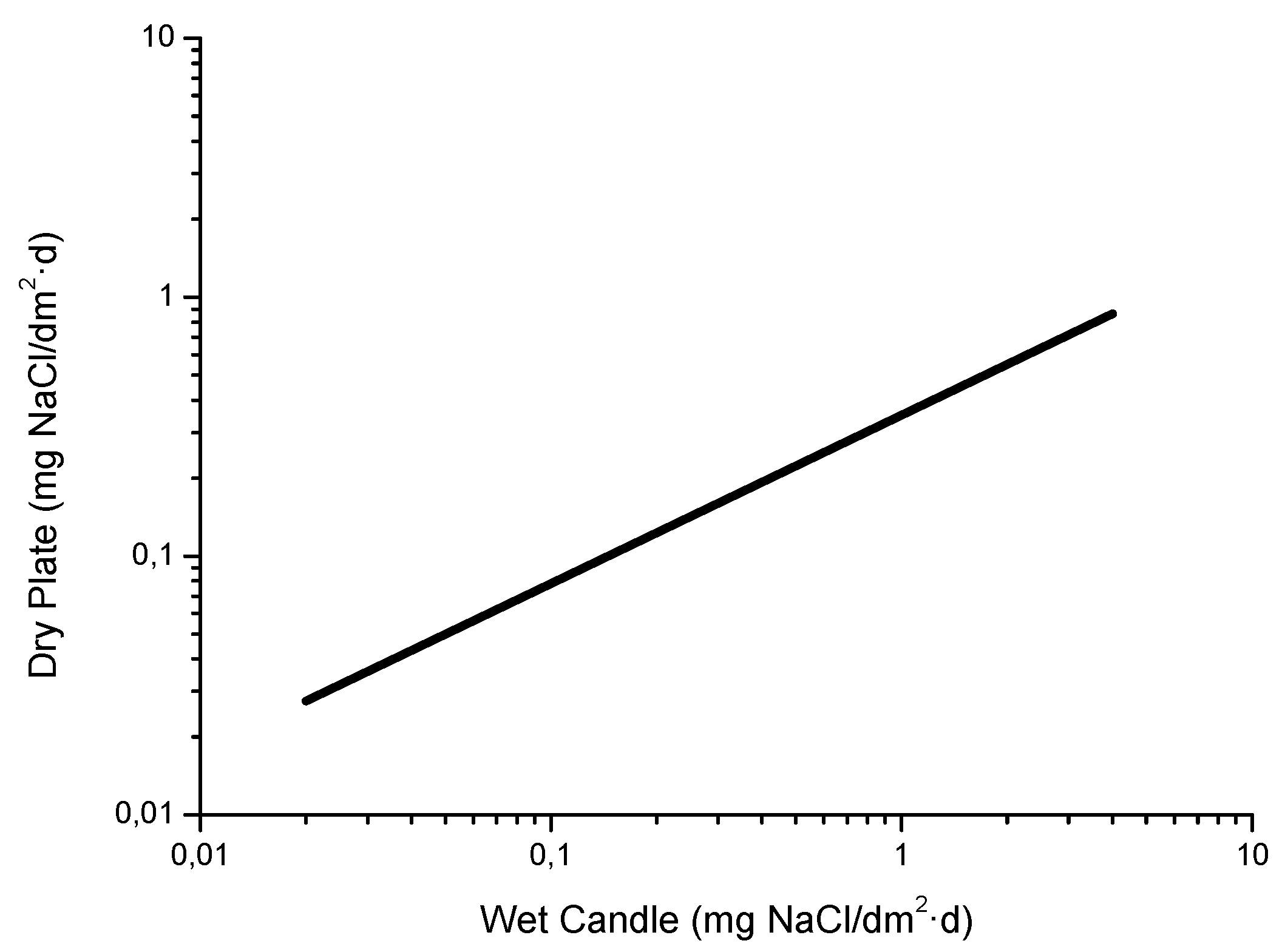
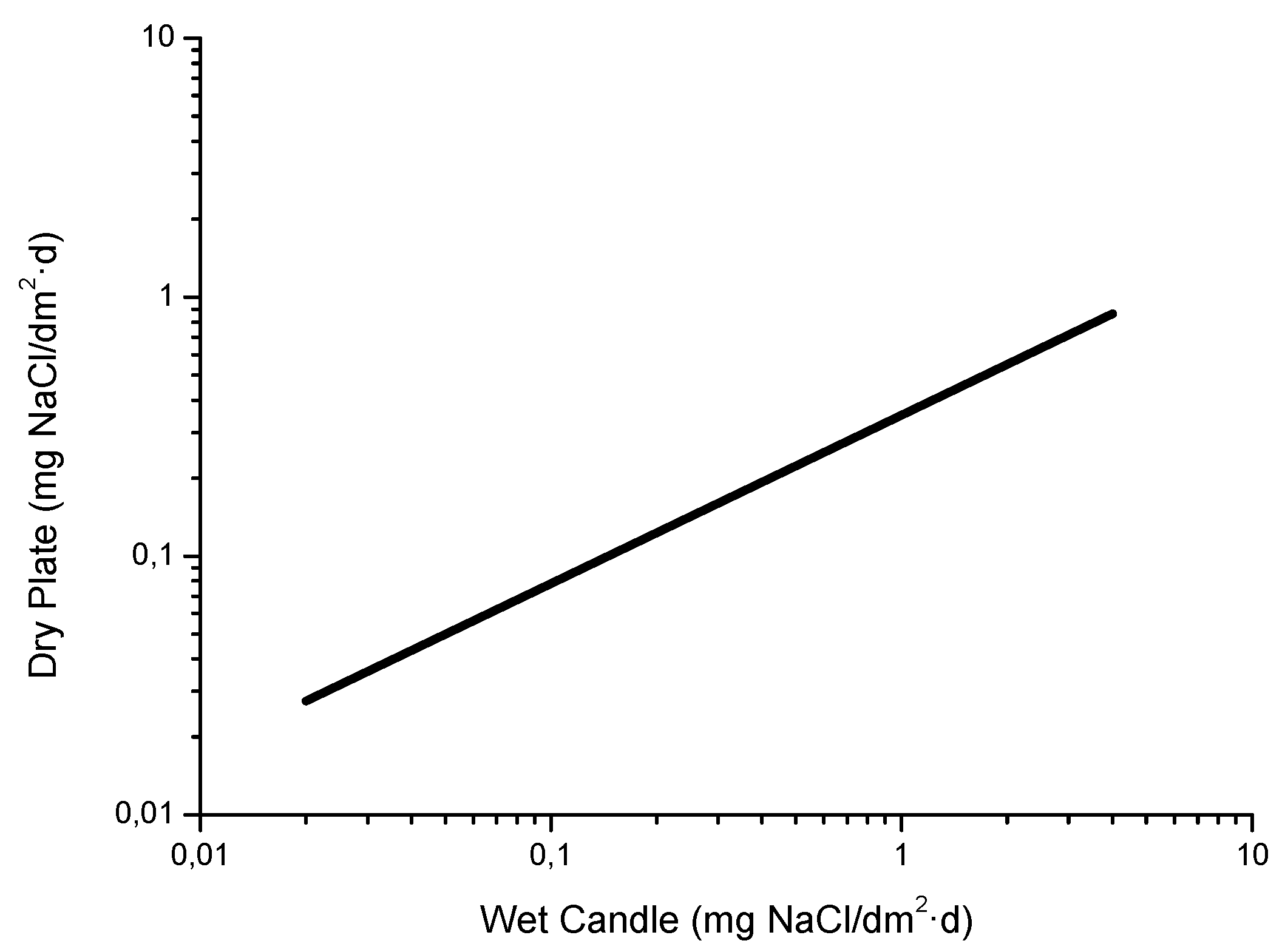
4.5. Measurement of Atmospheric Salinity
- [Cl−]wc = salinity determined by the wet candle method
- [Cl−]dc = salinity determined by the dry cloth method
- Is only valid for salinity values of a considerable magnitude.
4.6. Salt Lake Atmospheres
4.7. Deicing Salts
5. Atmospheric Corrosion Products
5.1. Most Significant Corrosion Products in Steel Corrosion in Marine Atmospheres
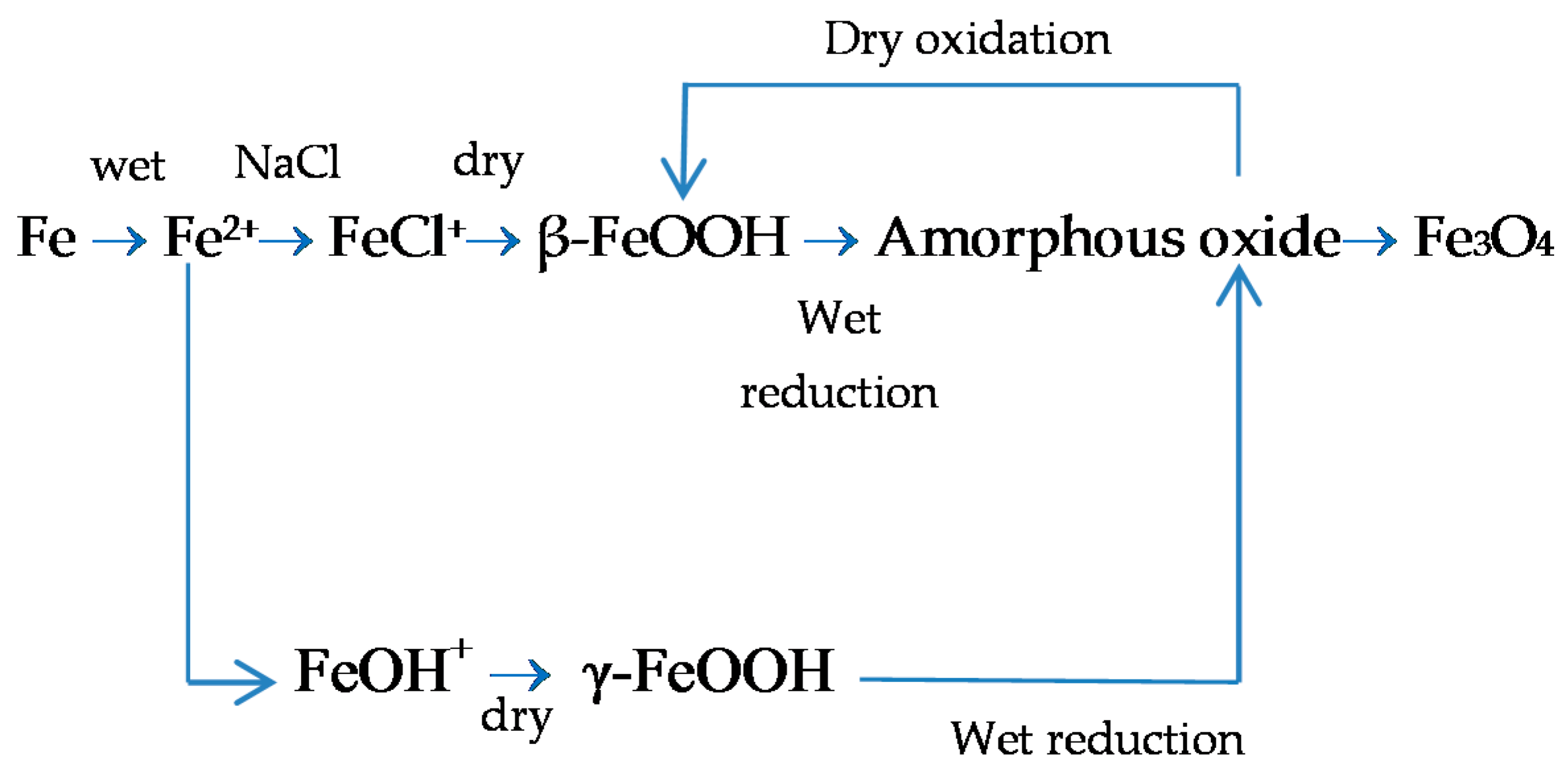
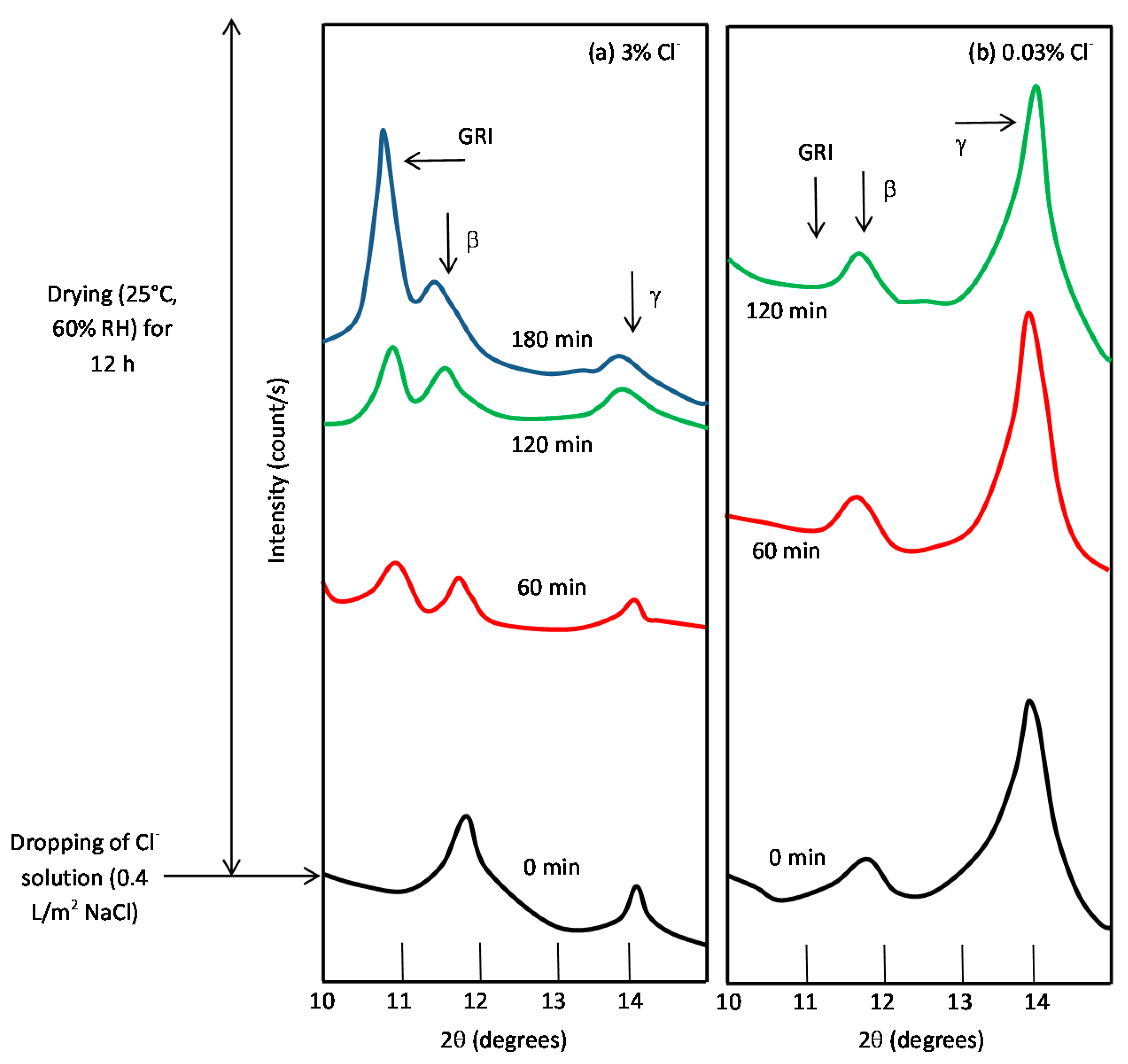
5.1.1. Green Rust 1 (GR1 or GR(Cl−))
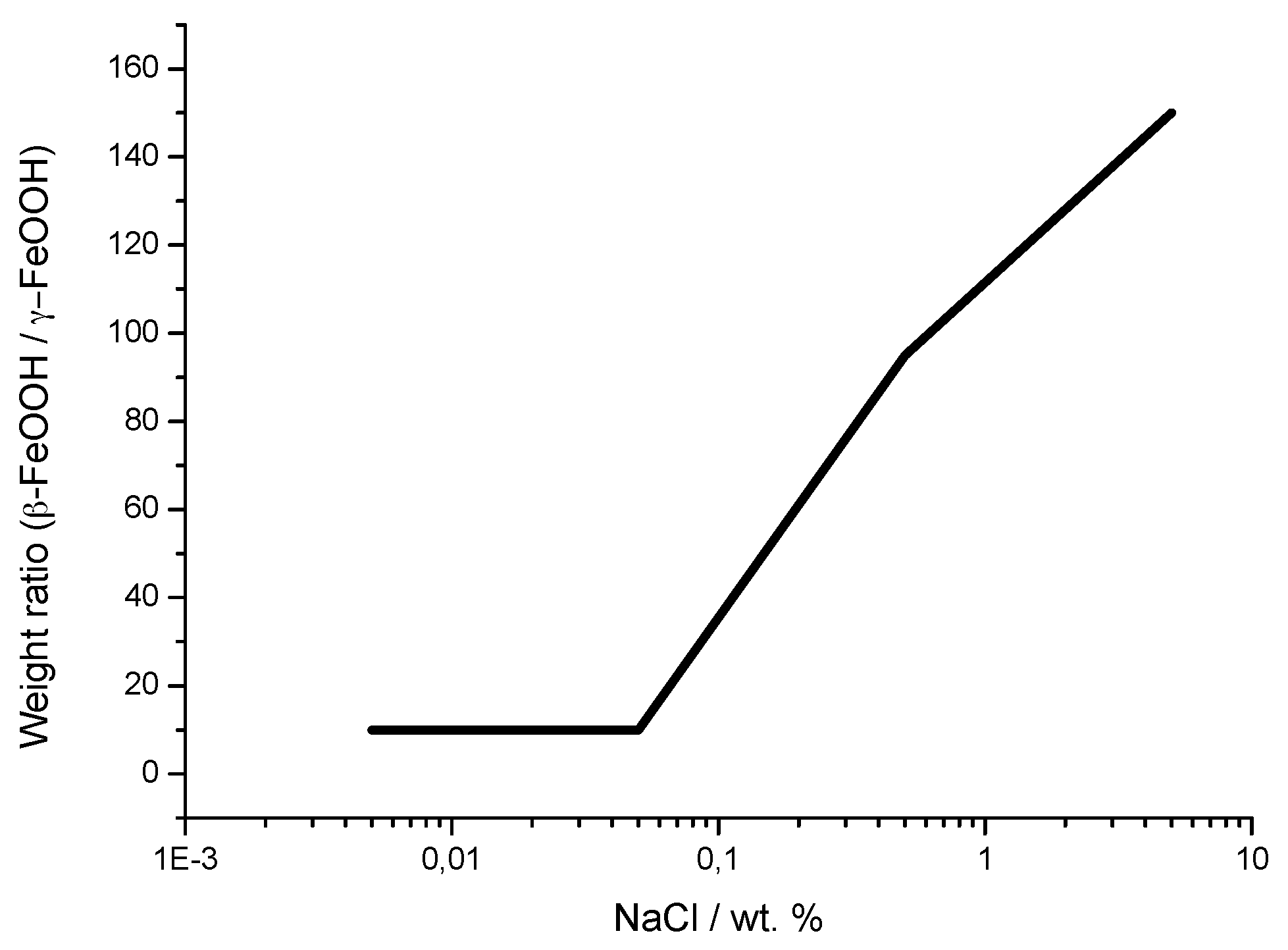
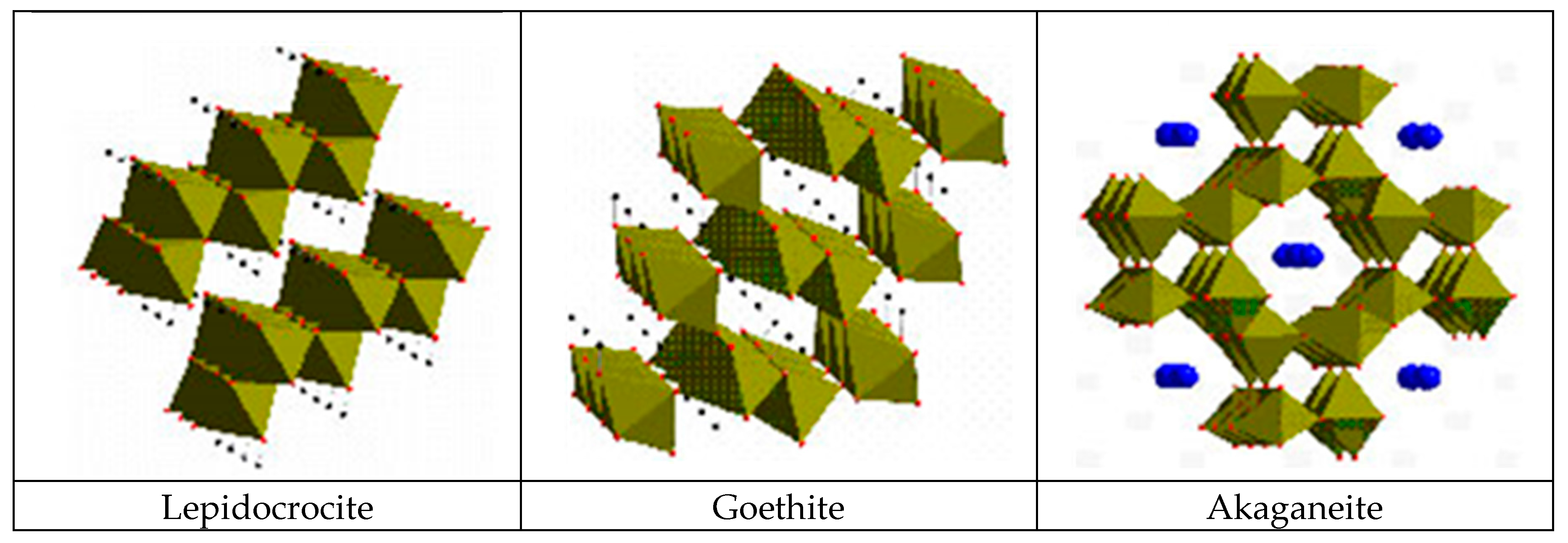
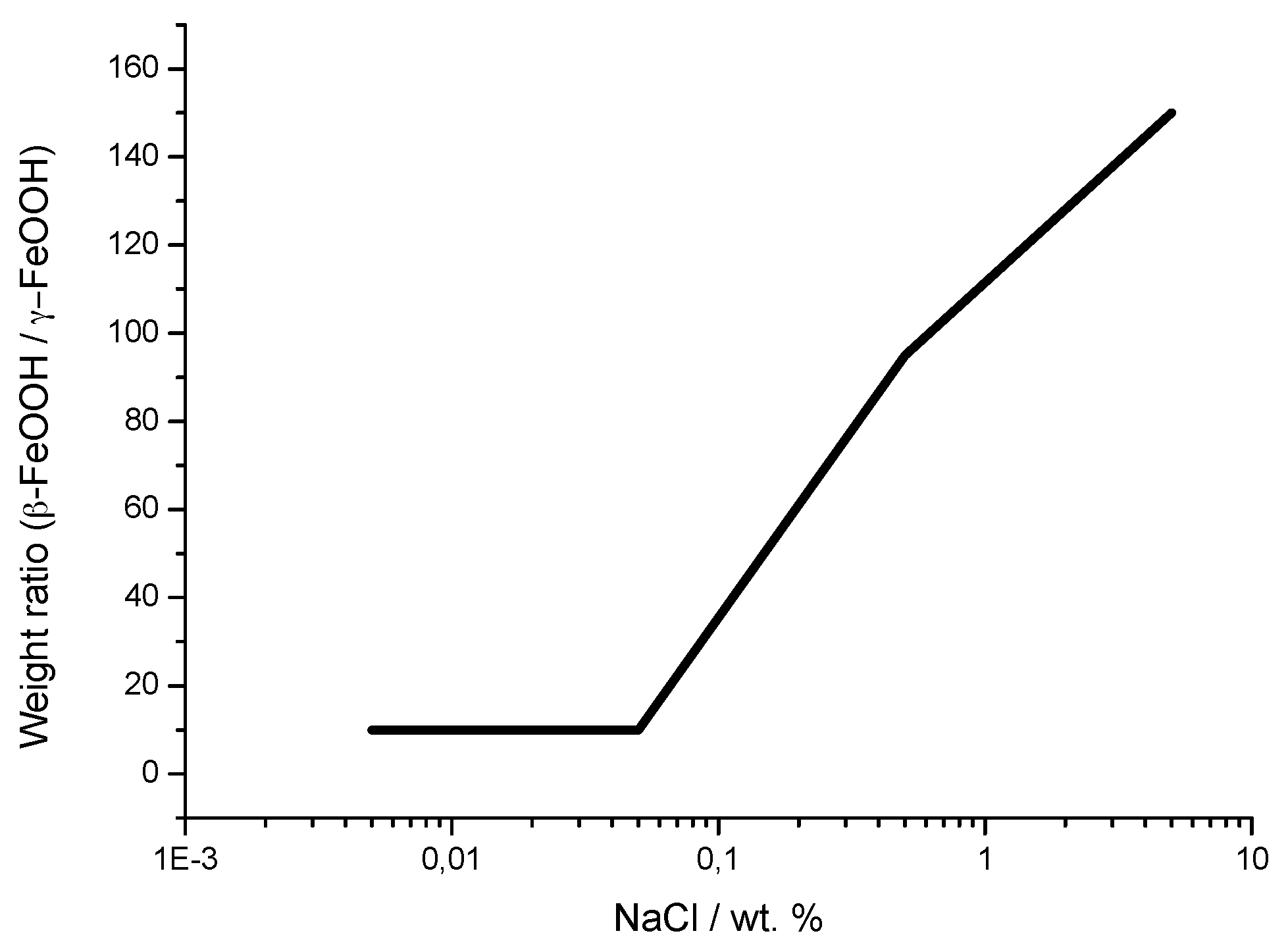
5.1.2. Akaganeite ((β-FeOOH) or β-FeO(OH,Cl−))
5.1.3. Magnetite (Fe3O4)/Maghemite (γ-Fe2O3)
5.2. Other Characteristics of the Steel Atmospheric Corrosion Products
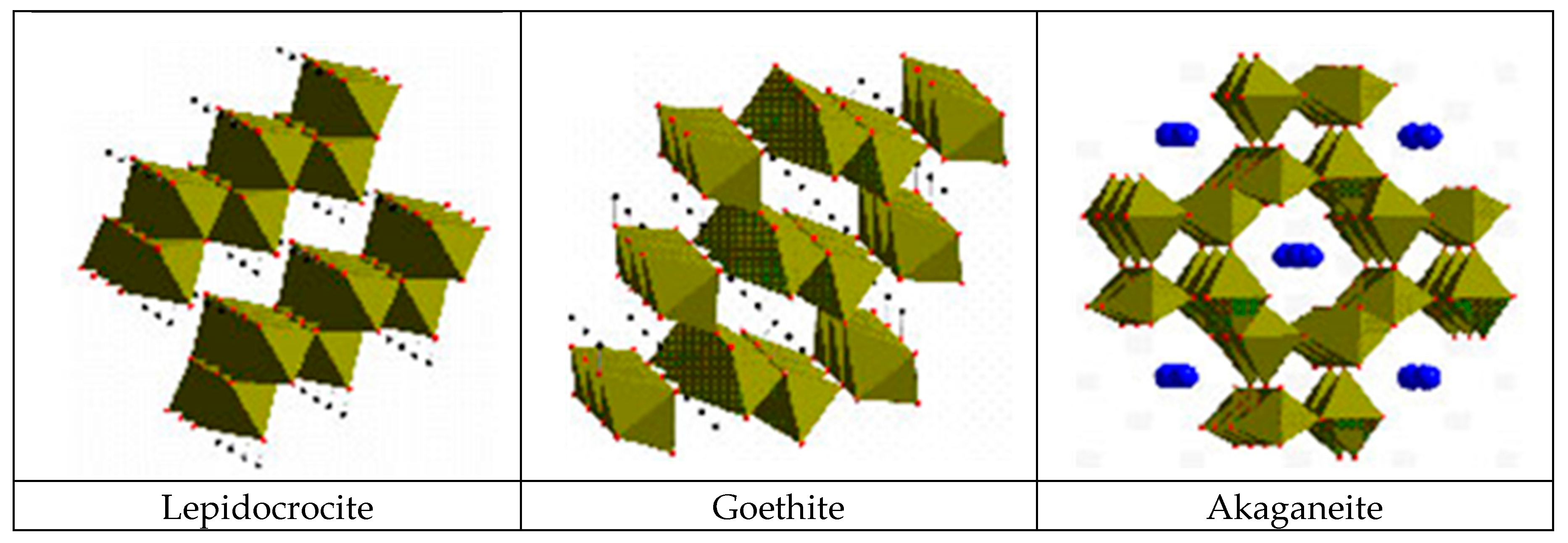
5.2.1. Towards a Greater Knowledge of the Structure of Iron Oxides
5.2.2. Morphology
- (a)
- Globular: hemispheric-shaped aggregated formations like small mounds.
- (b)
- Acicular: aggregates with a similar appearance to needles, hairs, or threads.
- (c)
- Laminar: this can appear in a wide range of different formations in which laminas grow perpendicularly to the surface: bar shape, worm nest shape, bird’s nest shape, flower petal shape, feather shape, etc.
- (d)
- Tubular: formations in which the crystalline aggregates are constituted by prisms, tubes, or rods, etc.
- (e)
- Toroidal
- (f)
- Geode-type: unusual or singular oolitic or globular morphology constituted by fish-egg-like spherical formations.
5.2.3. Grain Size (Granulometry)
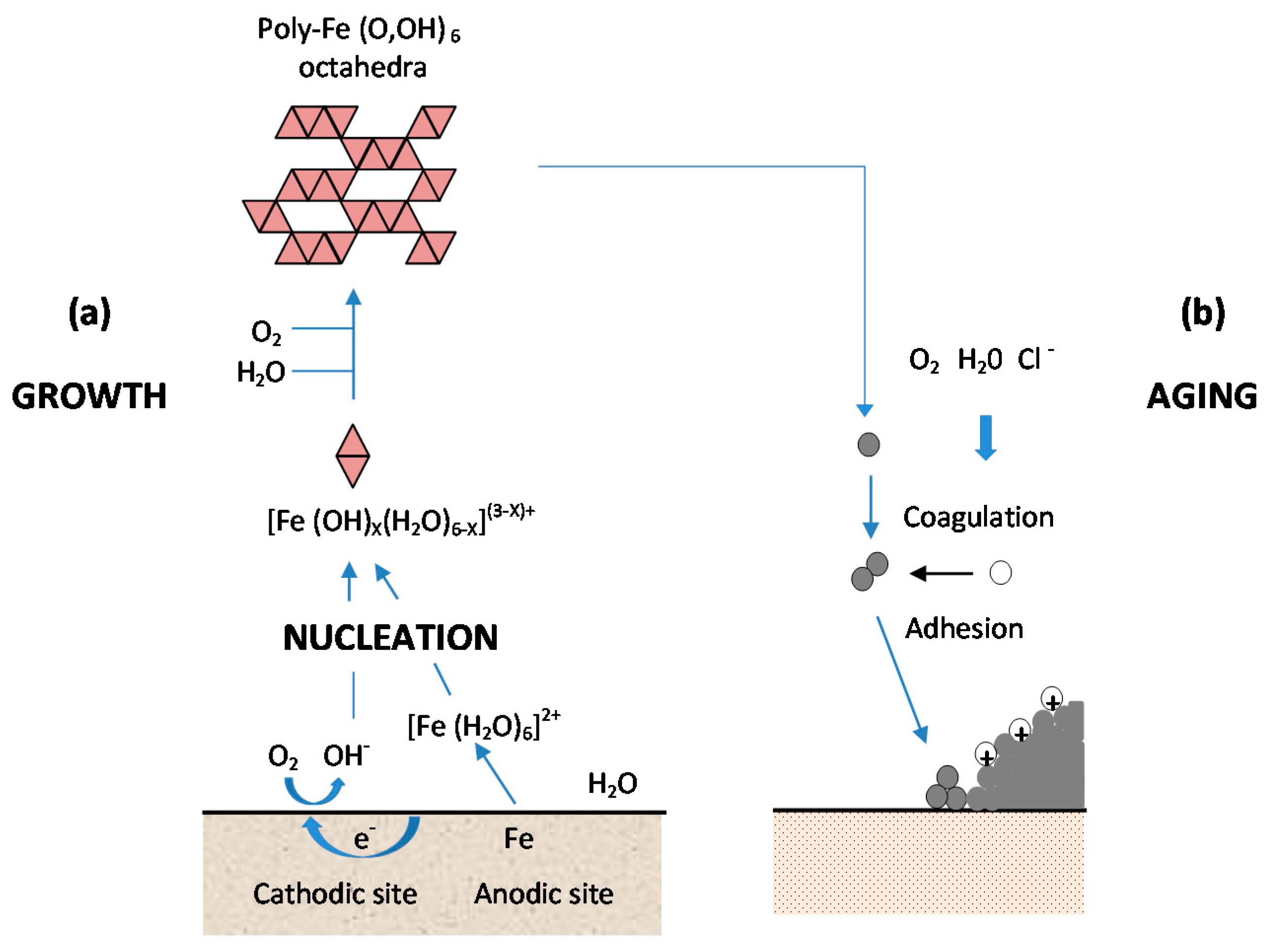
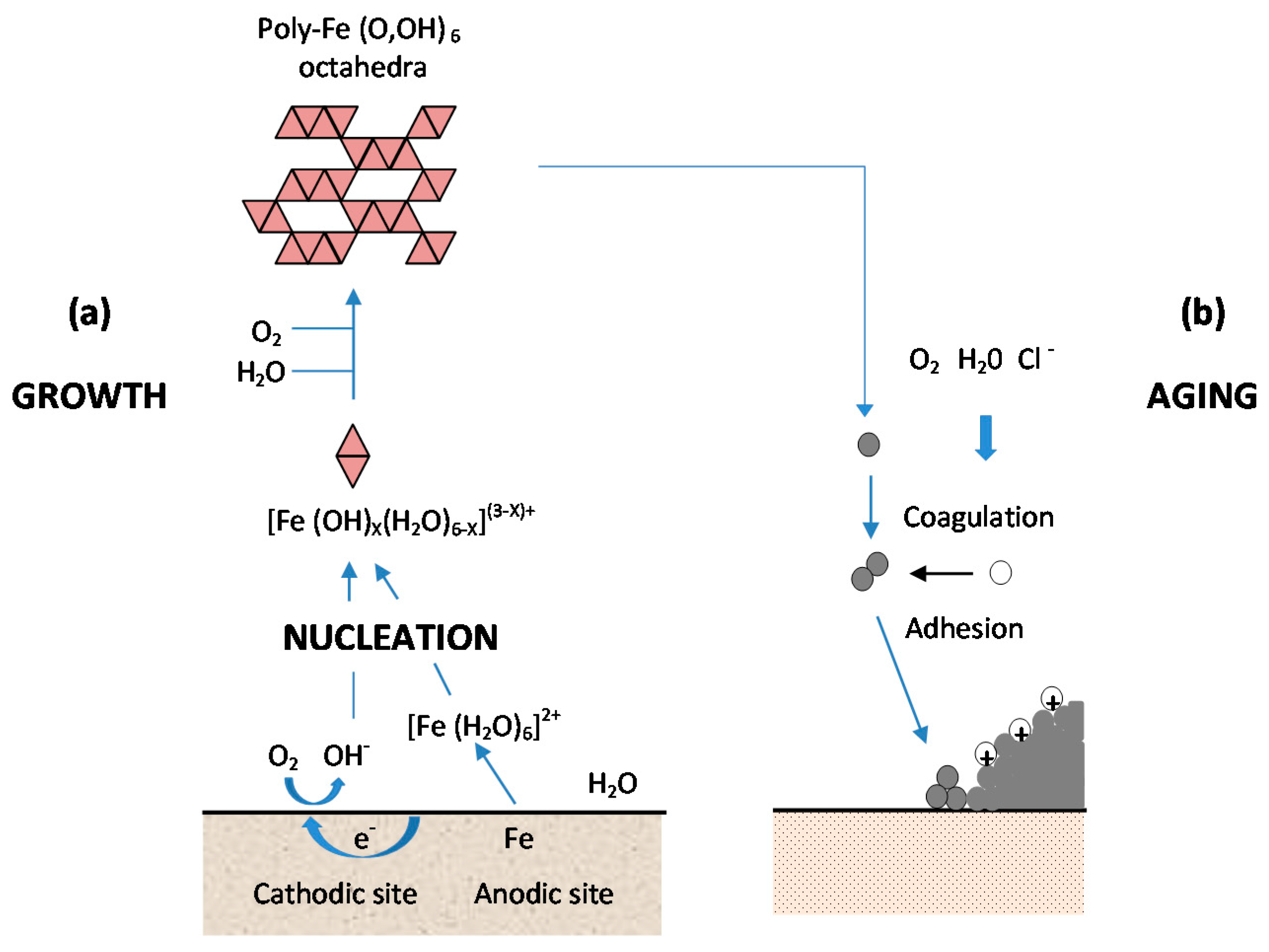
- (a)
- Nucleation corresponds to the first step of precursor condensation and solid formation. On an atomic scale, iron forms cations that are coordinated by six water molecules [Fe(H2O)6]2+ [139]. In a neutral solution, metal cations react with OH−, O2, and H2O resulting in the formation of hydroxo cations [Fe(OH)X(H2O)6−X](3−X)+.
- (b)
- Then the growth process follows, where Fe(O,OH)6 octahedra units as cations or smaller sized growing nuclei accumulate to form larger particles. On a colloidal scale, polymerisation of these Fe(O,OH)6 octahedra leads to the formation of fine particles of hydroxides, oxyhydroxides or oxides.
- (c)
- These particles grow into grains or layers through a long period of ageing processes affected by repeated wet and dry cycles. Reaction conditions (concentration, acidity, temperature, nature of anions, etc.) have a strong influence on the structural or morphological changes of poly-octahedra during corrosion. Coagulation and adhesion processes ensue to generate corrosion products, which undergo ageing processes leading the system to stability. During ageing the particles may undergo modifications such as increases in size, changes in crystal type, changes in morphology, etc. [140]. Thus, according to Ishikawa et al. [141], steel rusts can be regarded as agglomerates of colloidal nanoparticles of ferric oxyhydroxides (goethite, akaganeite and lepidocrocite), spinels (magnetite/maghemite), and poorly recrystallised iron oxides (amorphous substances). Voids of different sizes form between the fine particles in the rust layer.
6. The Rust Layer
6.1. Organoleptical Properties
6.1.1. Colour
6.1.2. Texture
6.2. Properties More Related with the Protective Capacity of Rust Layers
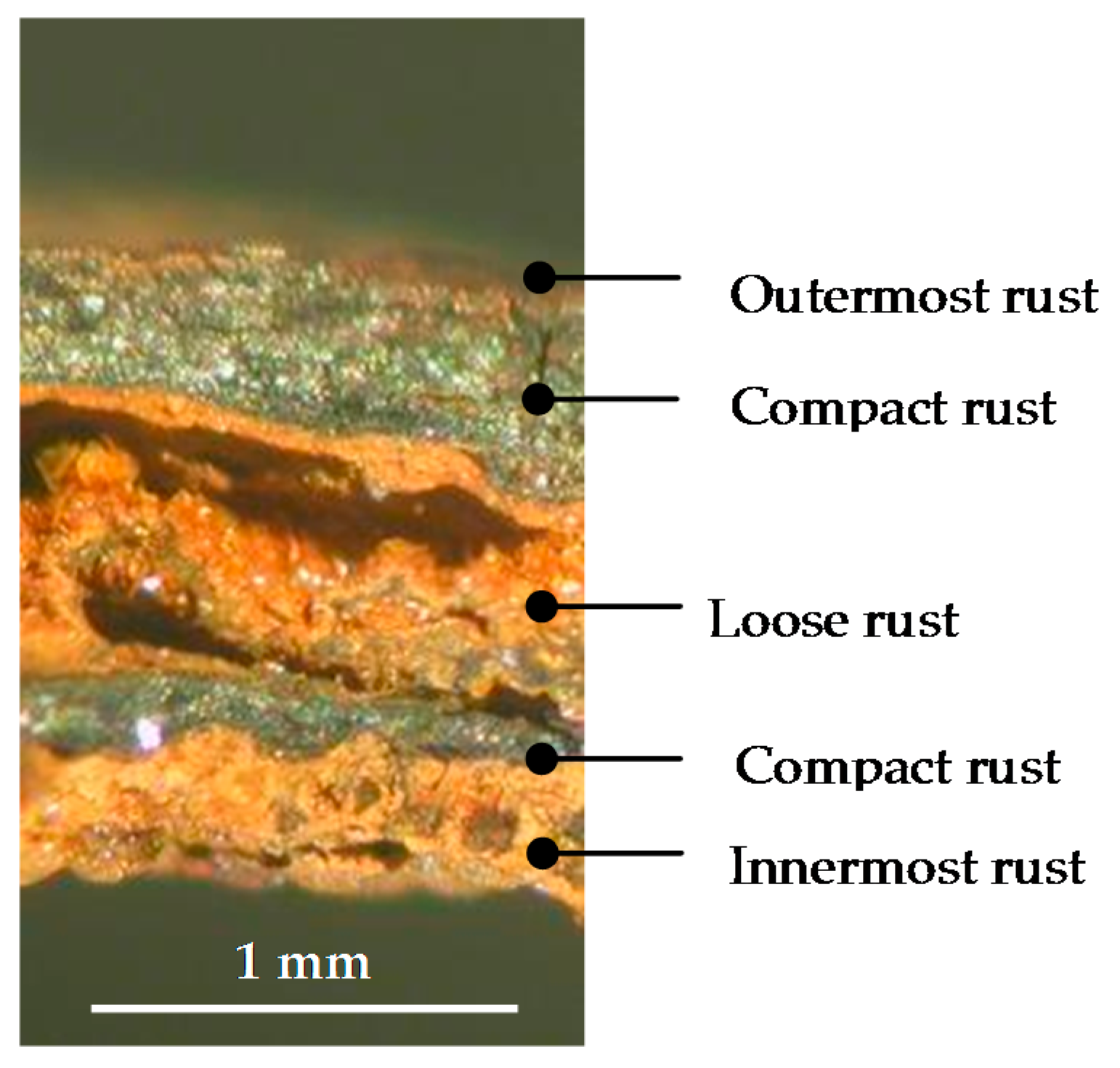
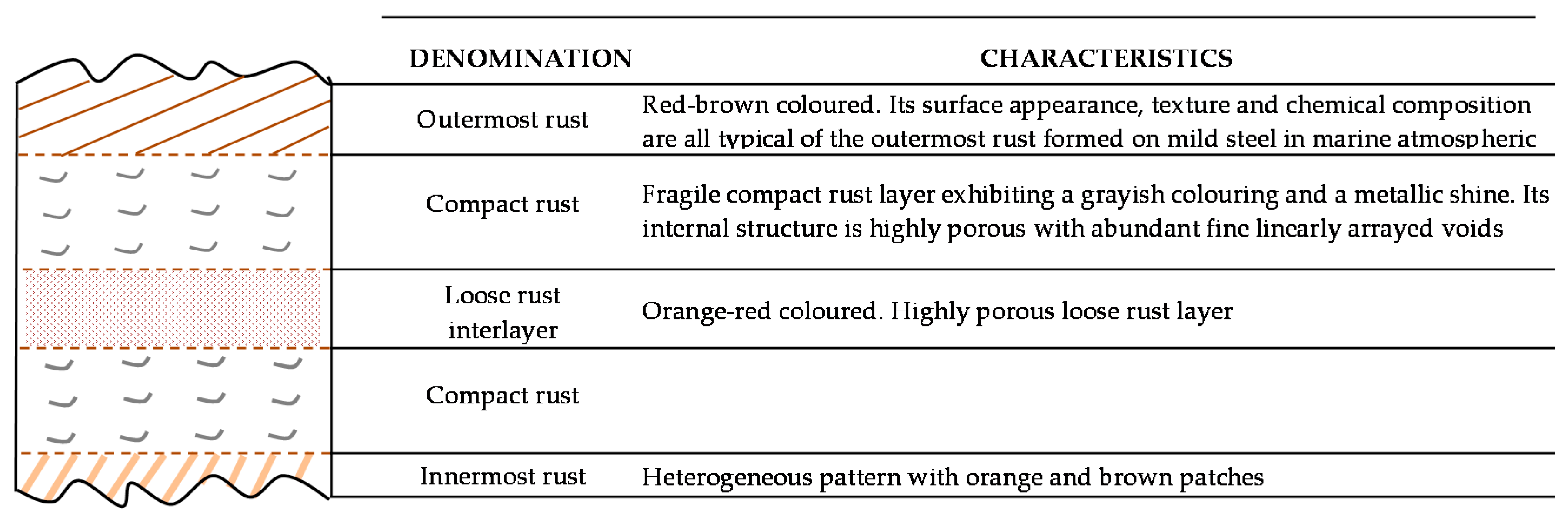
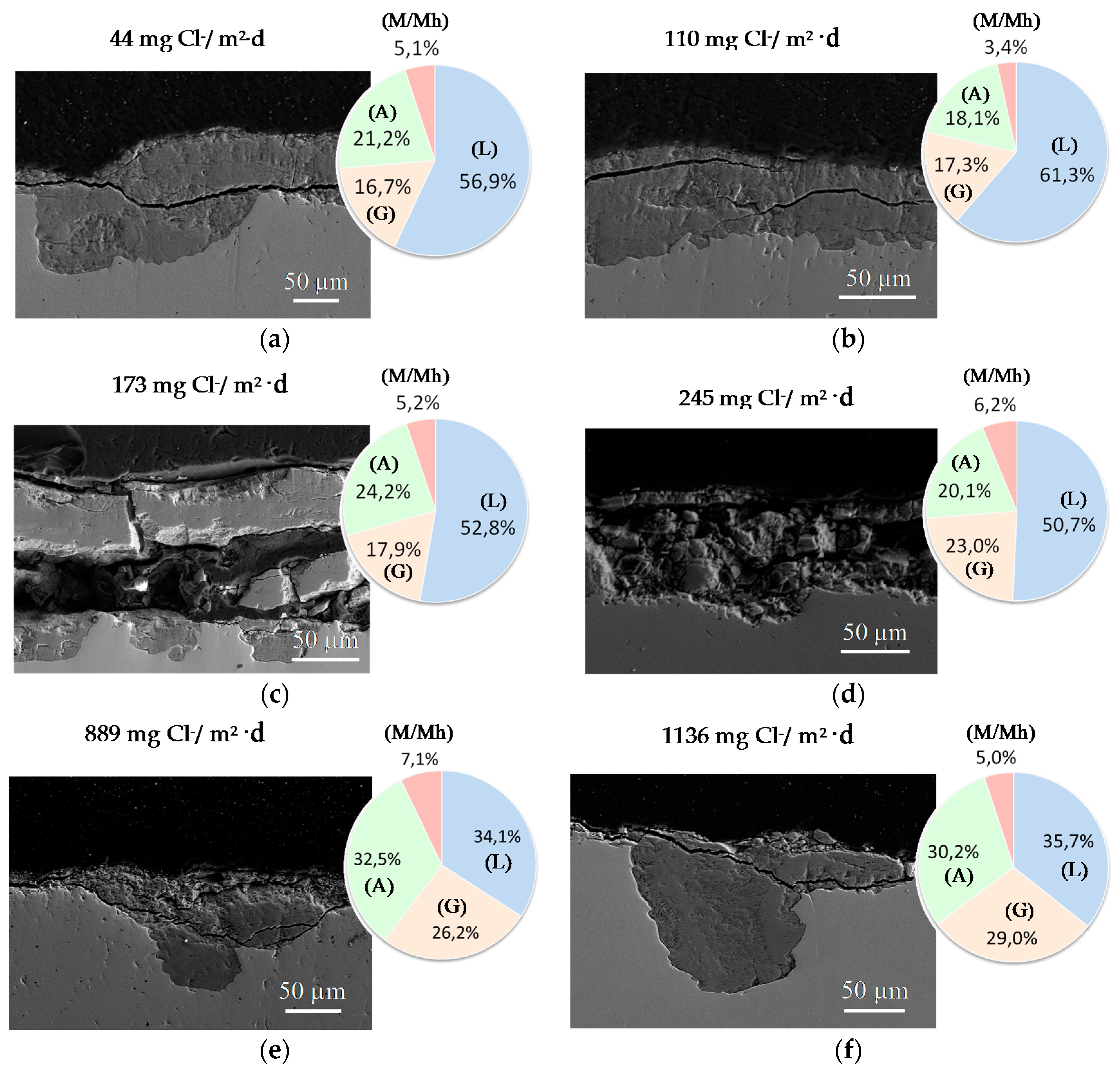
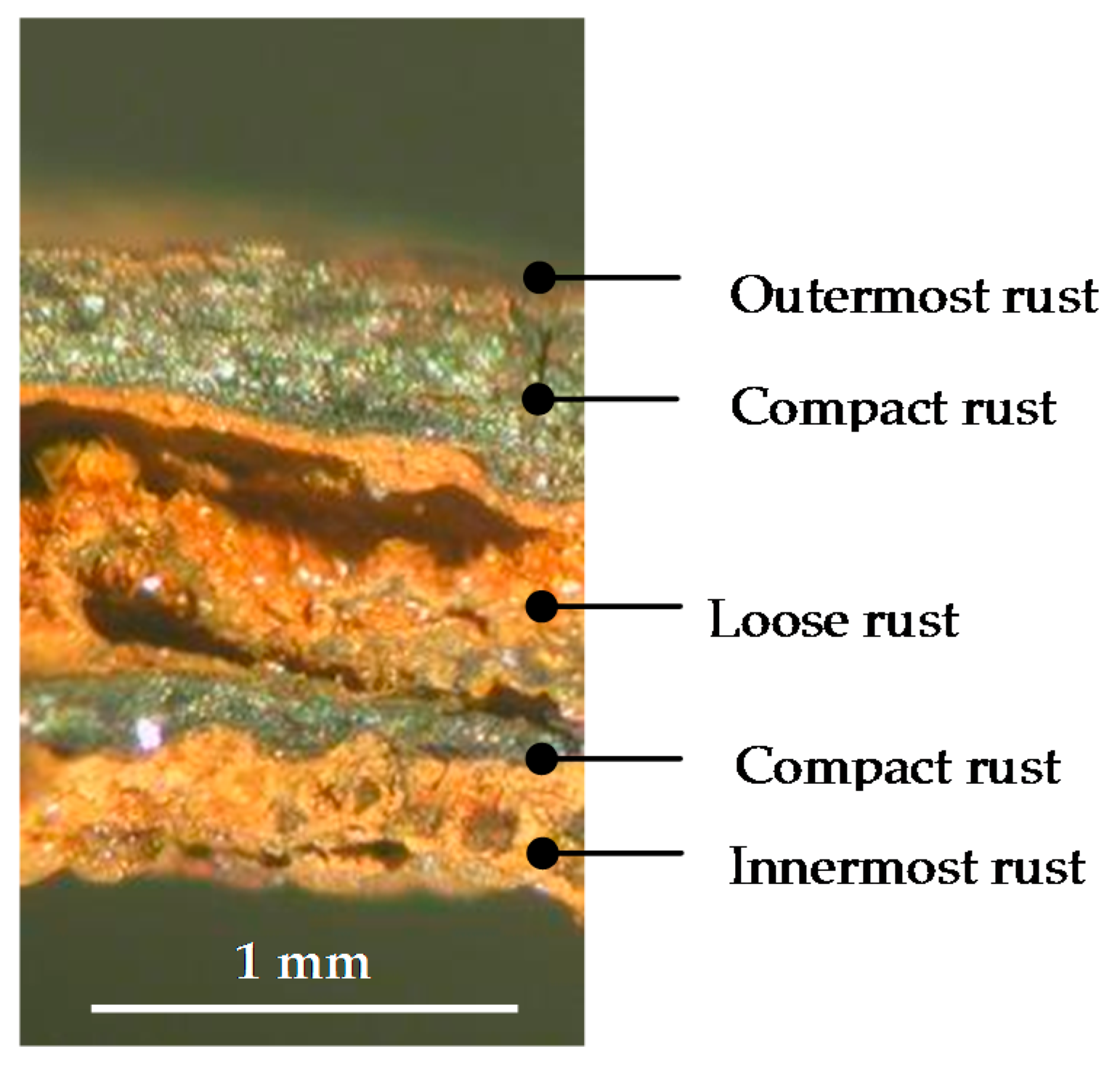
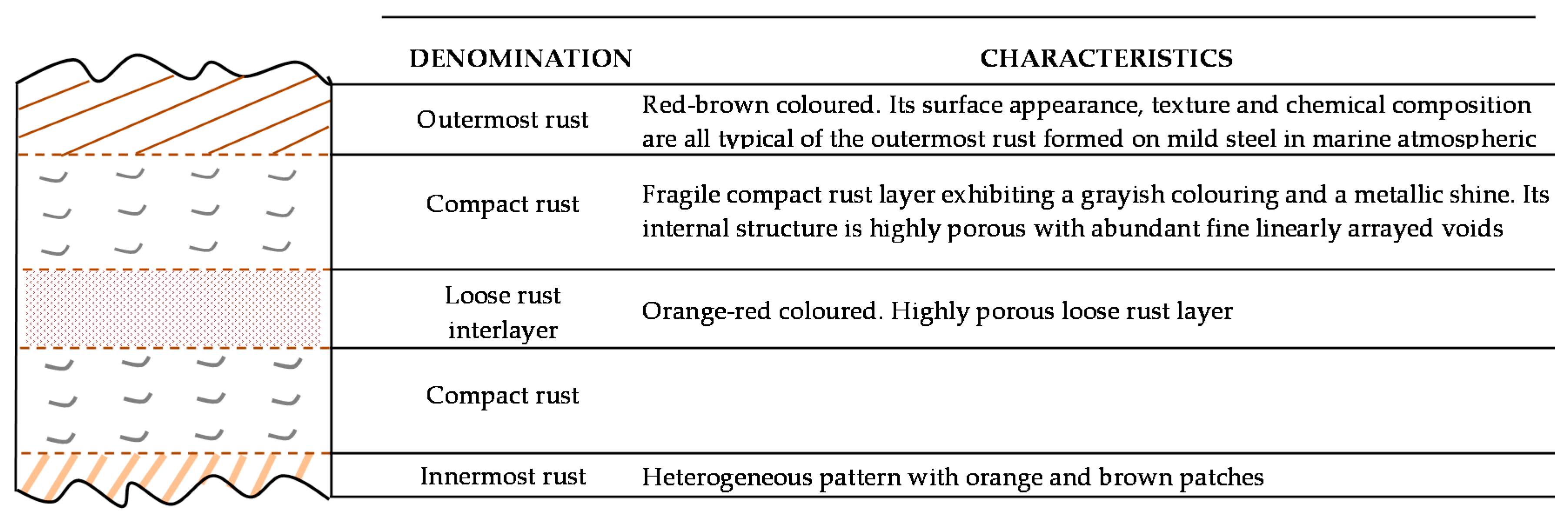
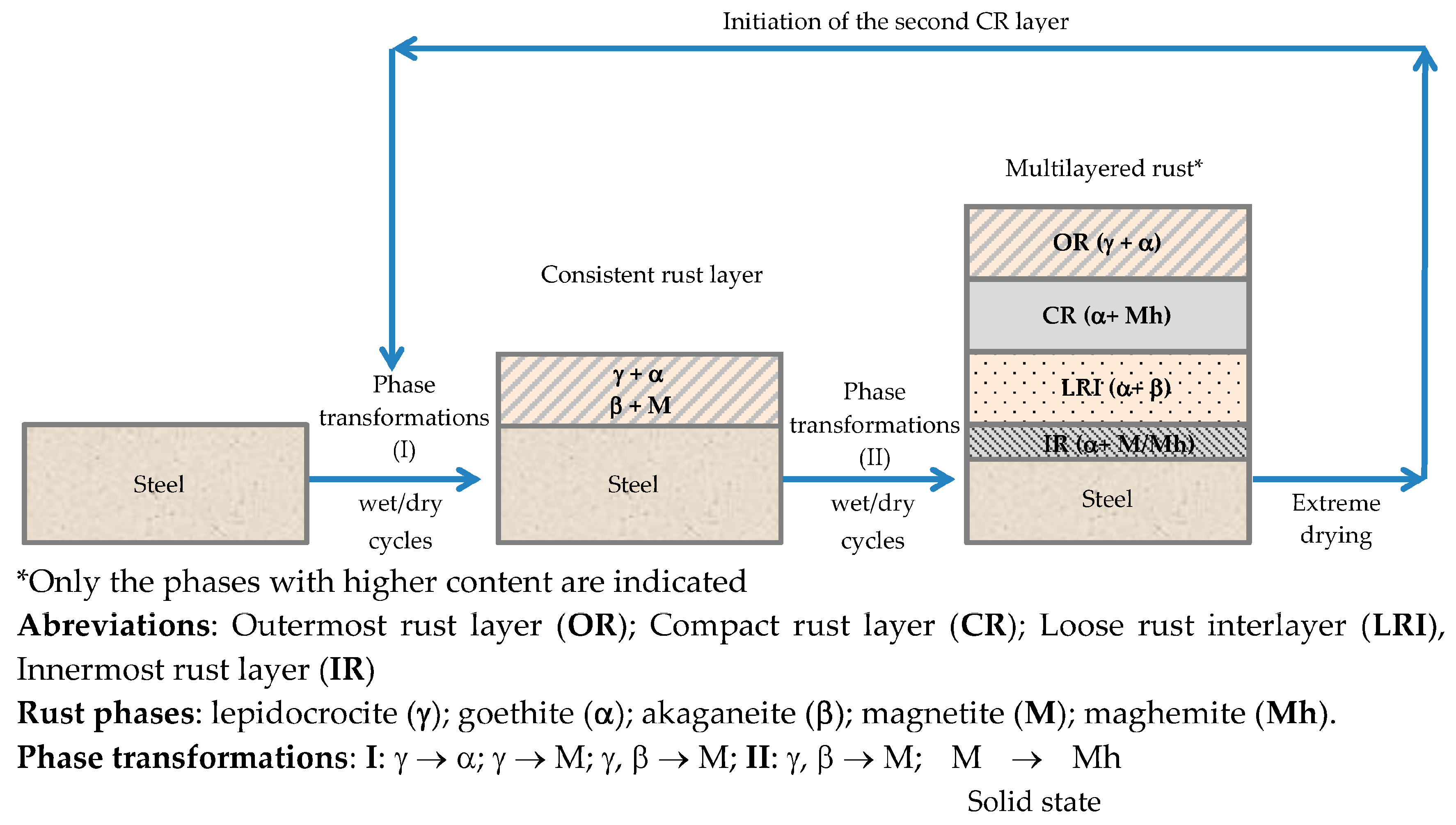
6.2.1. Stratification of Rust Layers
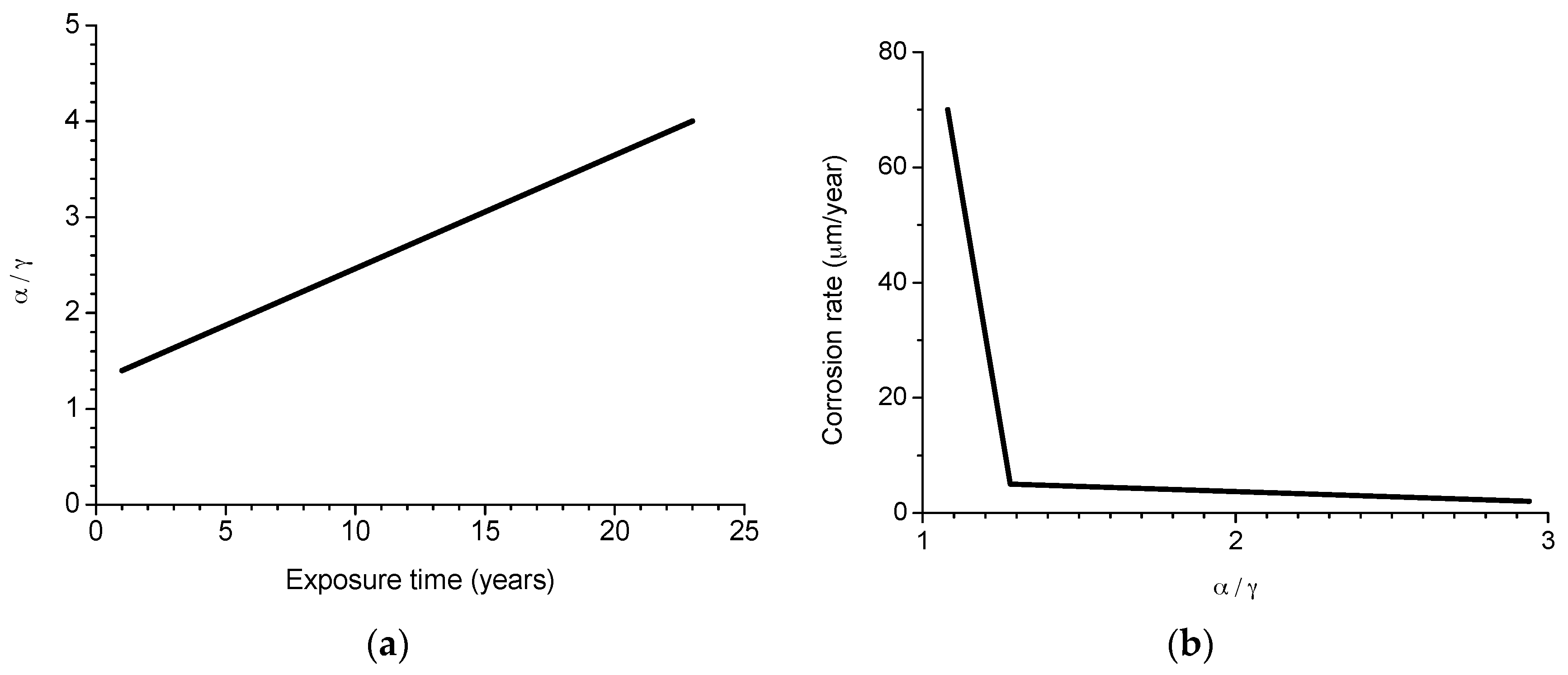
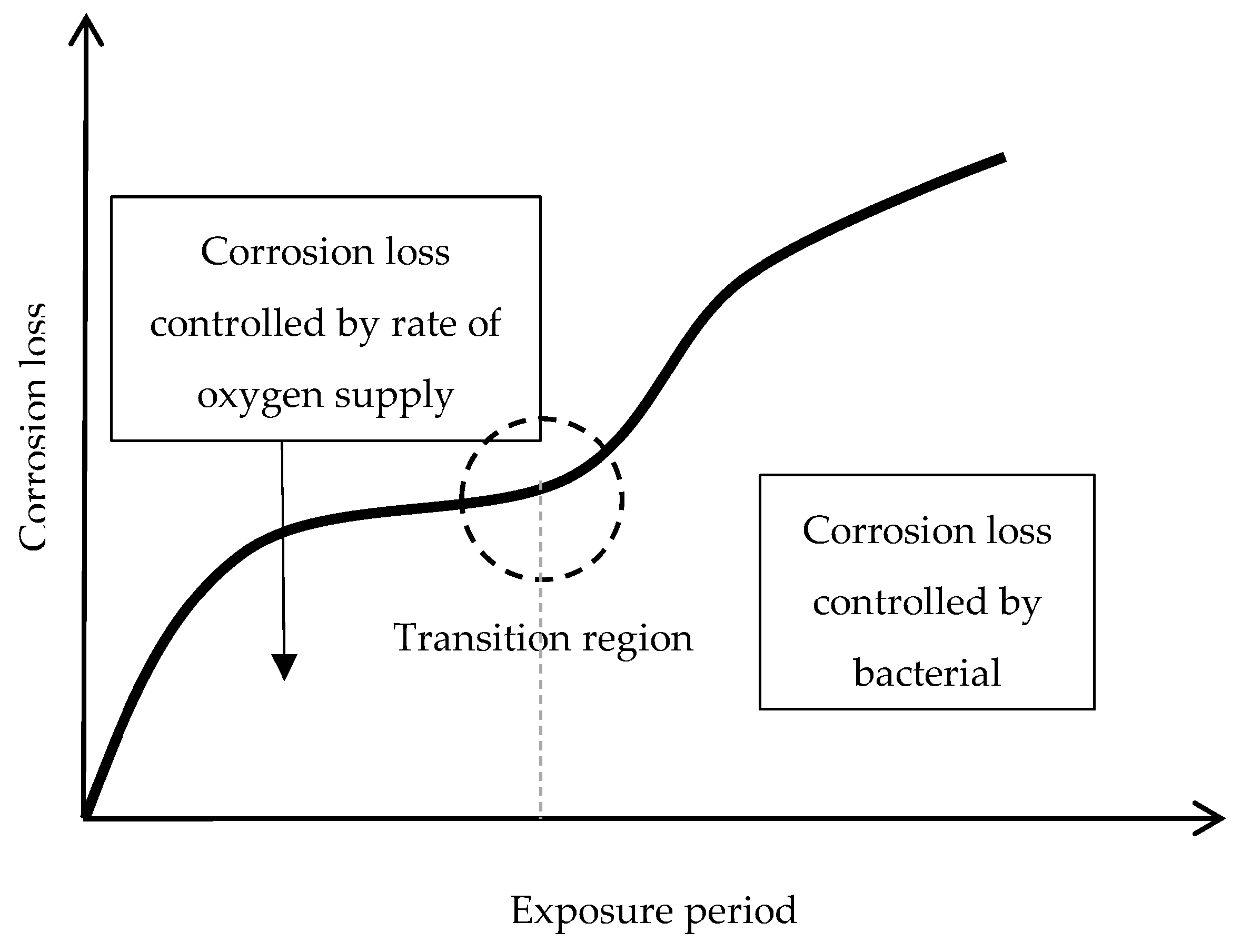
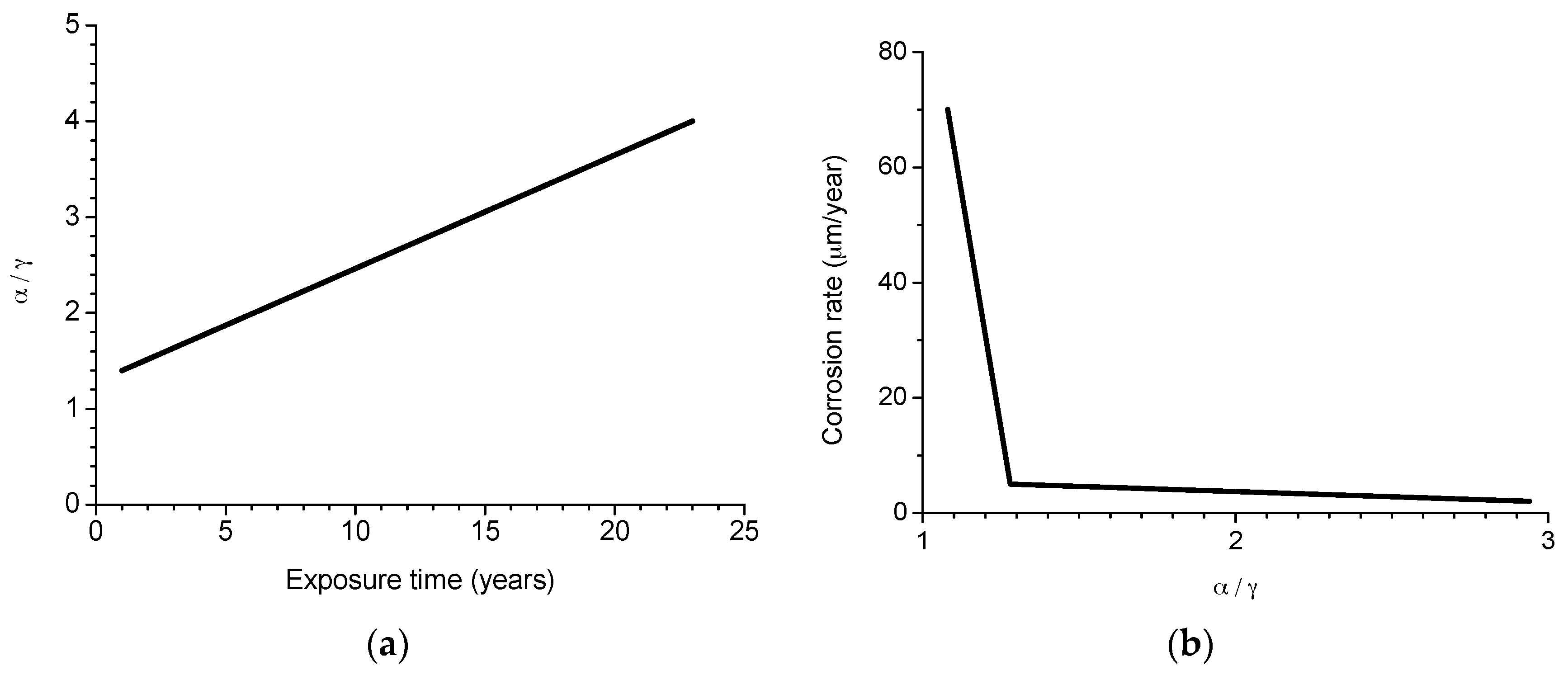
6.2.2. Stabilisation of Rust Layers and Steady-State Corrosion Rate
- y = corrosion rate, µm/y
- x = time, years
- 1/t1 = decrease constant
- y0 = steady state corrosion rate, µm/y
- A1 + y0 = corrosion rate at x = 0, µm/y
6.2.3. Adhesion
- (a)
- Brown stains as a consequence of run-off processes
- (b)
- Leaching of soluble components of the rust layer (iron chlorides in marine atmosphere) by rainwater, and
- (c)
- Rust lost through abrasion and erosion, the latter particularly in high wind areas.
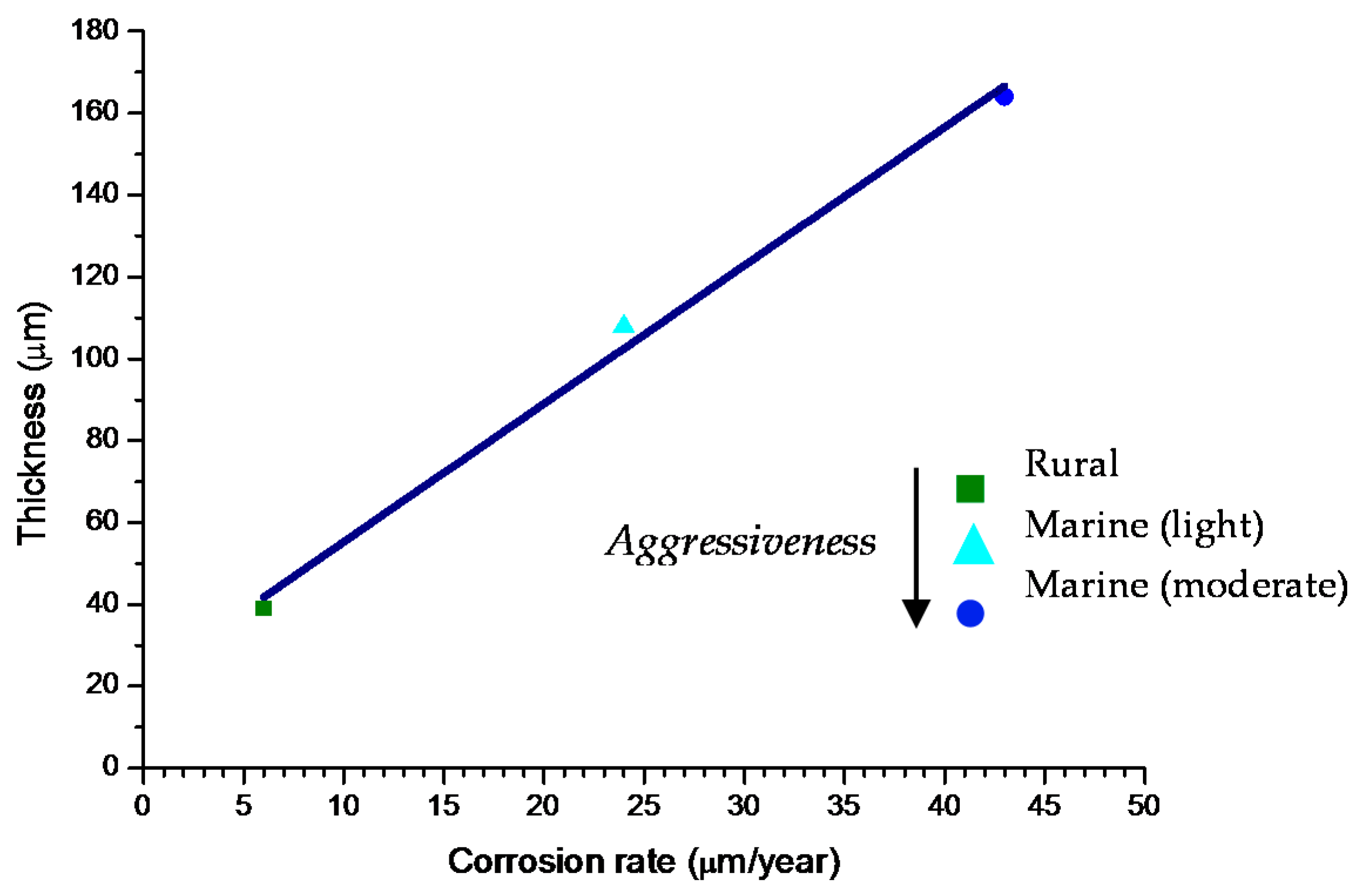
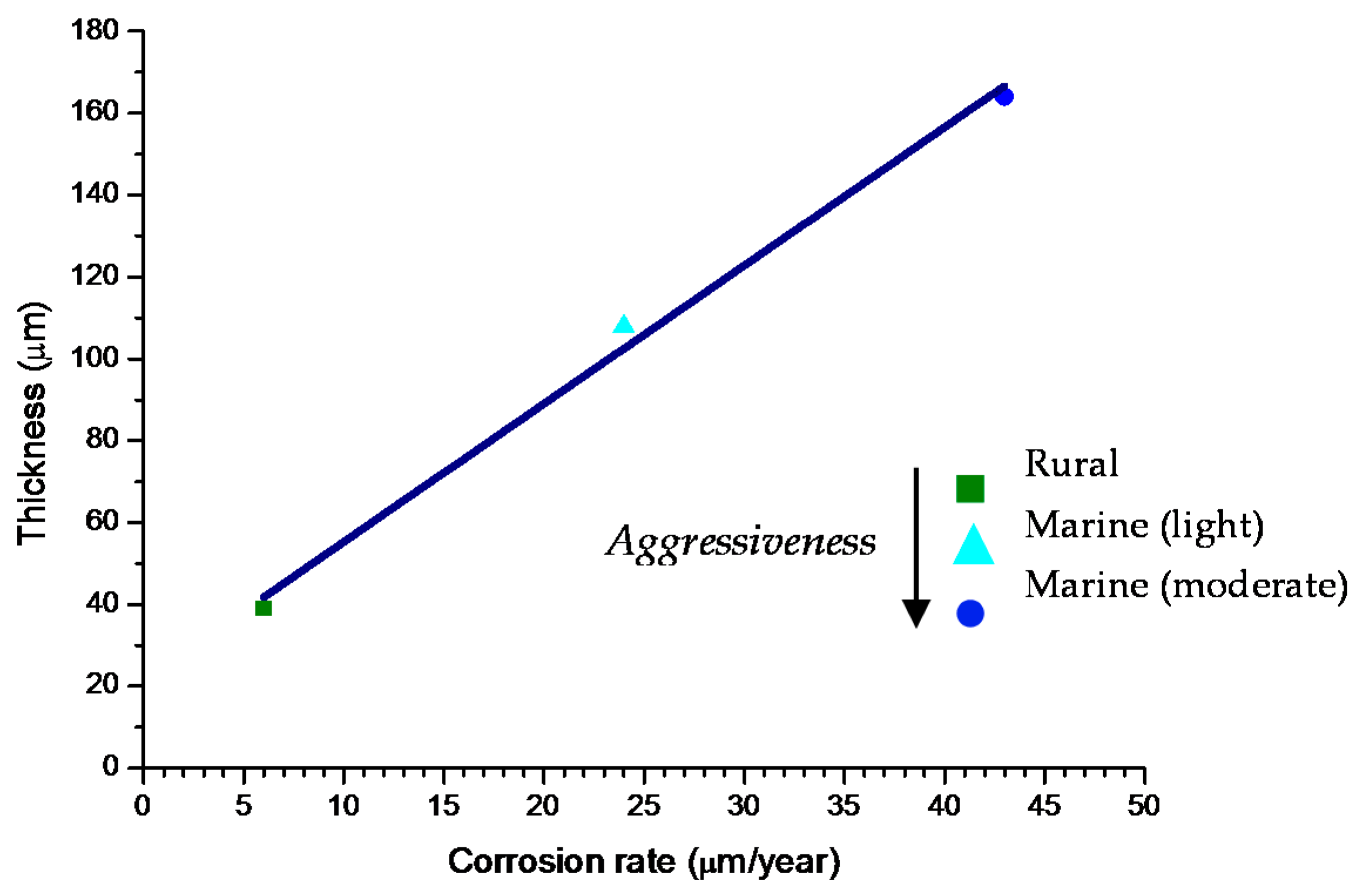
6.2.4. Thickness and Internal Structure
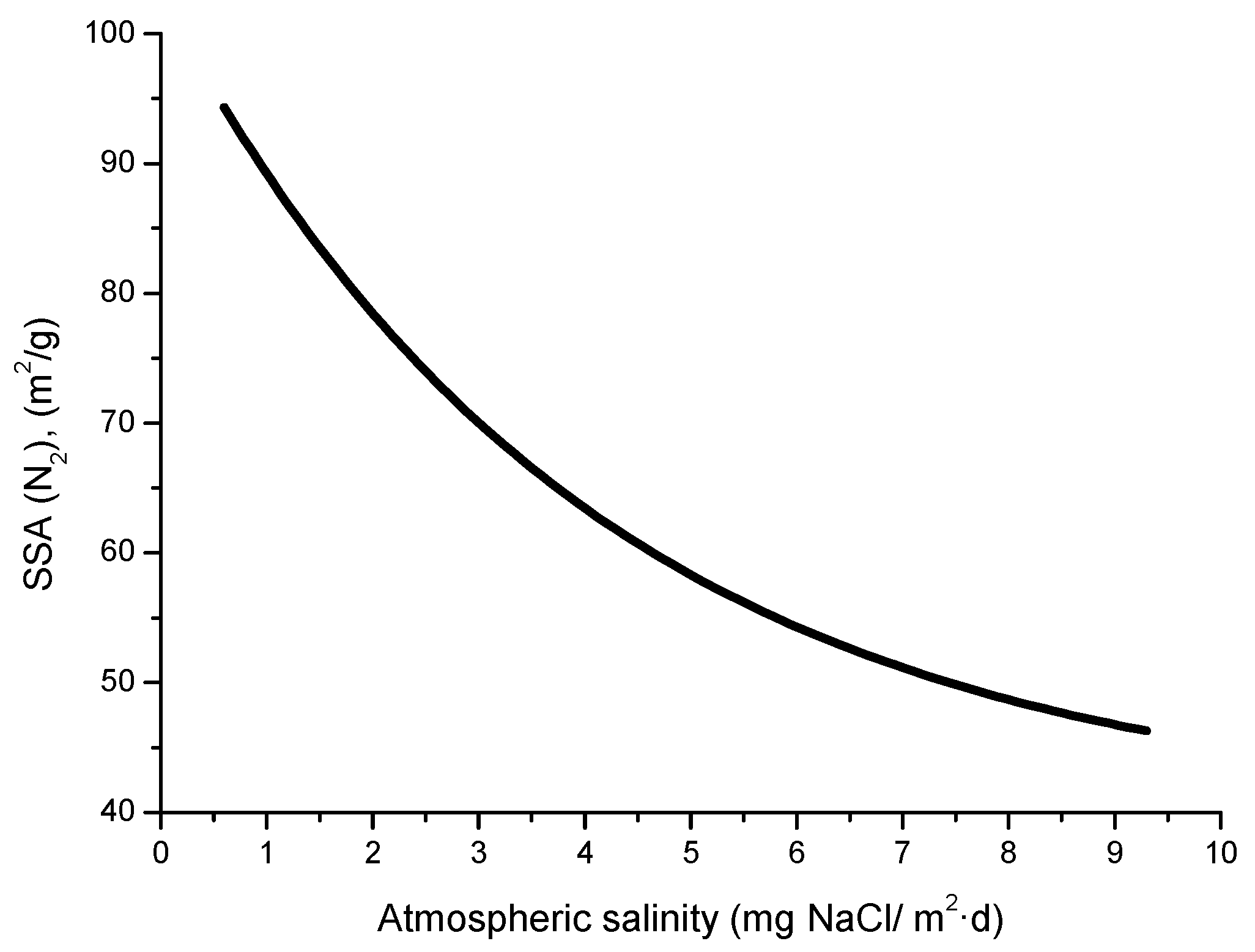
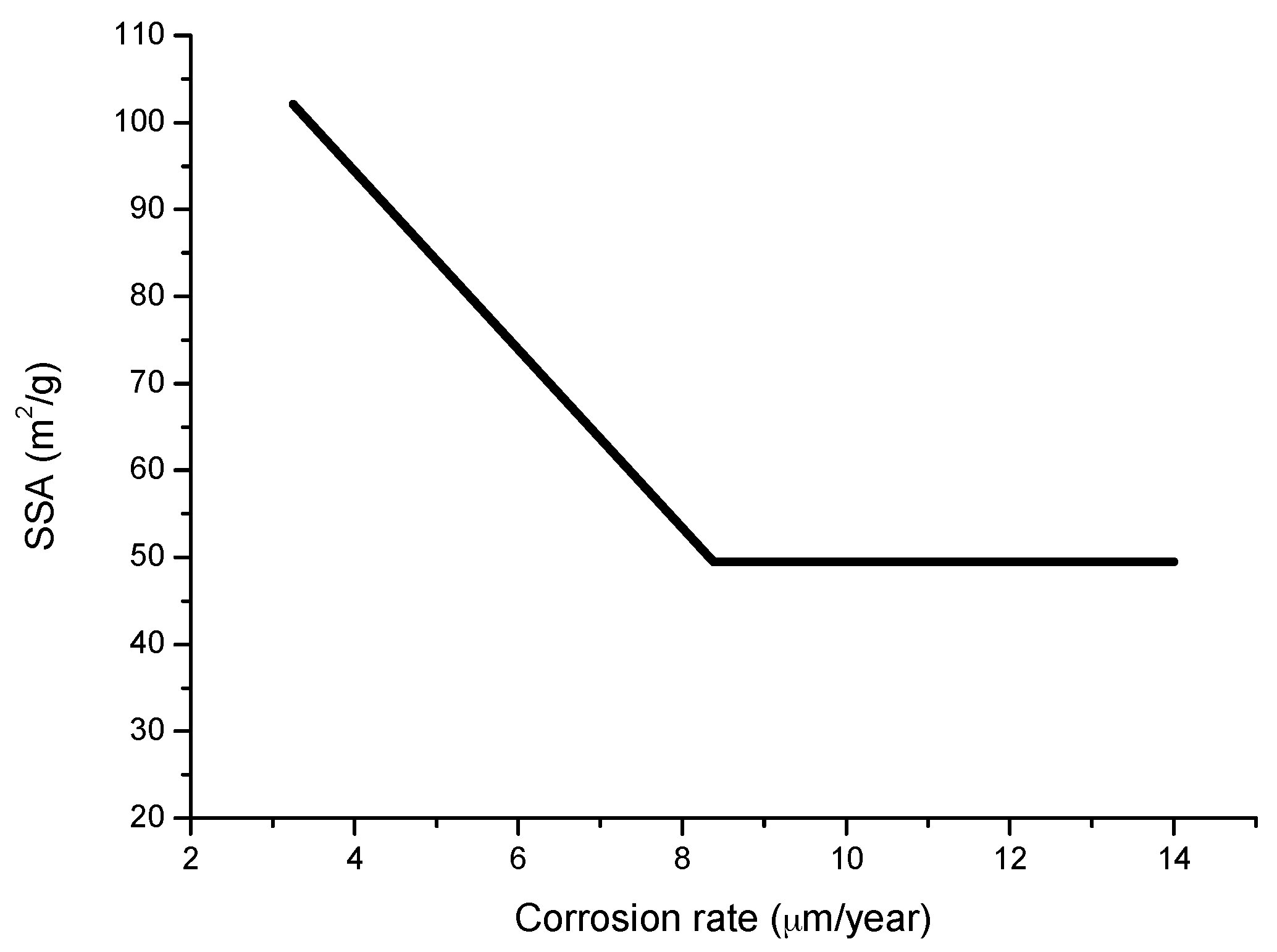
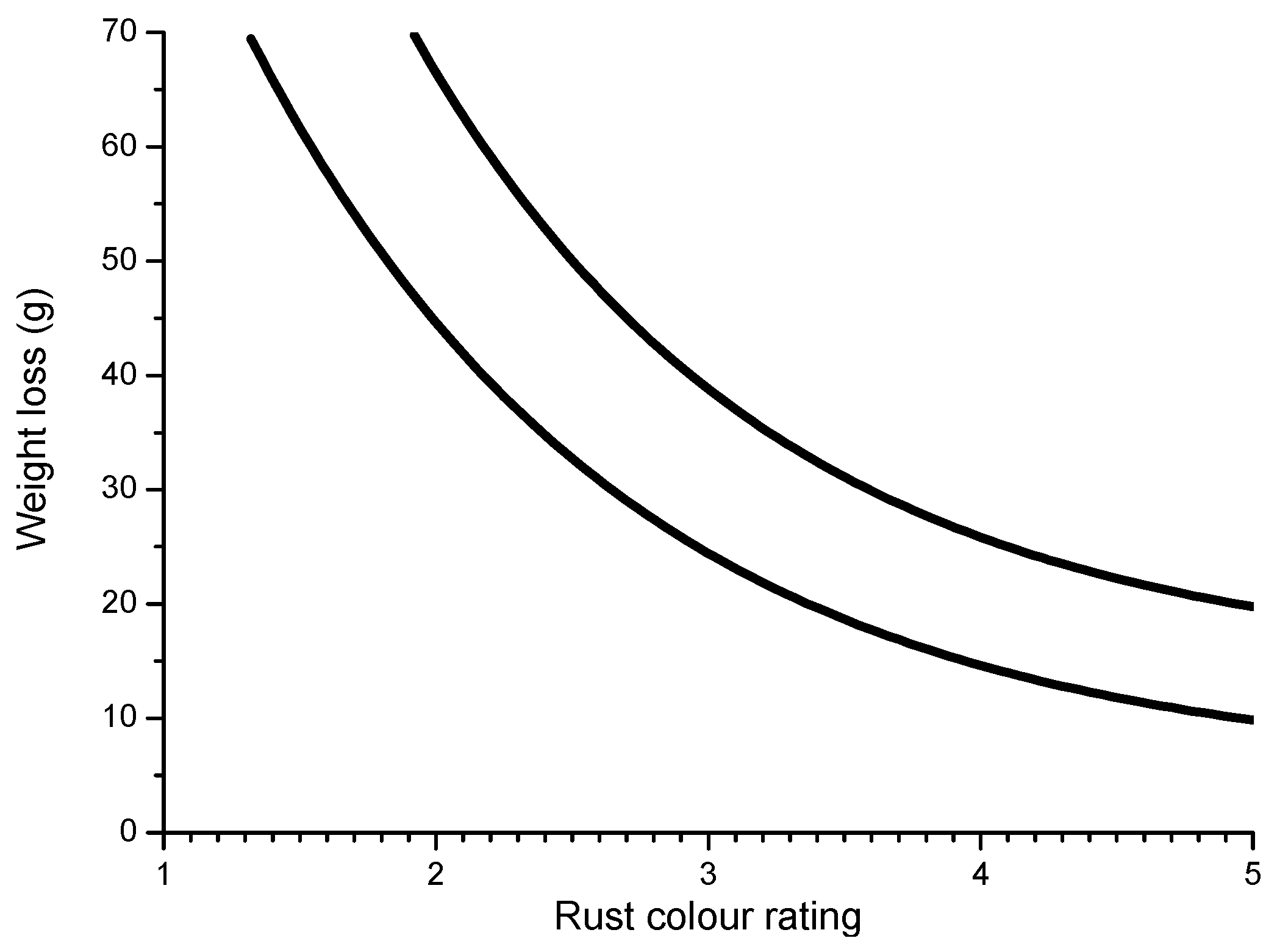
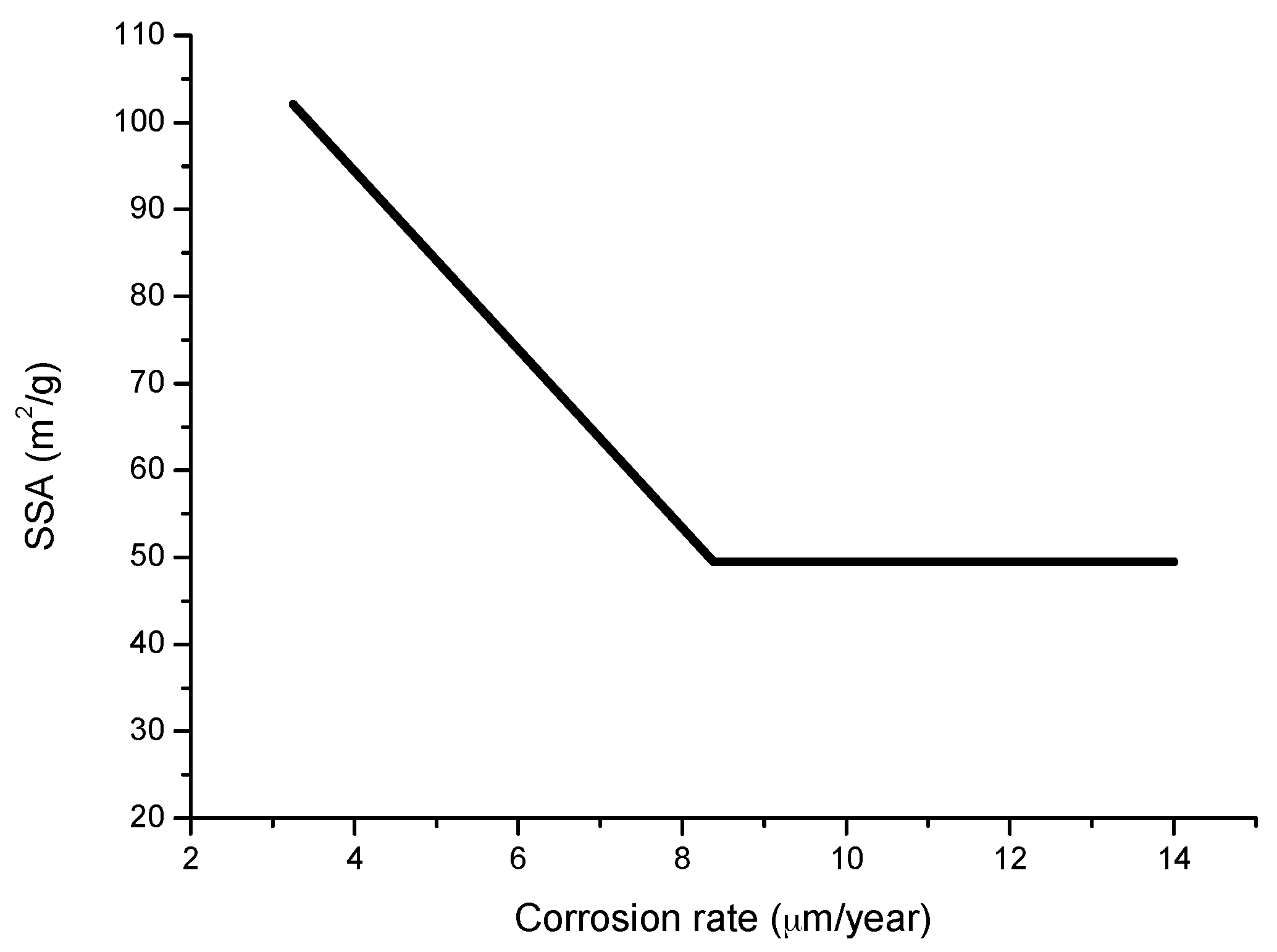
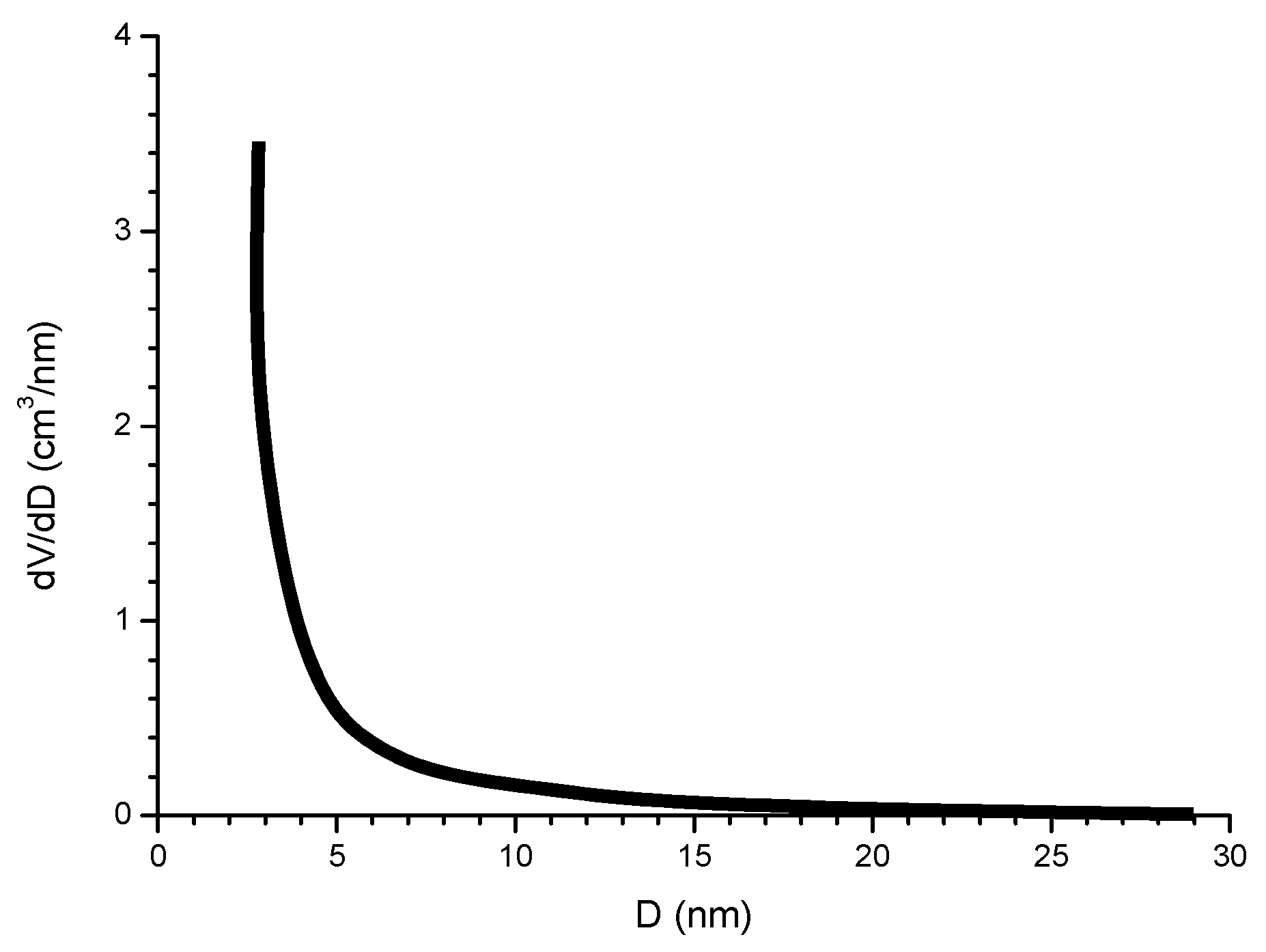
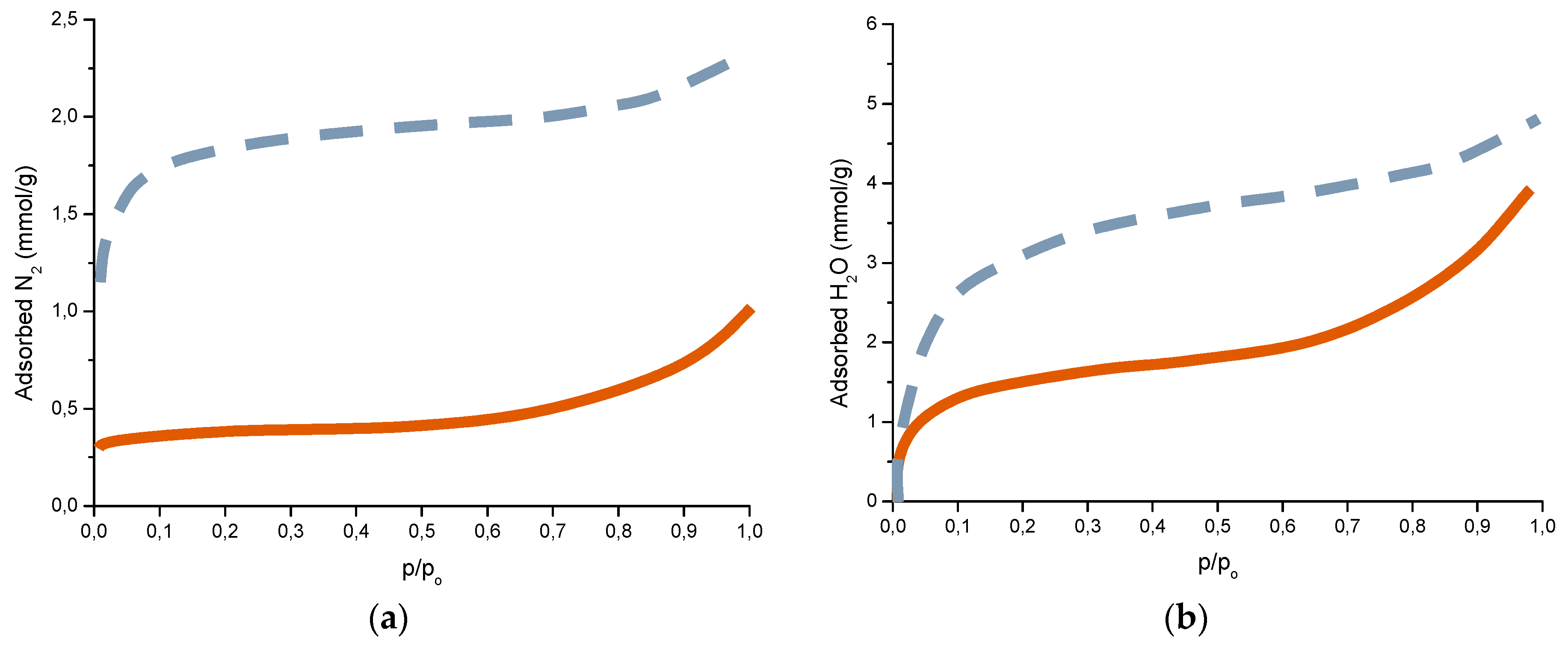
6.2.5. Porosity
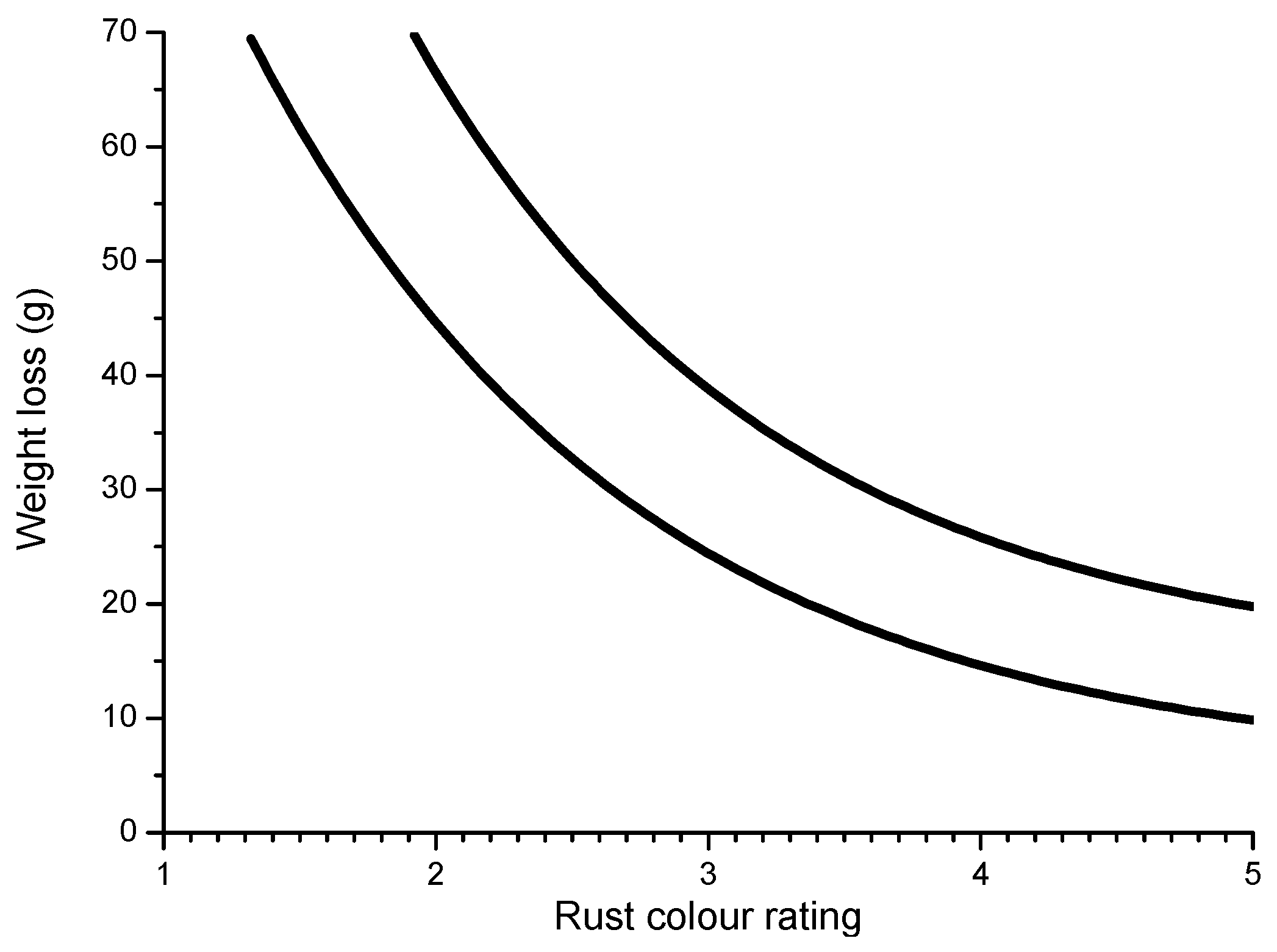
6.2.6. Indices to Evaluate the Protective Capacity of Rust Layers
7. Mechanisms of Steel in MAC
7.1. First Researches
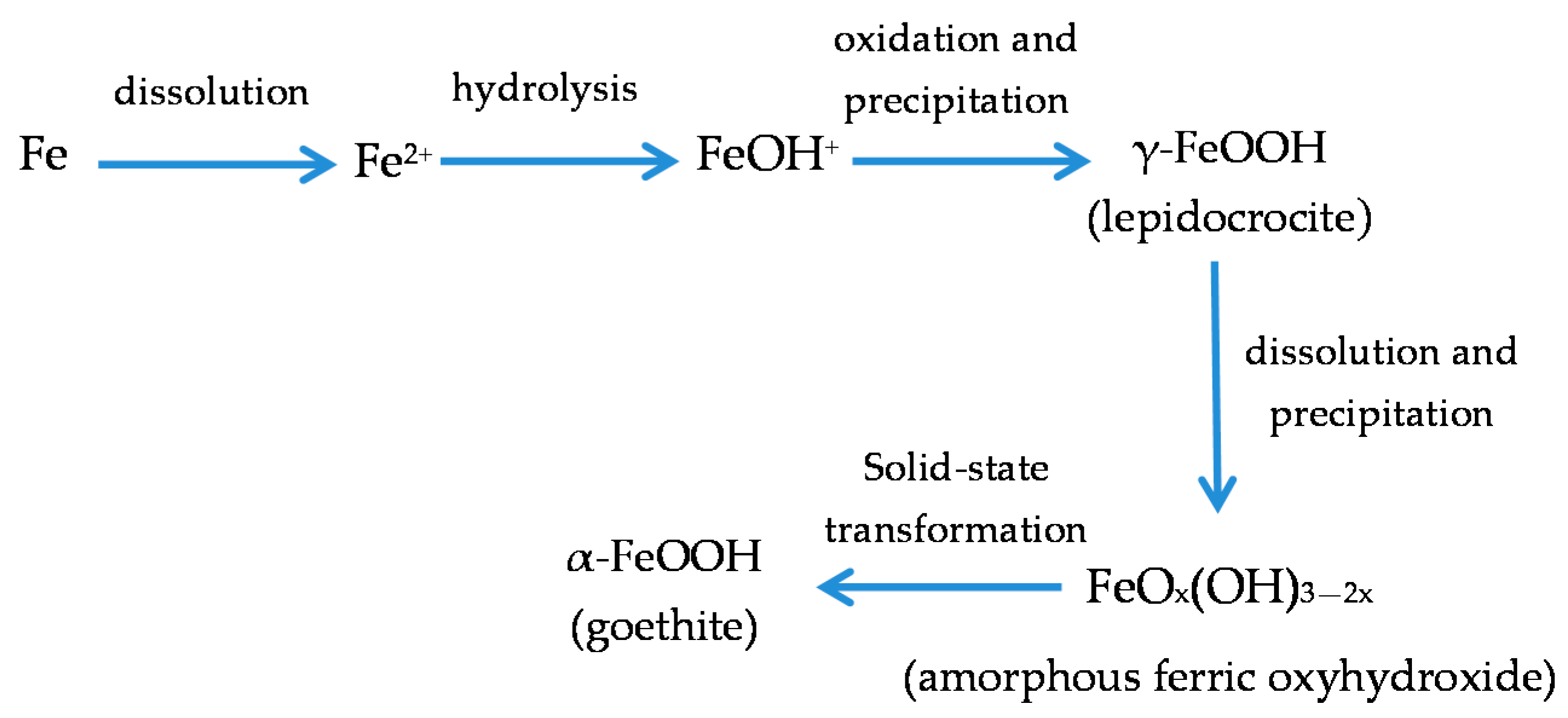
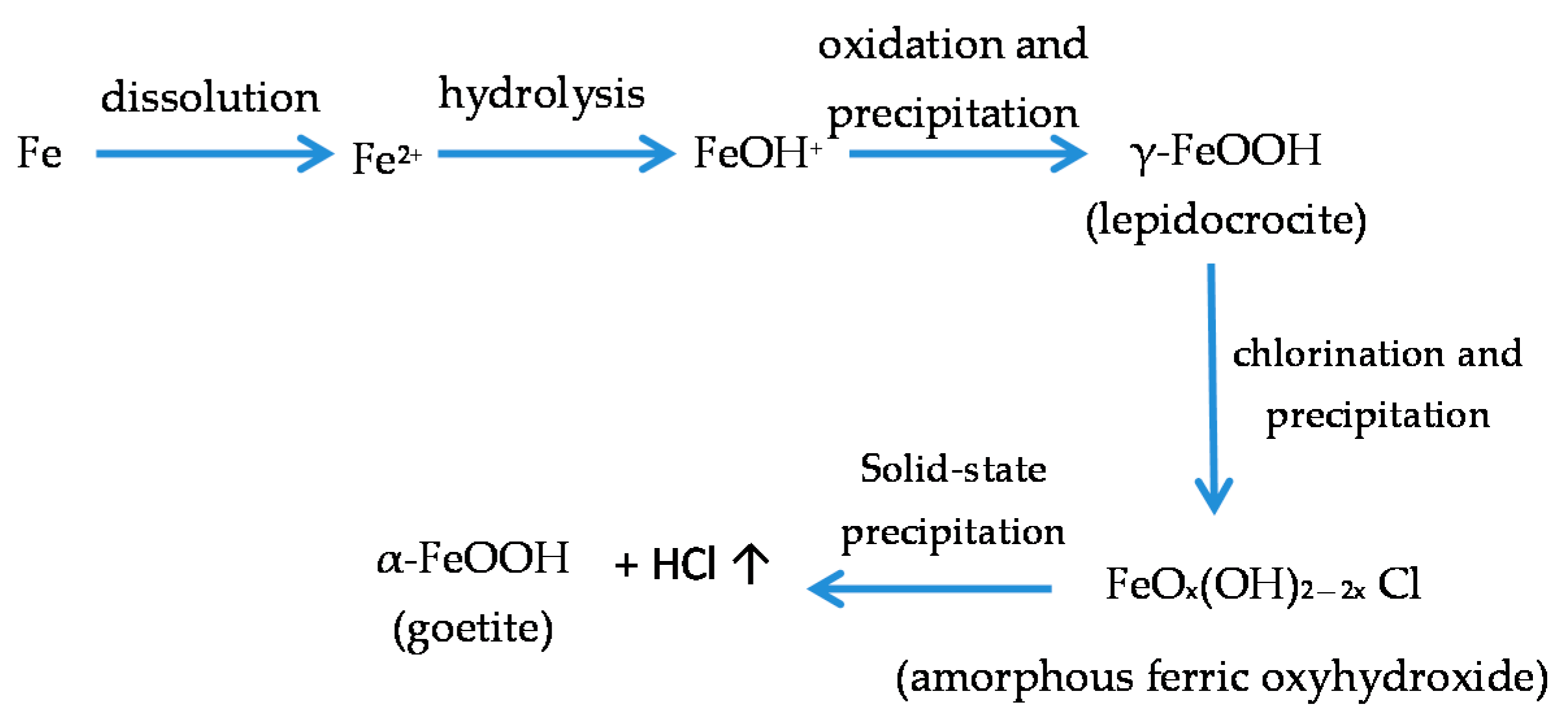
7.2. The Fundamental Role of Akaganeite in the Atmospheric Corrosion Process of Steel in Marine Atmospheres
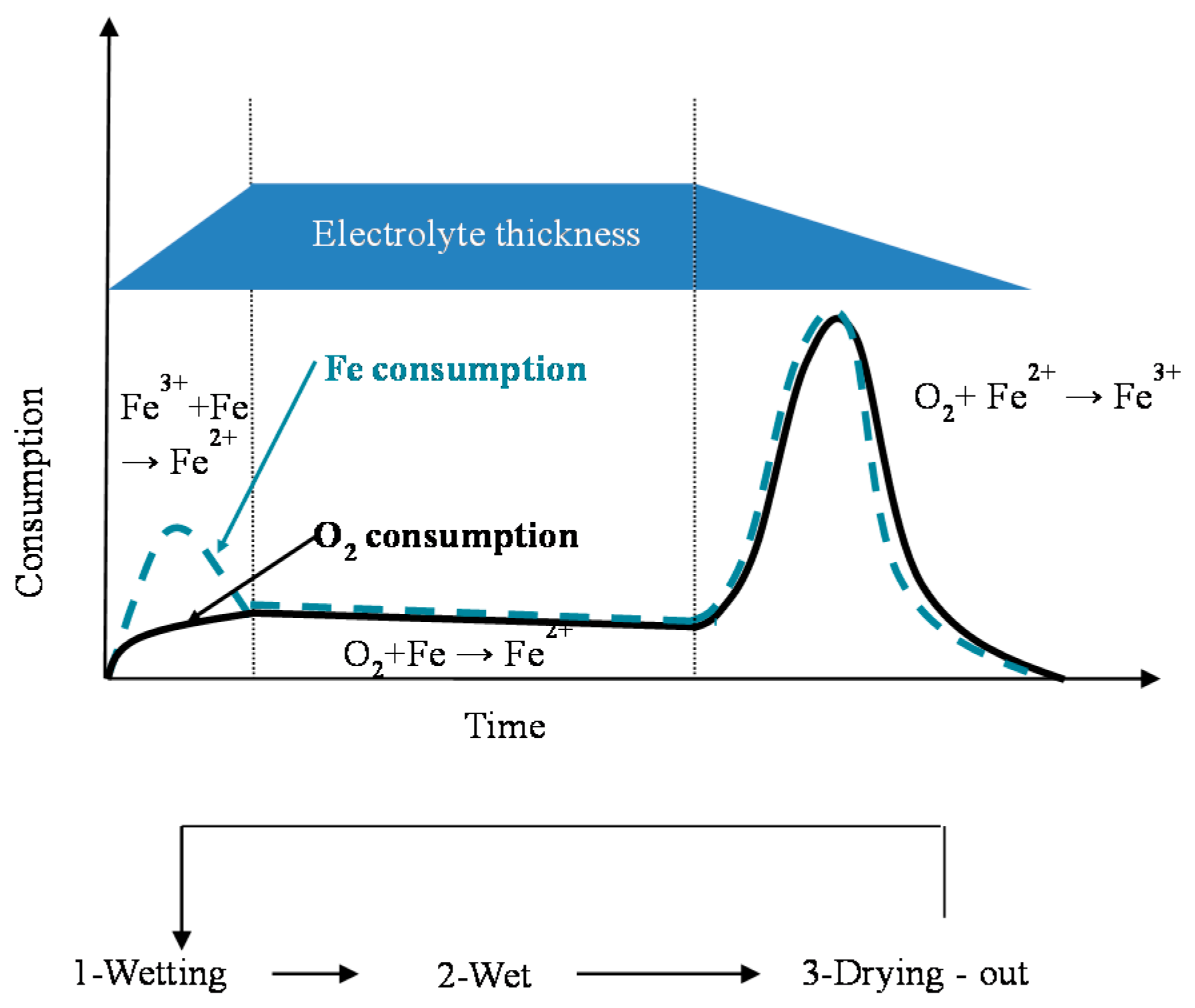
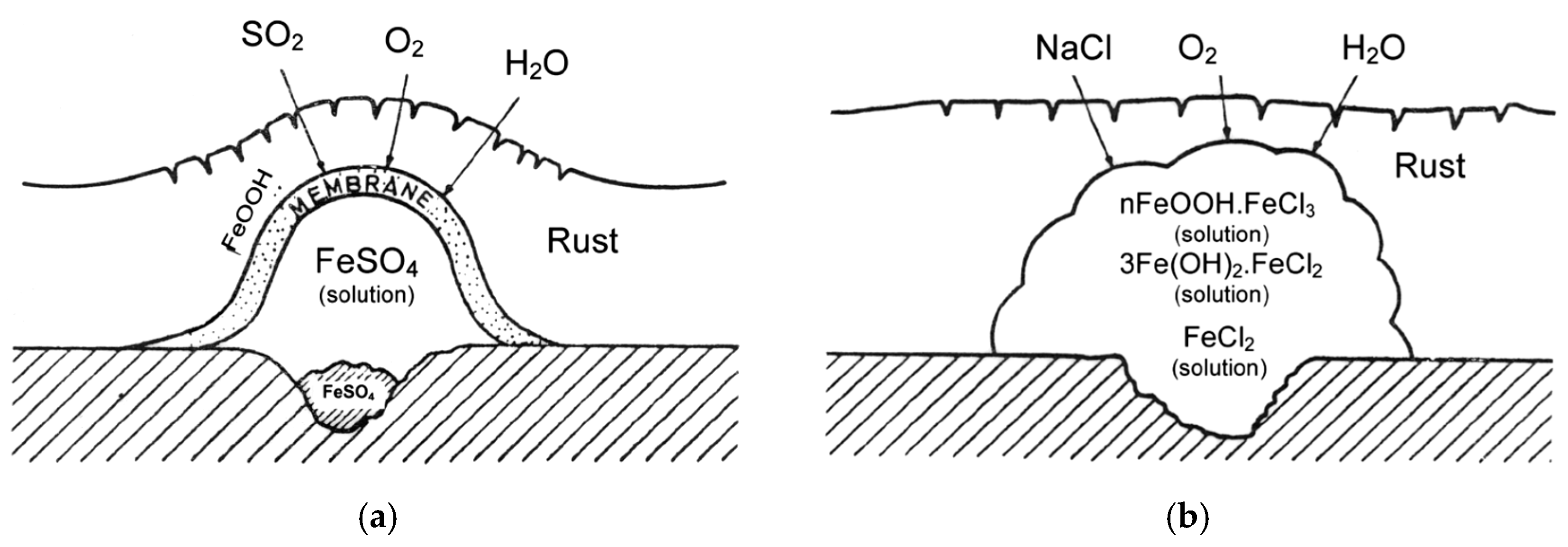
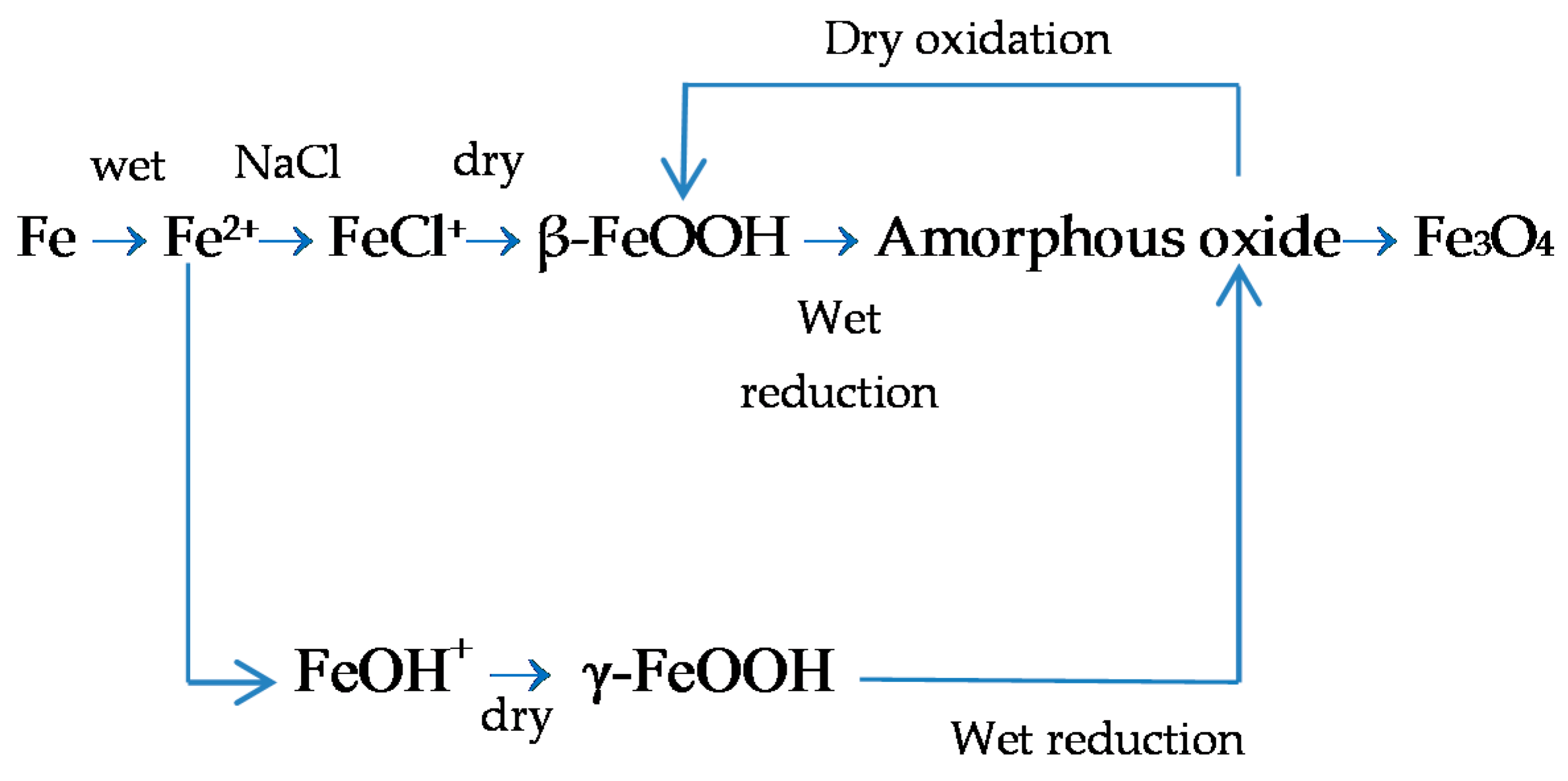
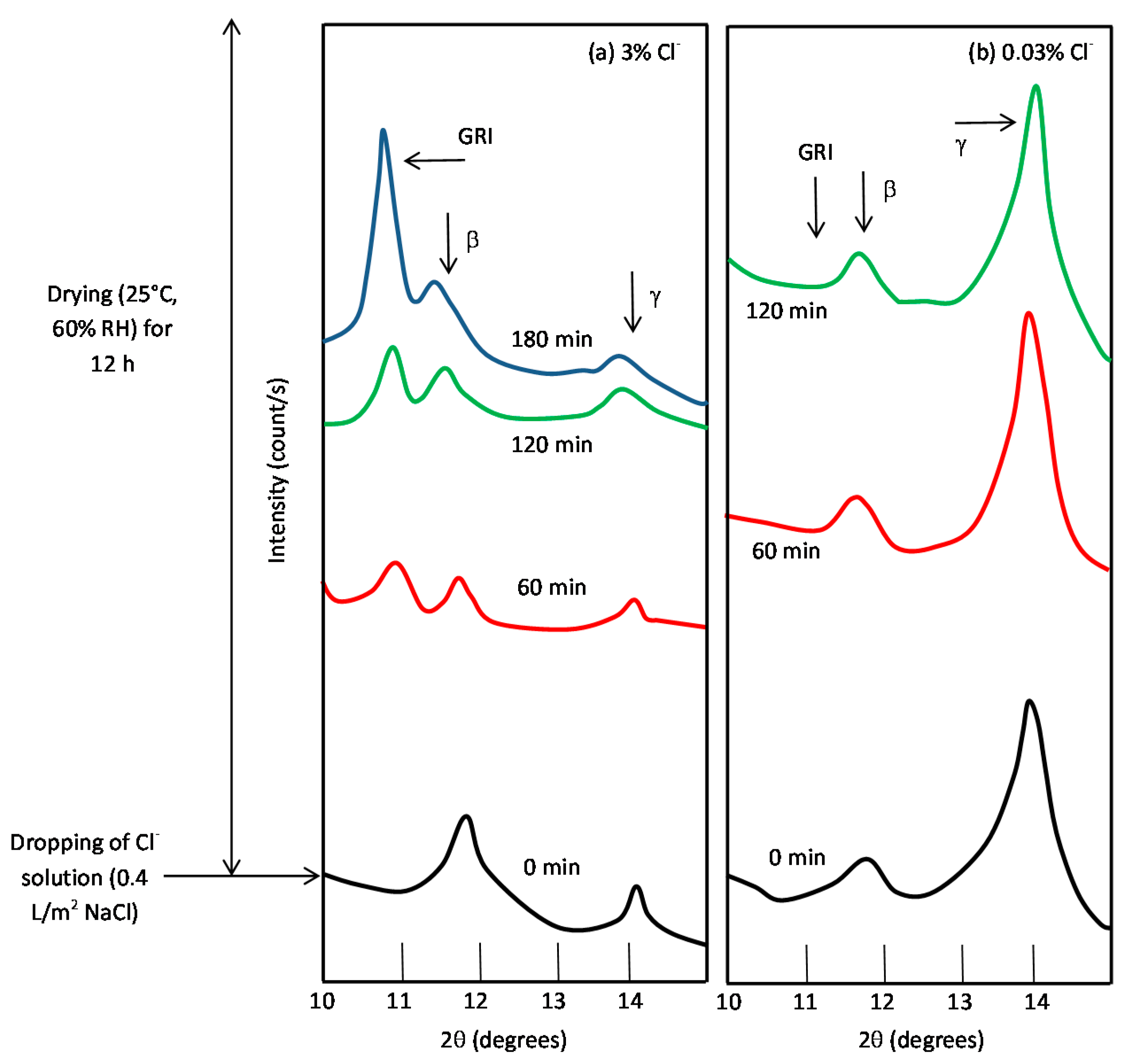
7.3. Initial Stages of MAC
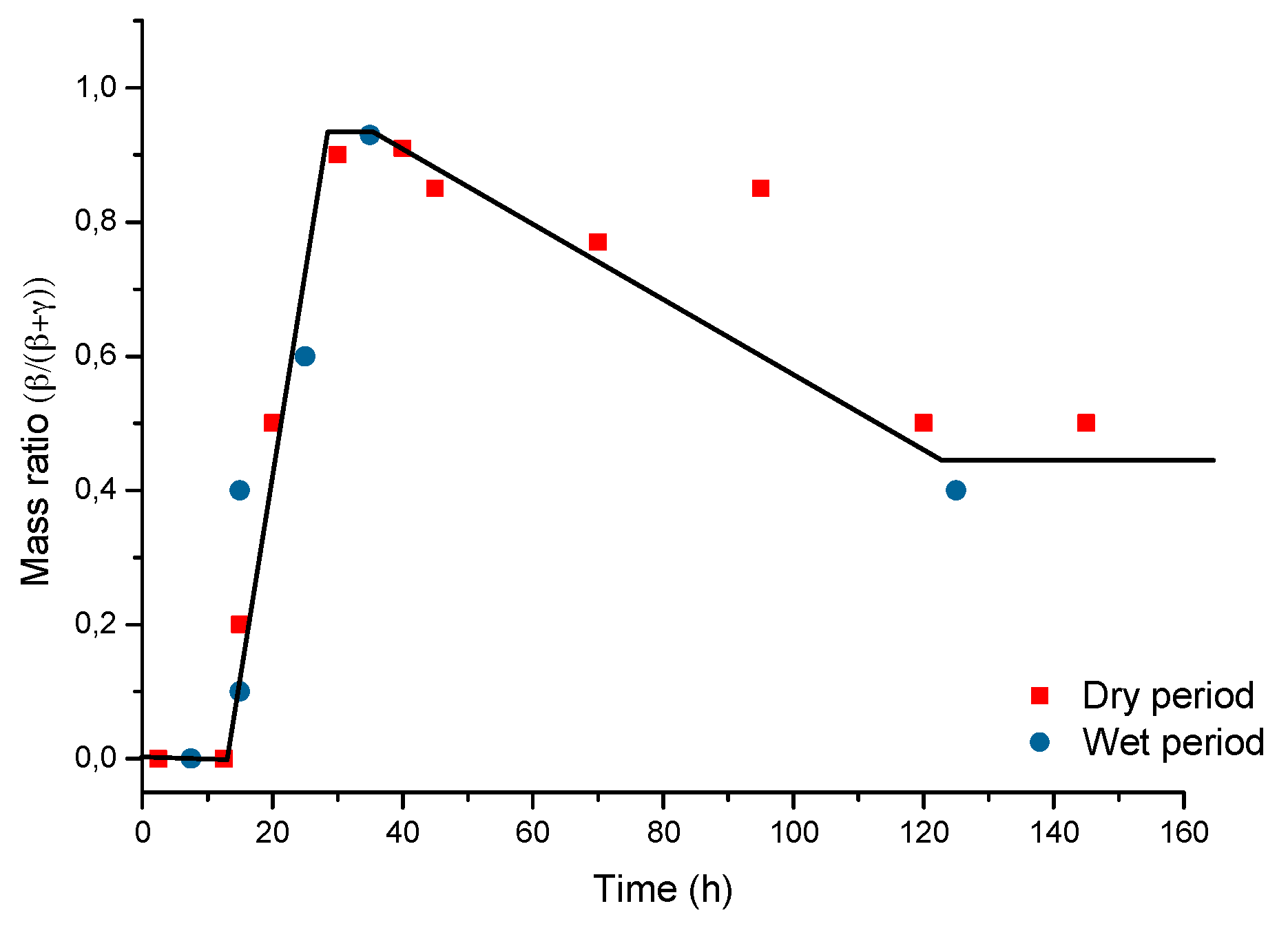
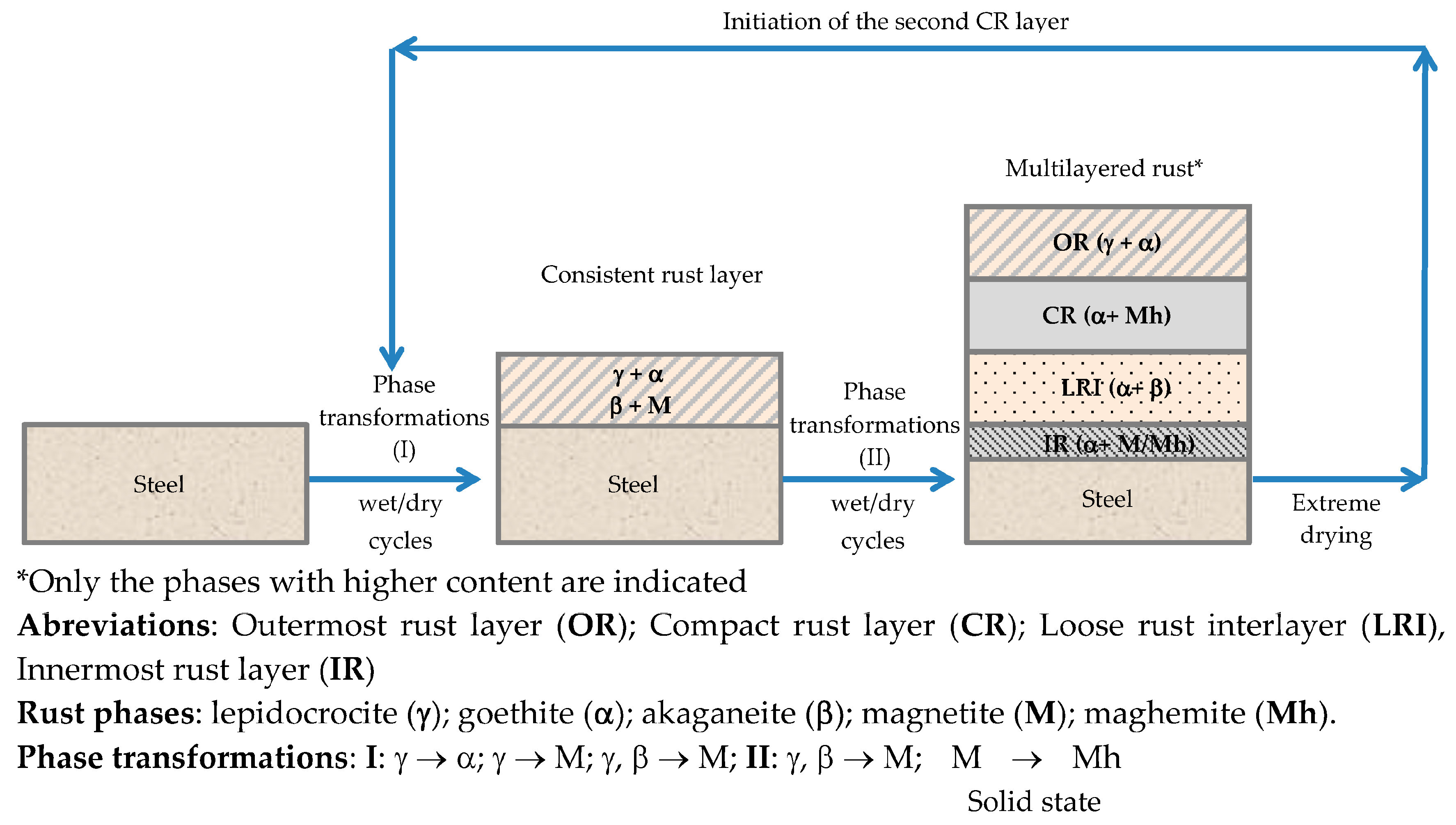
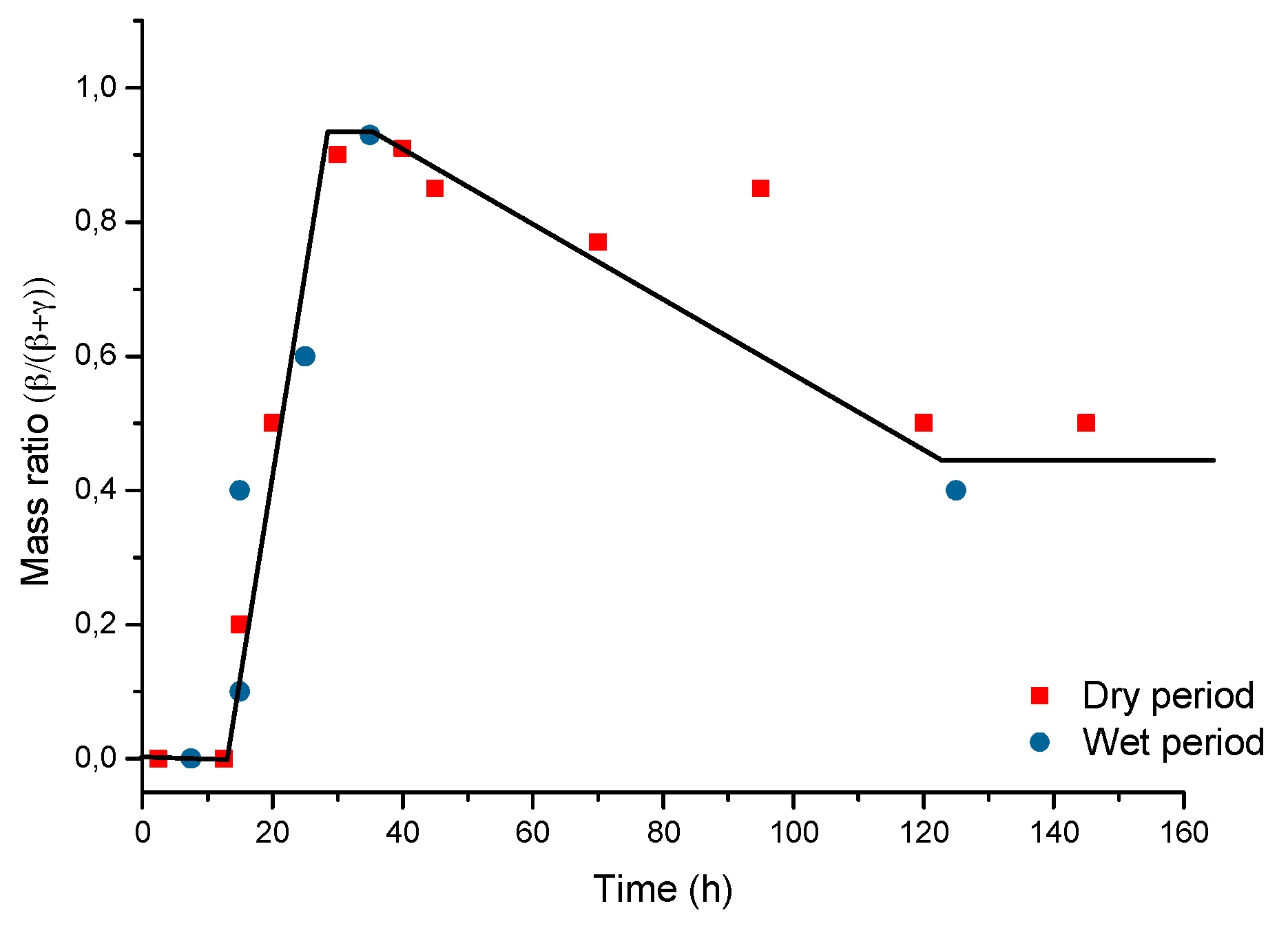
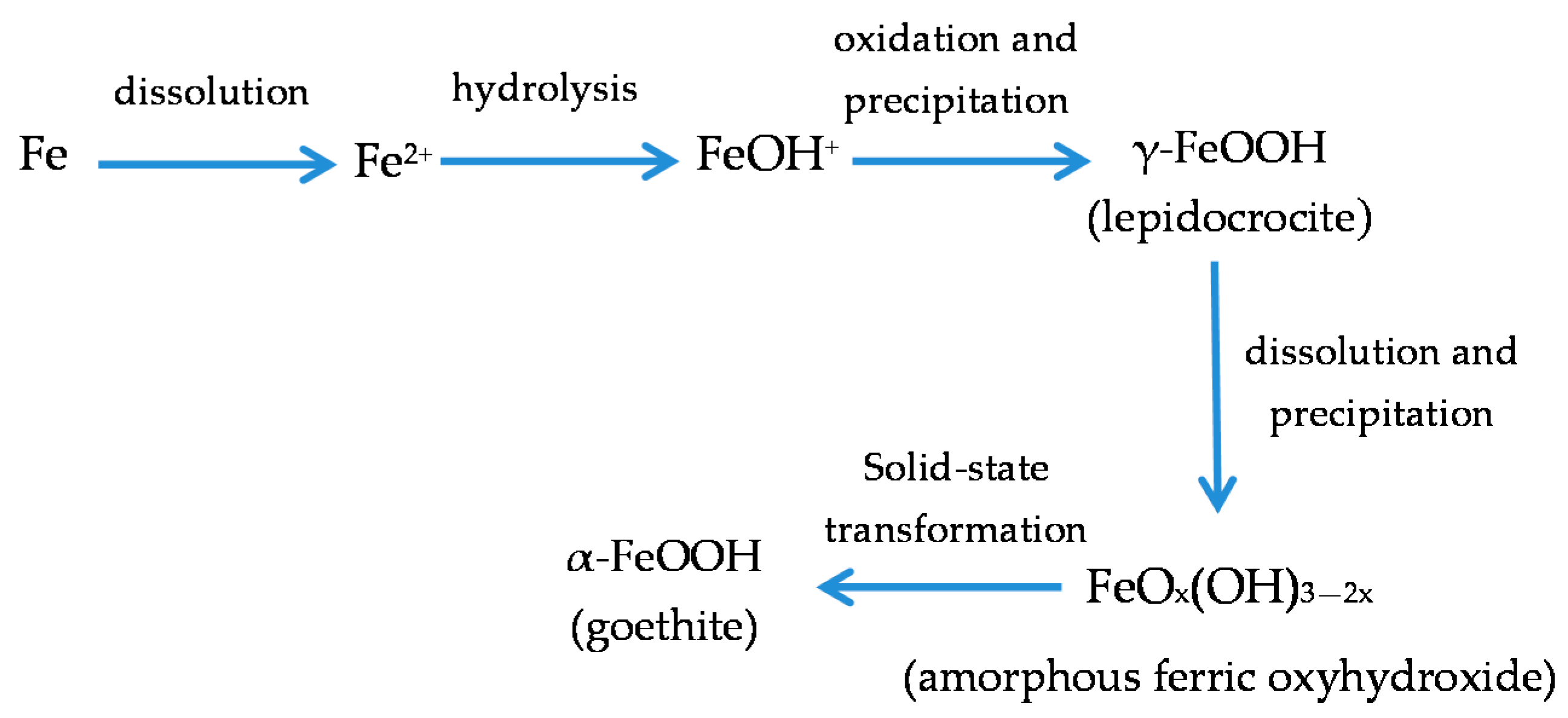
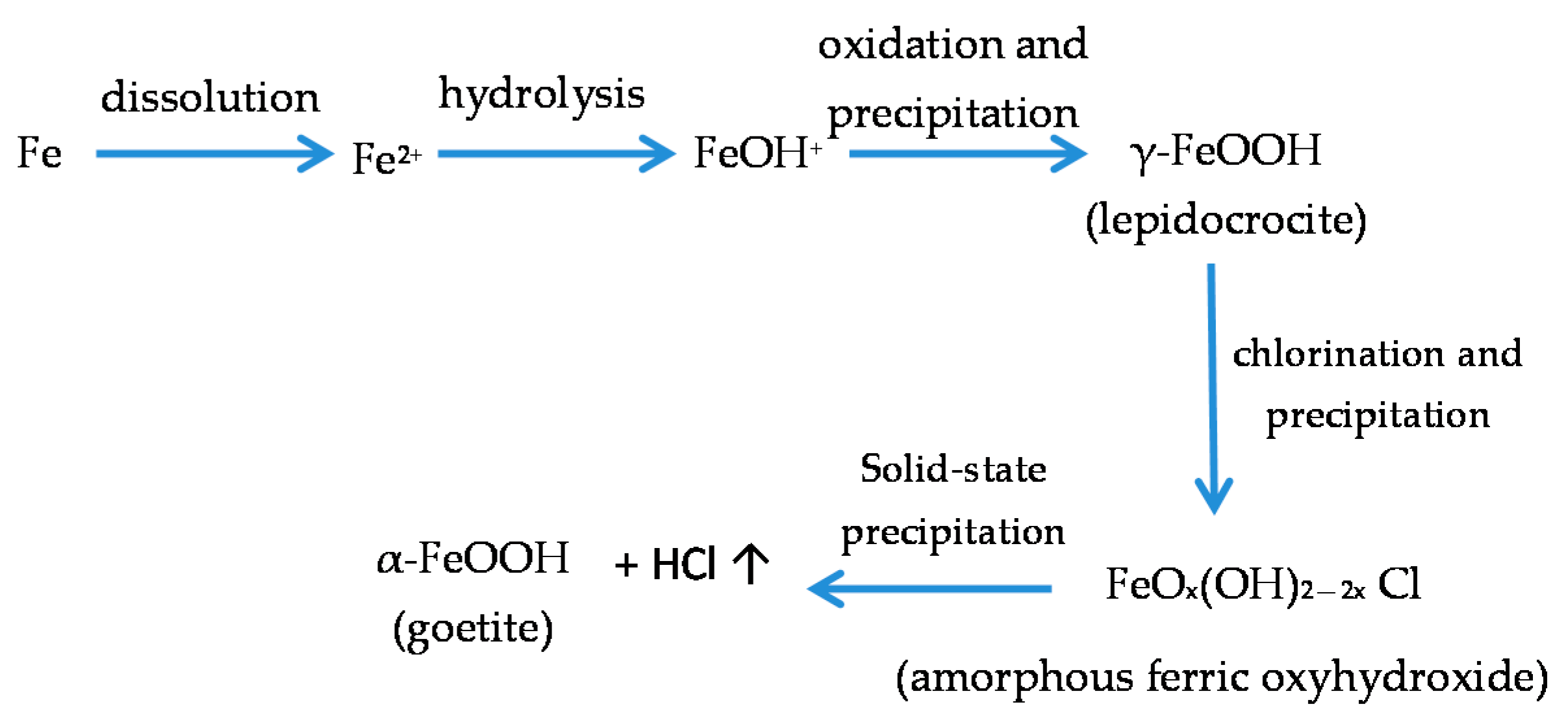
7.4. Formation and Growth of the Corrosion Layer
- (a)
- In the first stage of rusting the aerial oxidation of ferrous ions, dissolved from the steel into a slightly acidic thin water layer formed by rain on the steel surface, leads to the precipitation of lepidocrocite. Fine weather accelerates the precipitation and crystallisation of lepidocrocite by drying.
- (b)
- The lepidocrocite is formed on the steel surface and transformed to amorphous ferric oxyhydroxide and goethite during the atmospheric rusting process. The amorphous ferric oxyhydroxide transforms to goethite by deprotonation using hydroxyl ions provided by the rainwater.
- (a)
- the location at which precipitation of corrosion products occurs within the corrosion system (steel/rust/atmosphere), and
- (b)
- the structural evolution of the corrosion product layer during wet/dry cycles.
7.5. Proposal of an Overall Mechanism for the MAC Process of Steel
8. Coastal-Industrial Atmospheres
9. Long-Term Behaviour of Carbon Steel Exposed to Marine Atmospheres
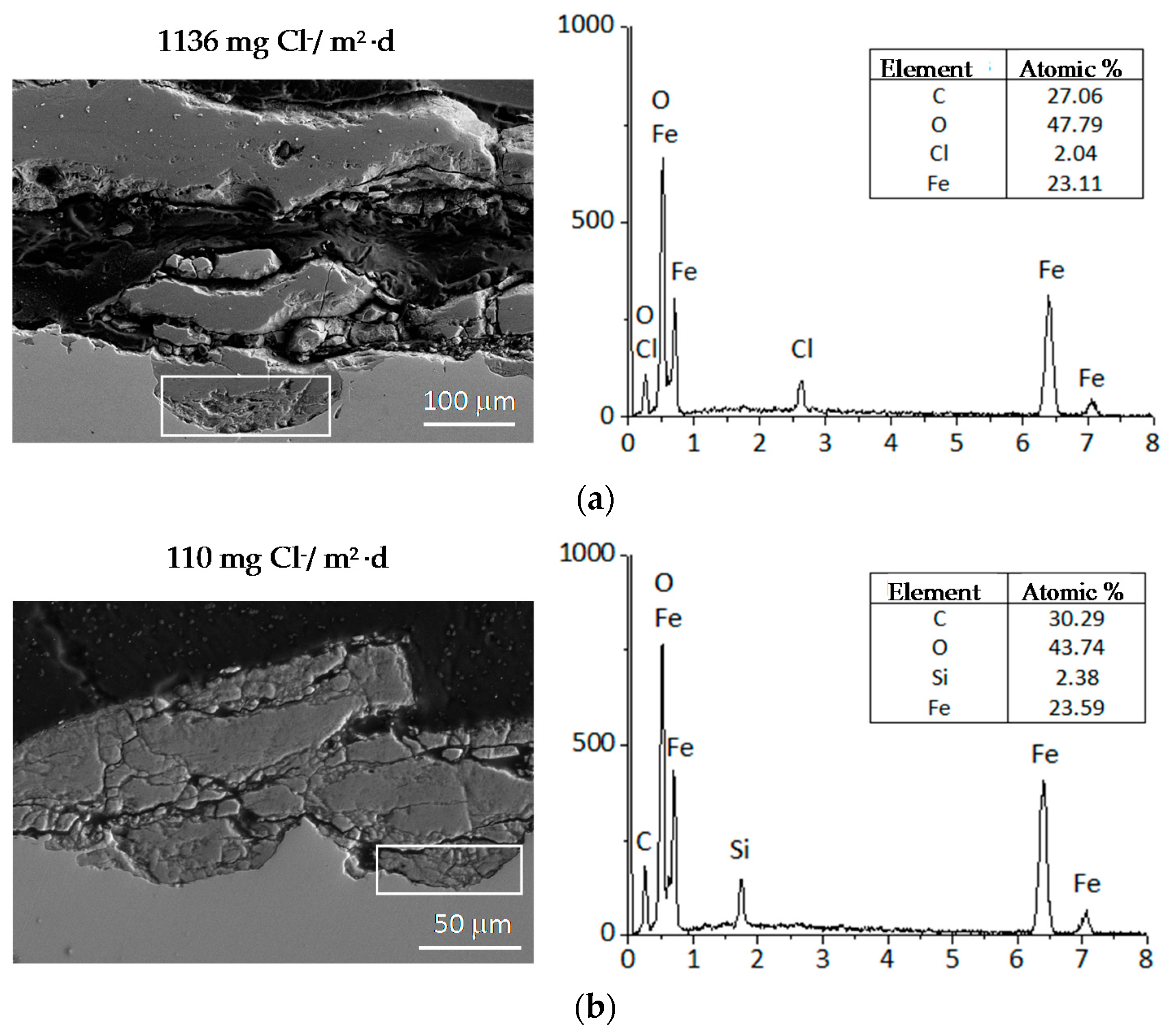
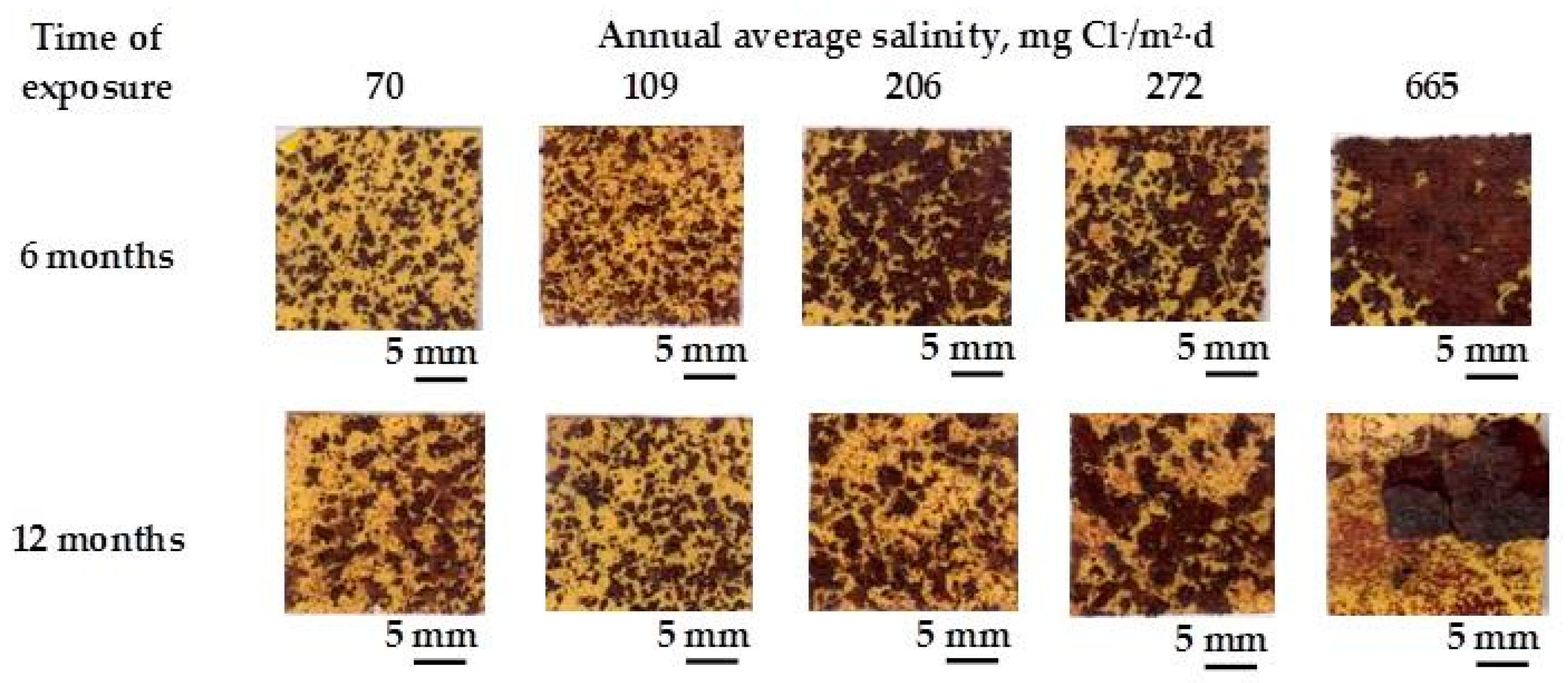
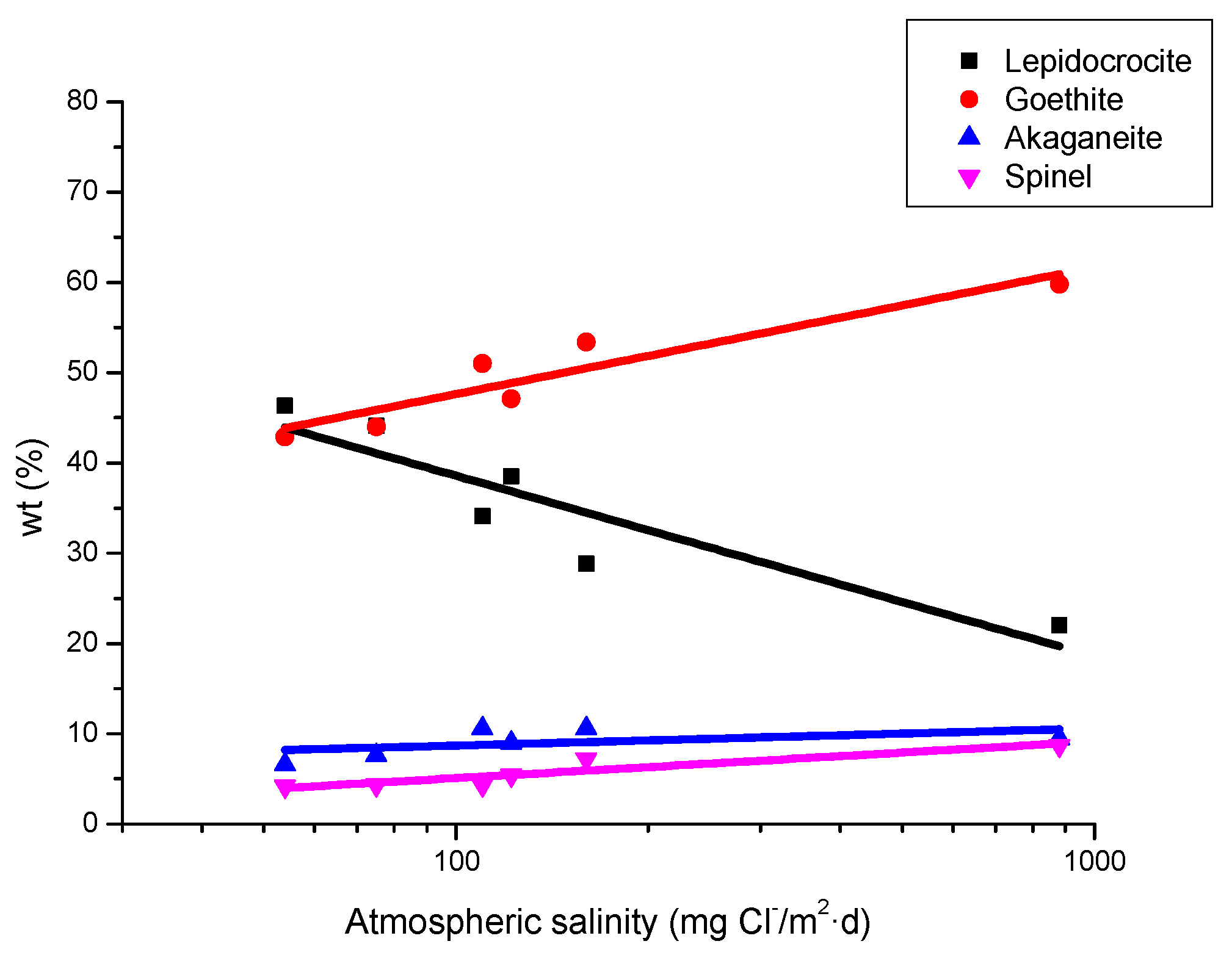
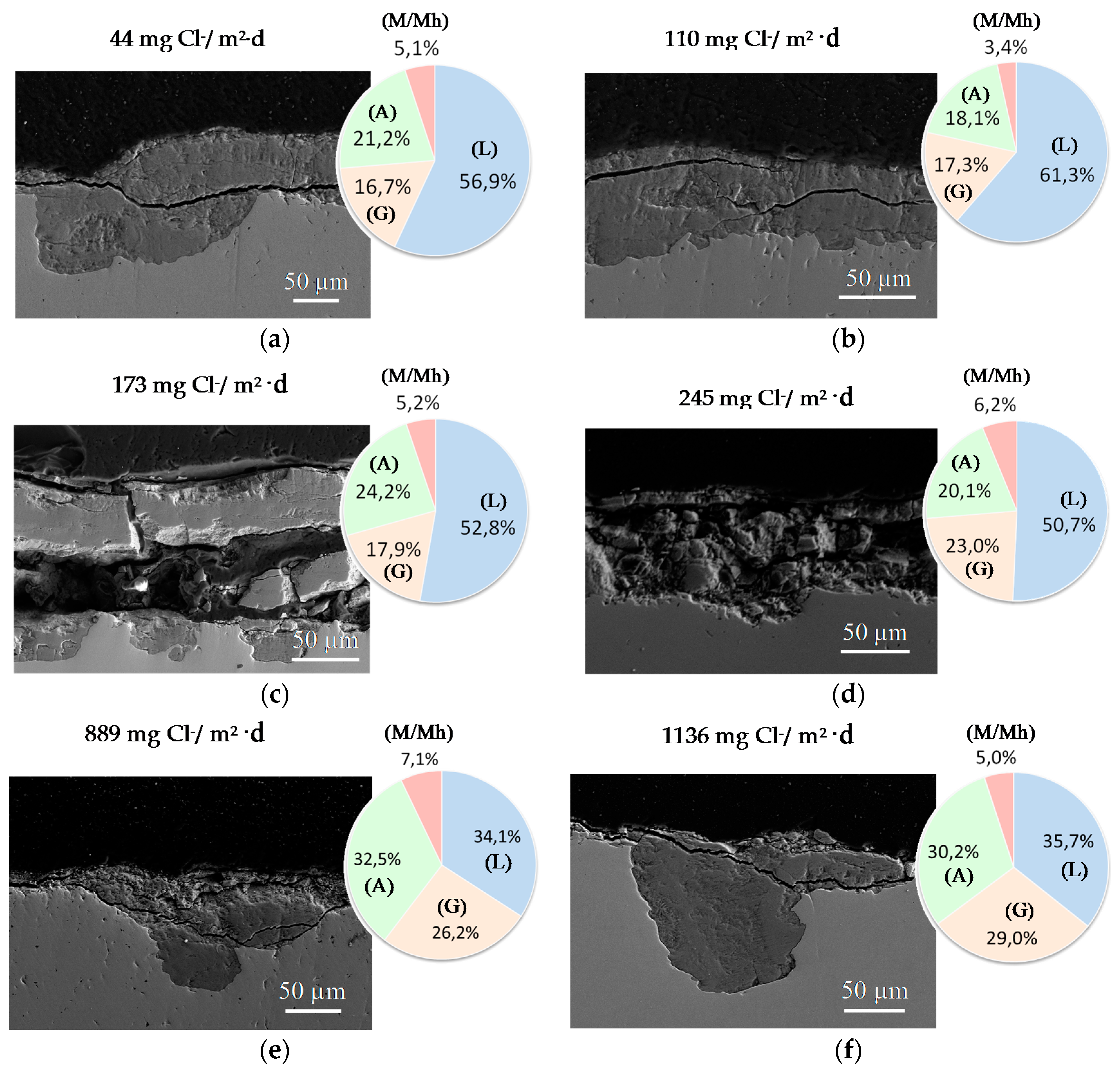
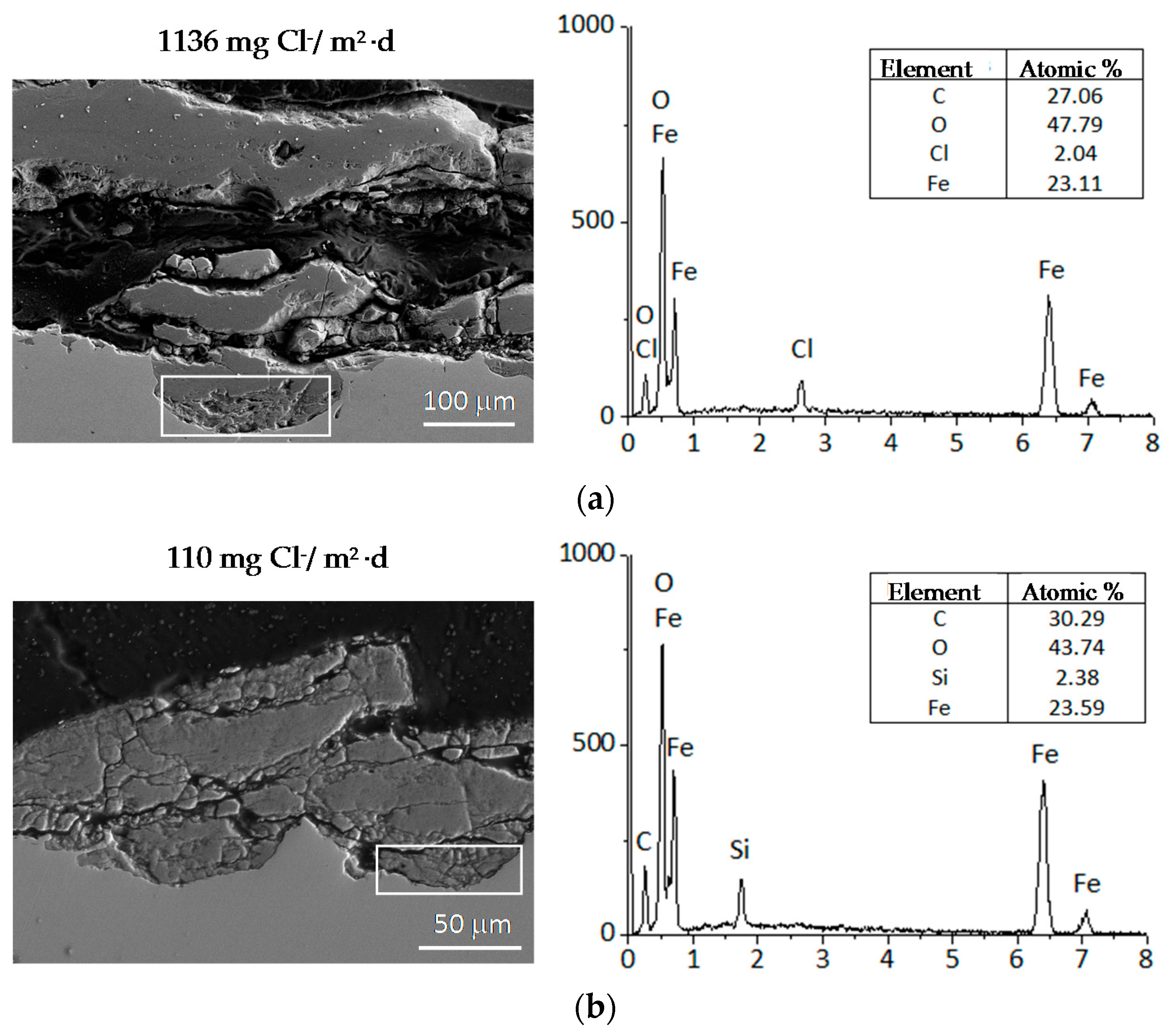
9.1. Nature of Corrosion Products
9.2. First Year Steel Corrosion
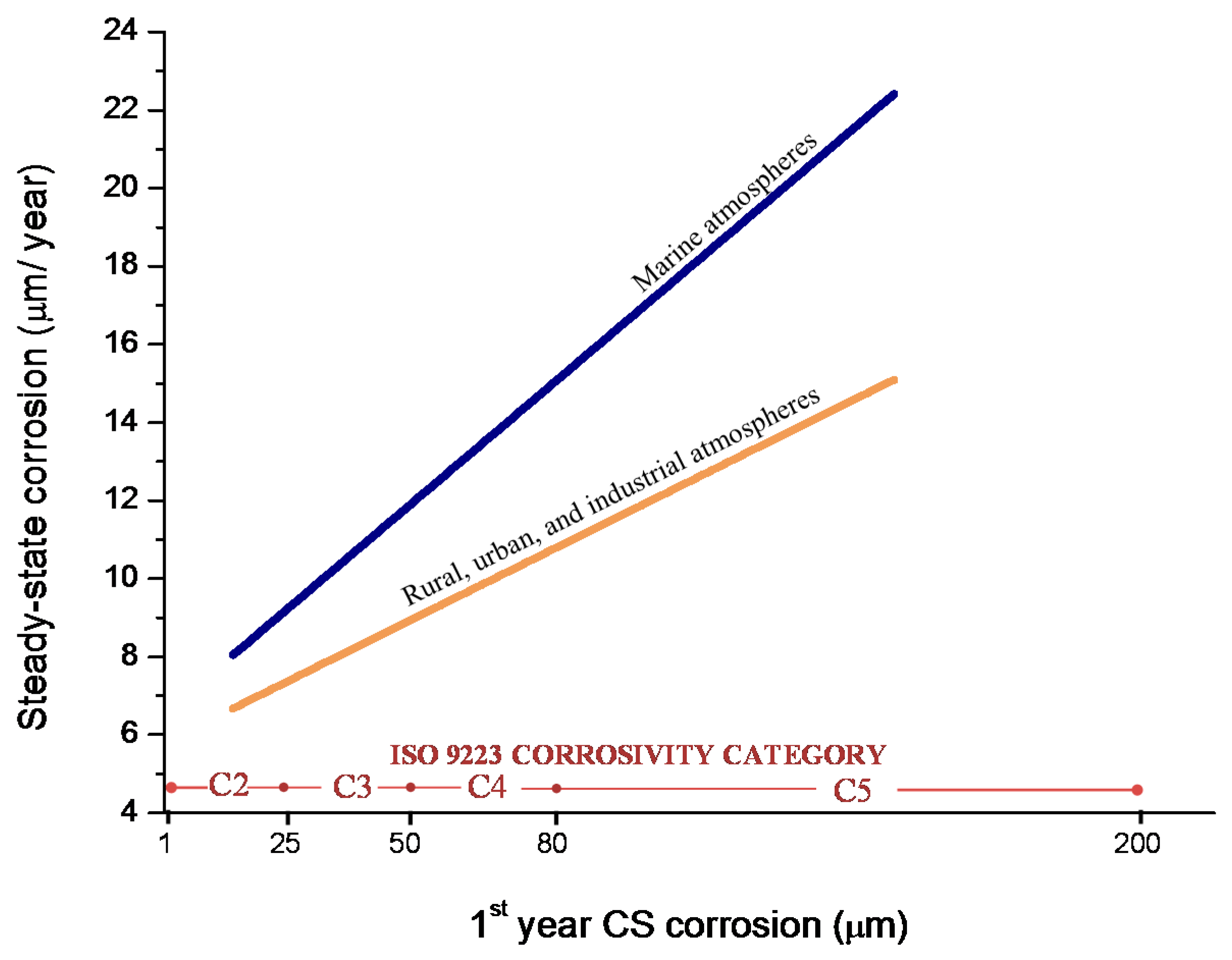
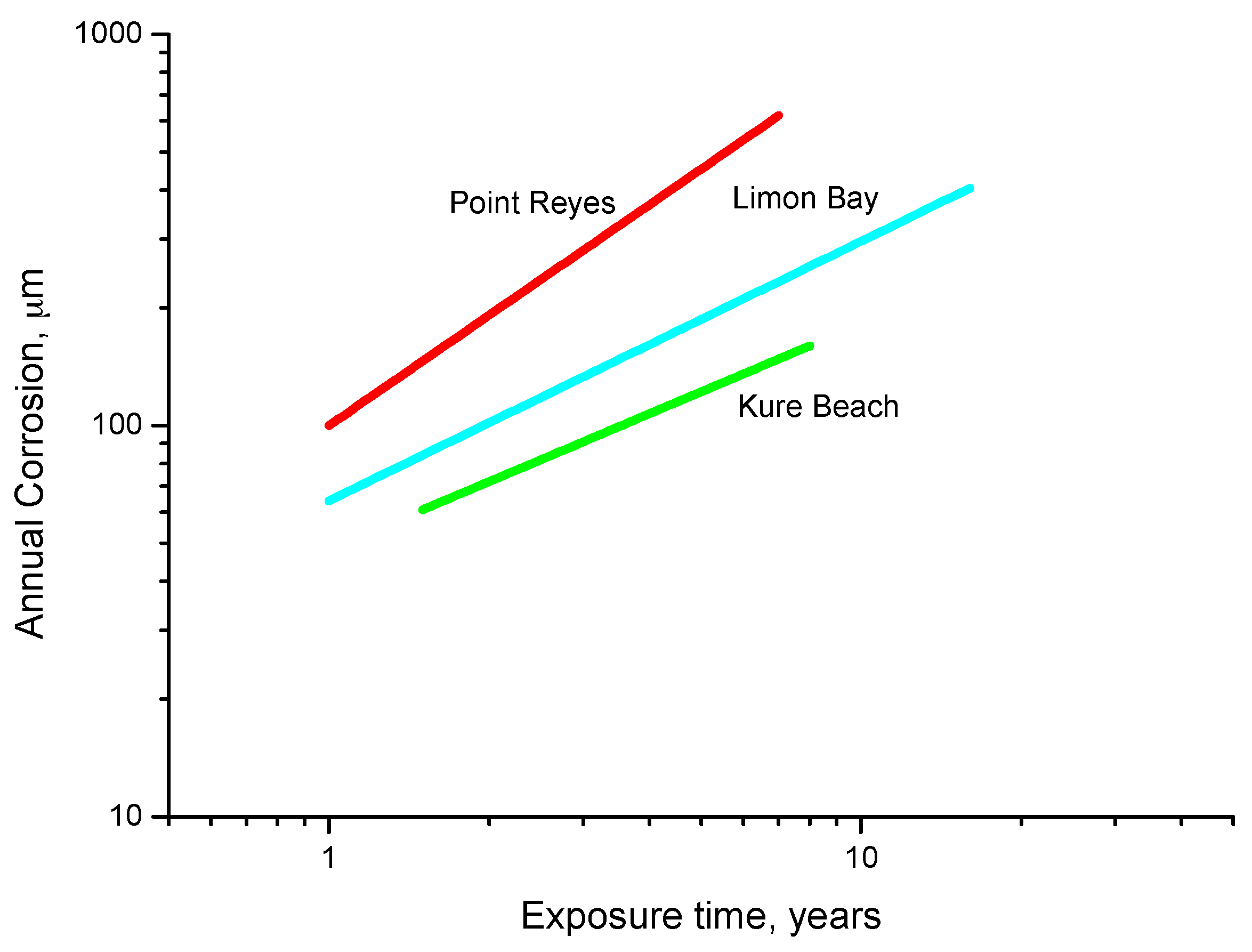
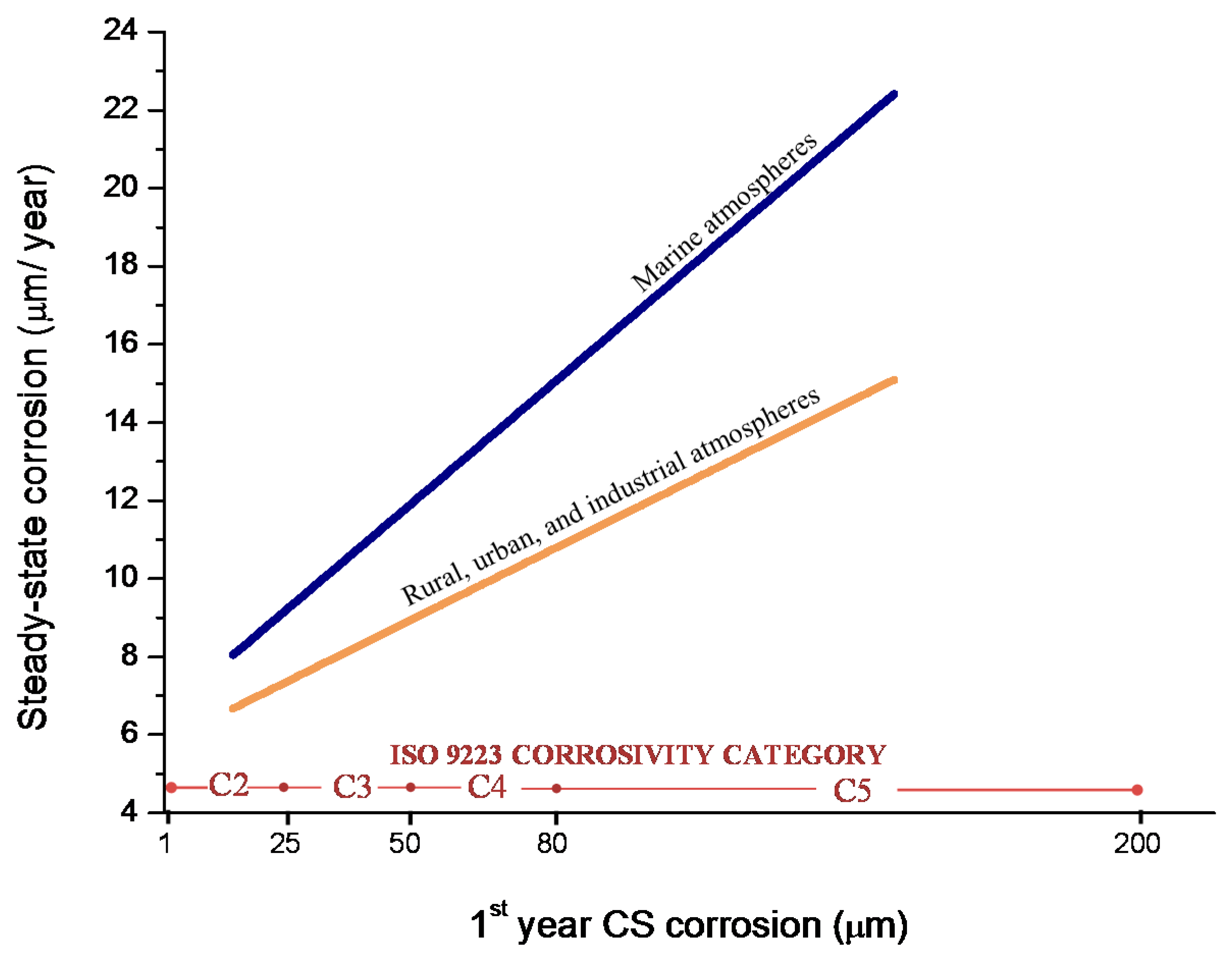
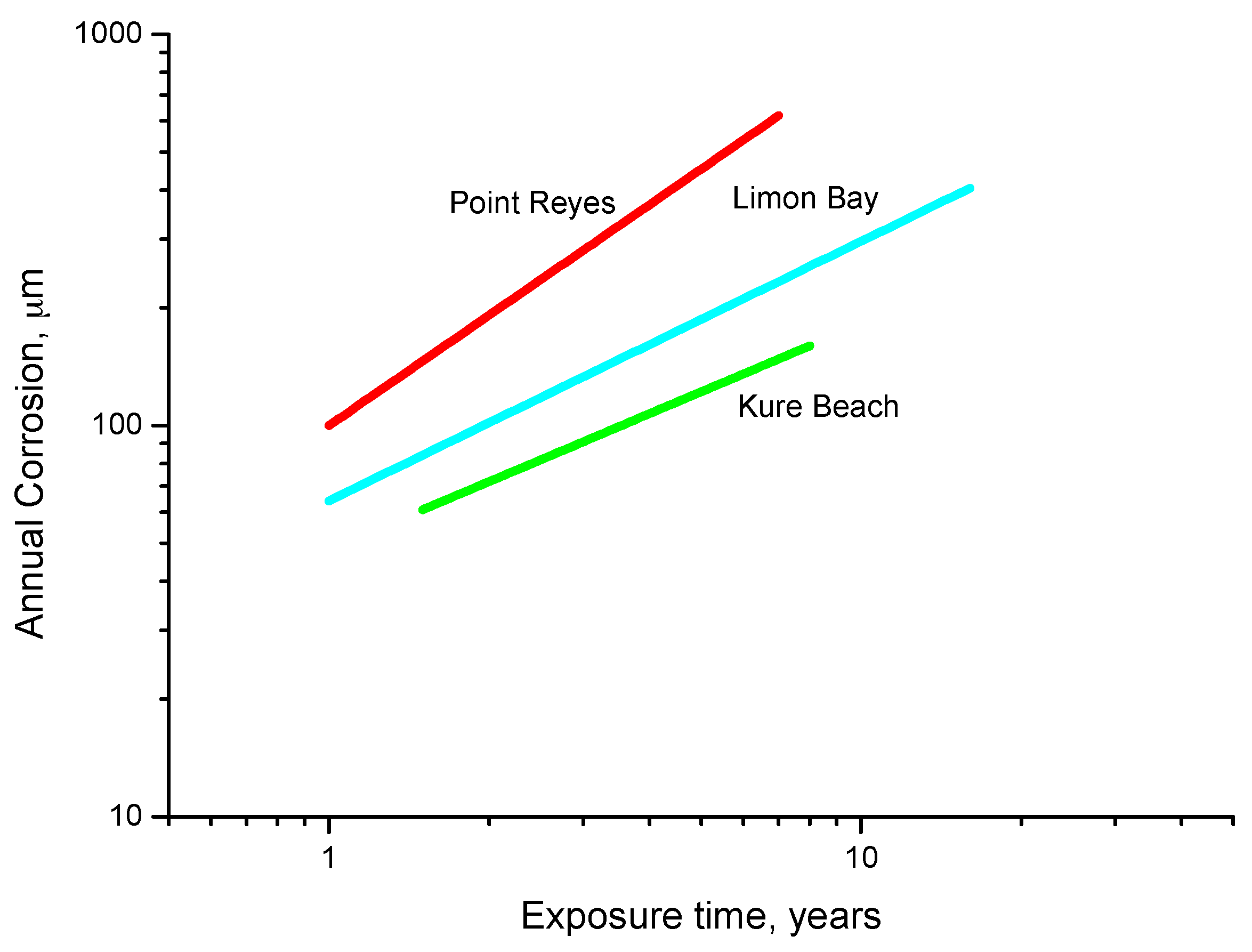
9.3. Long-Term Steel Corrosion
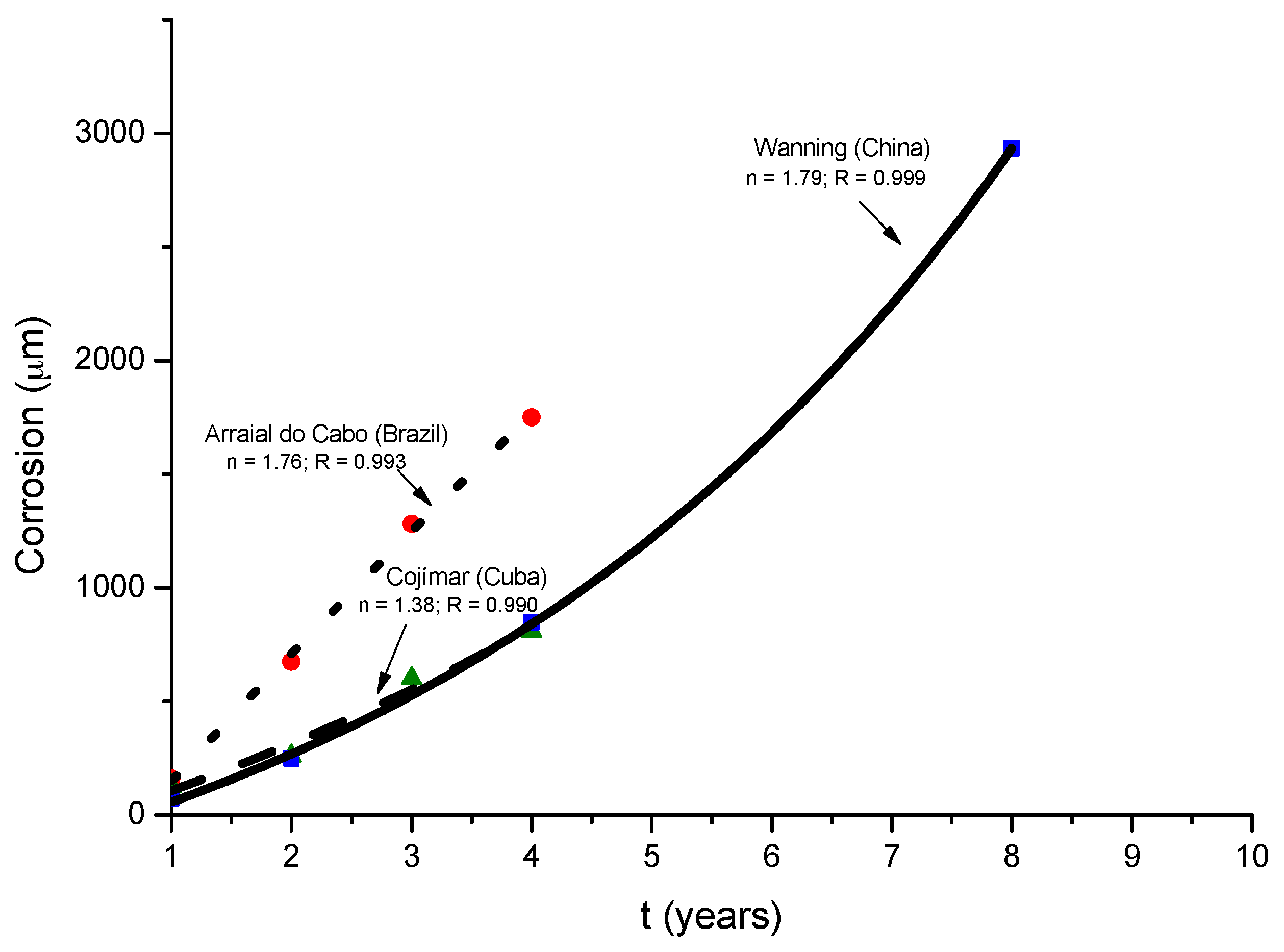
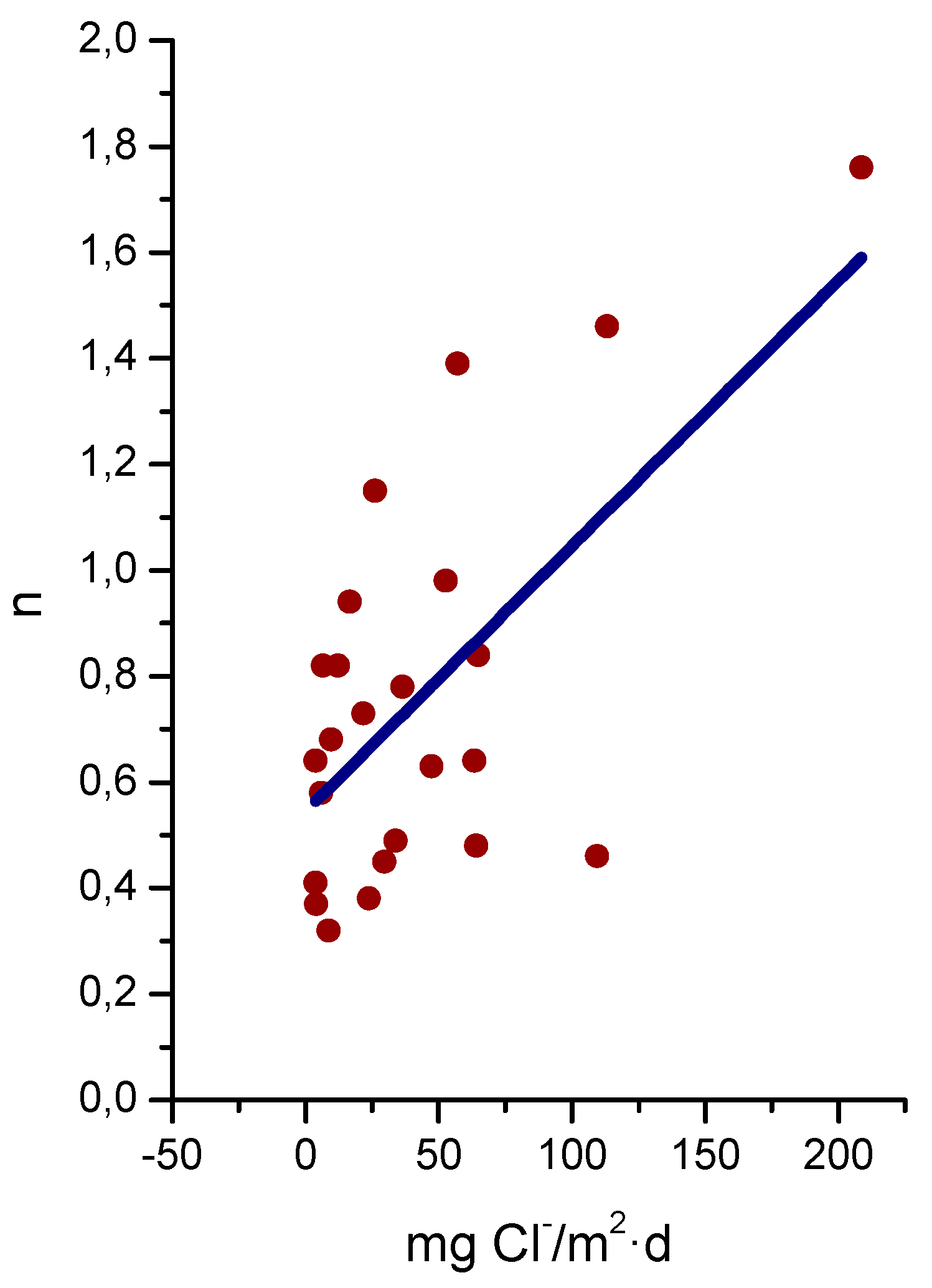
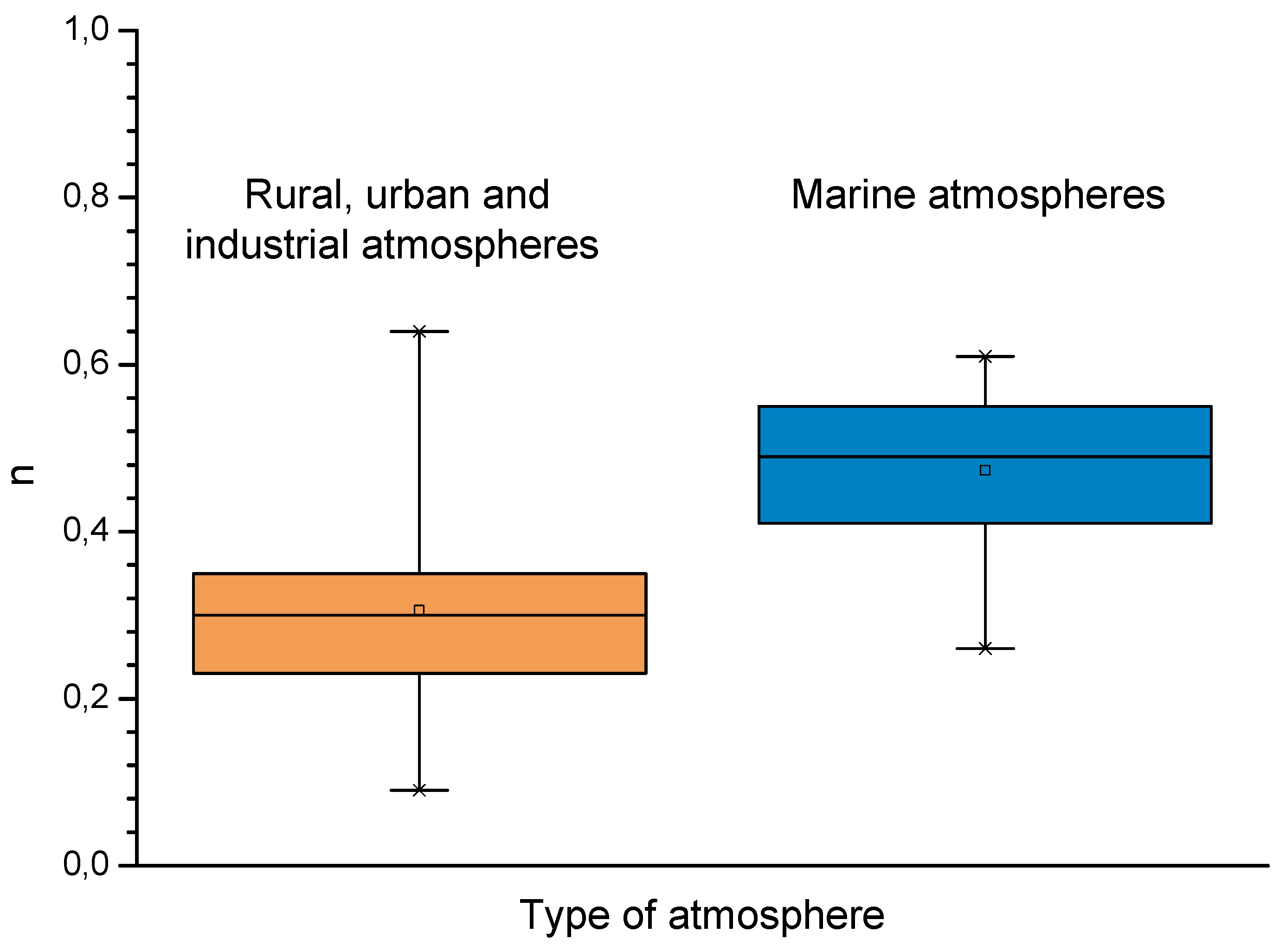
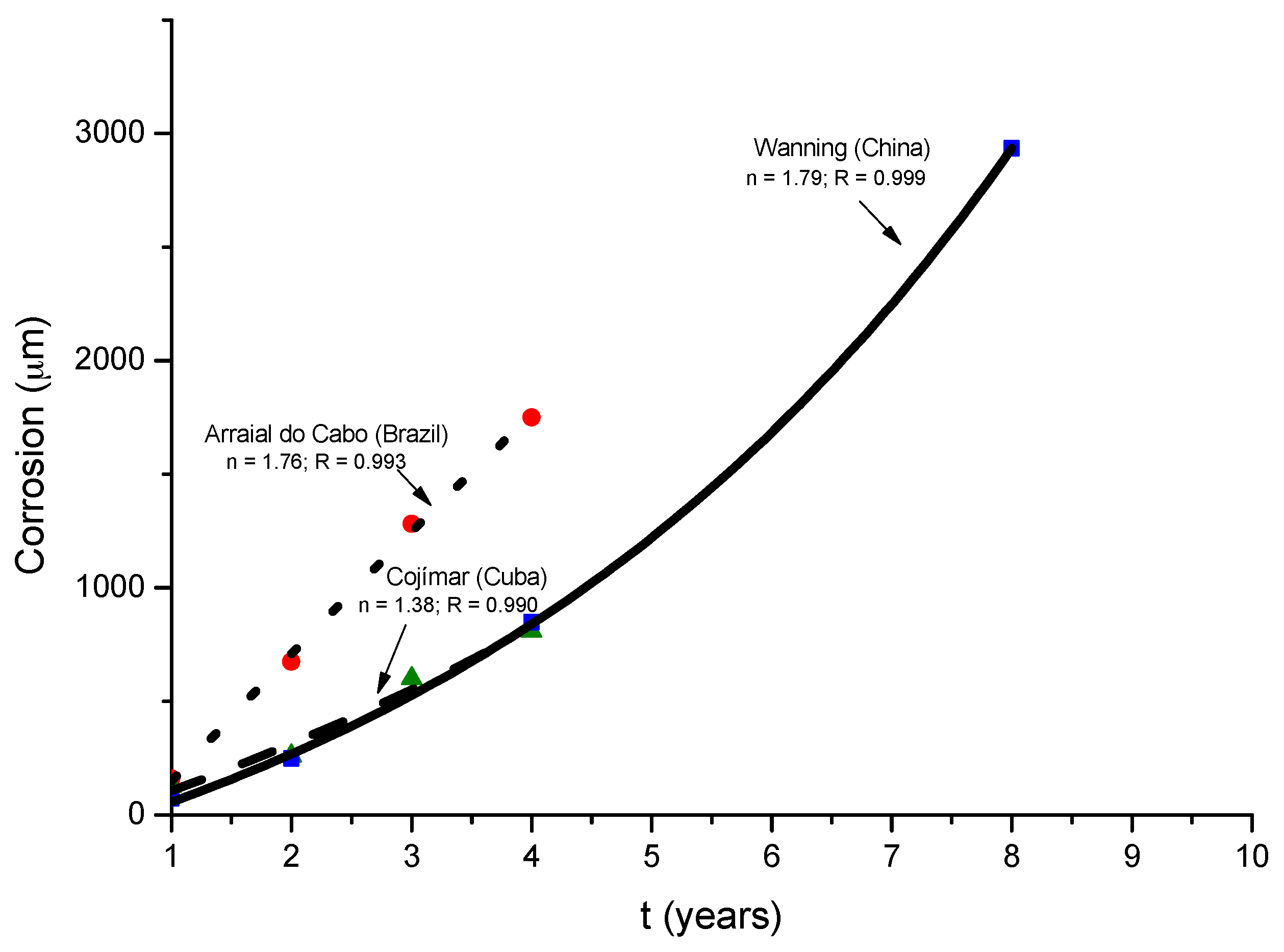
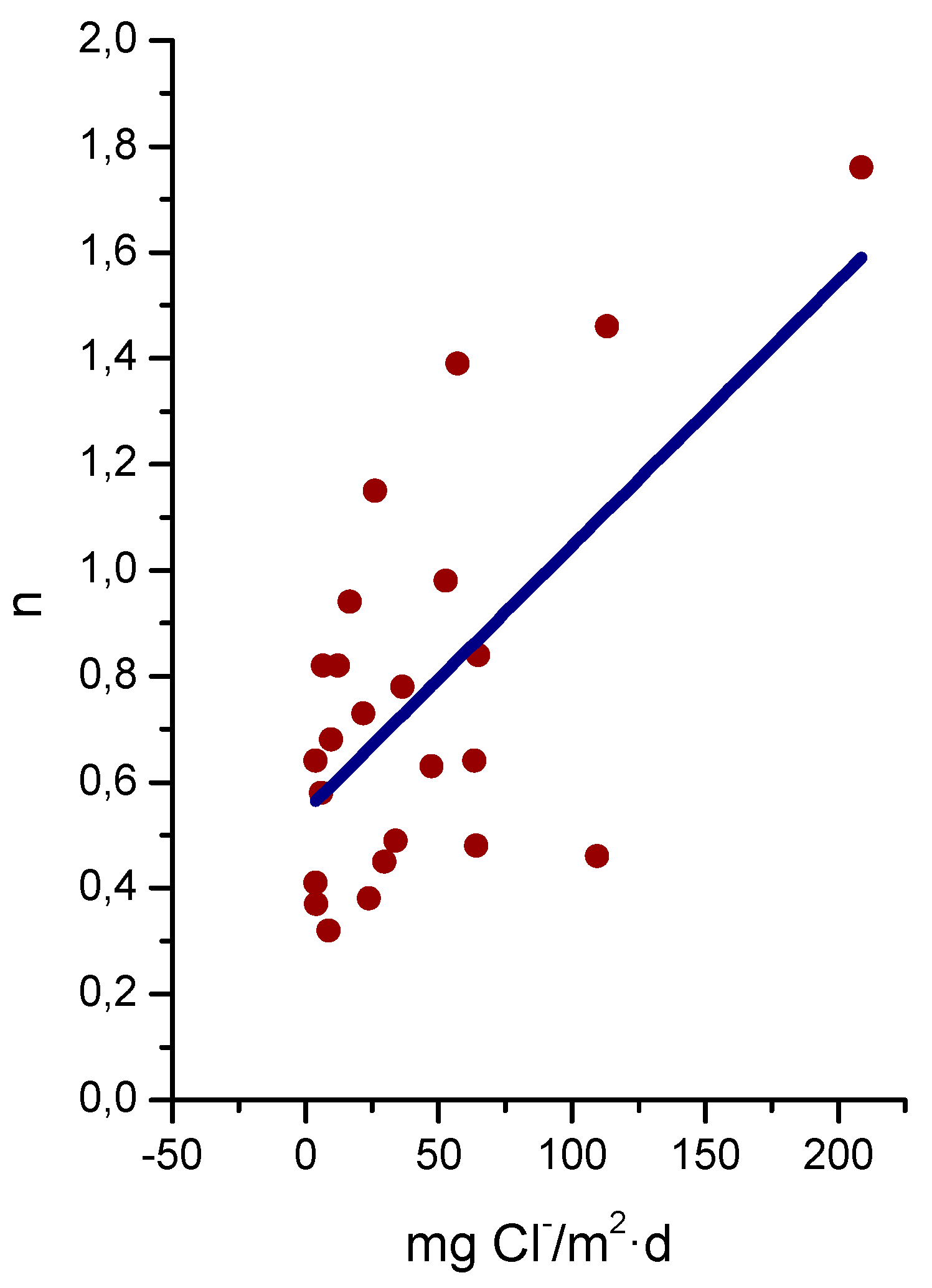
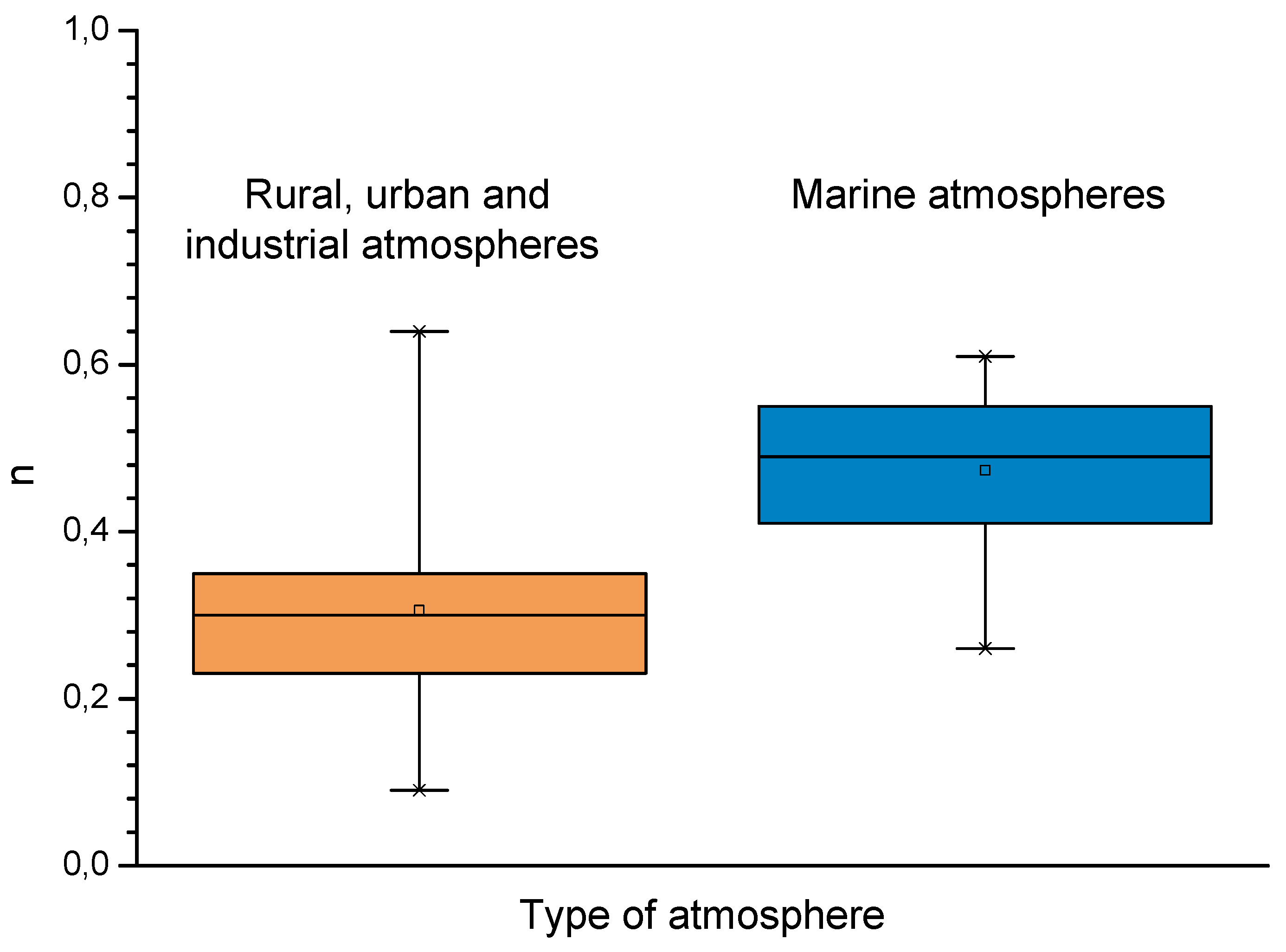
9.4. Modelling of the Long-Term Atmospheric Corrosion Process
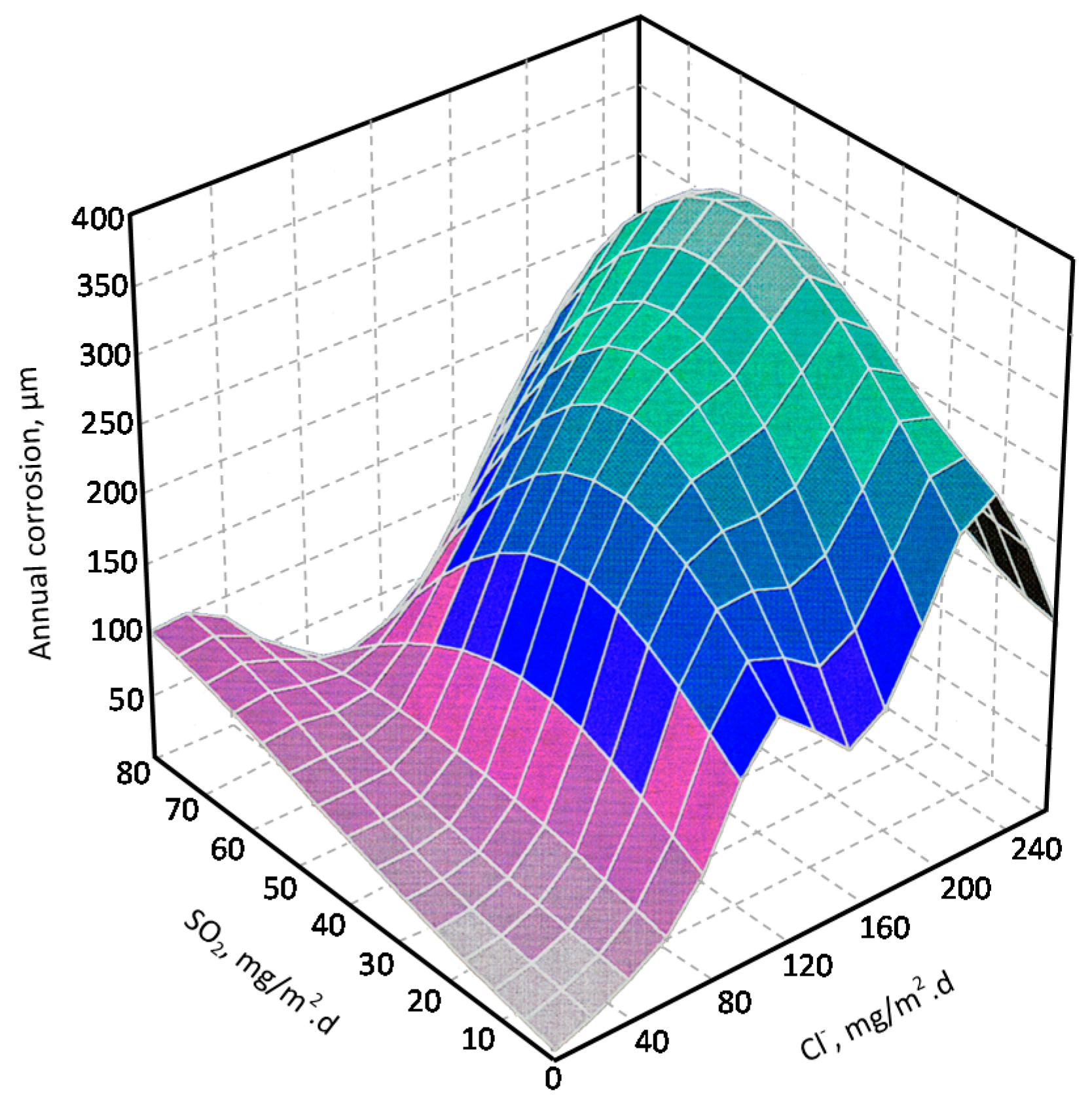
9.5. Towards an Estimation of the Marine Atmospheric Corrosion of Steel Based on Existing Environmental Parameters
9.5.1. Time of Wetness
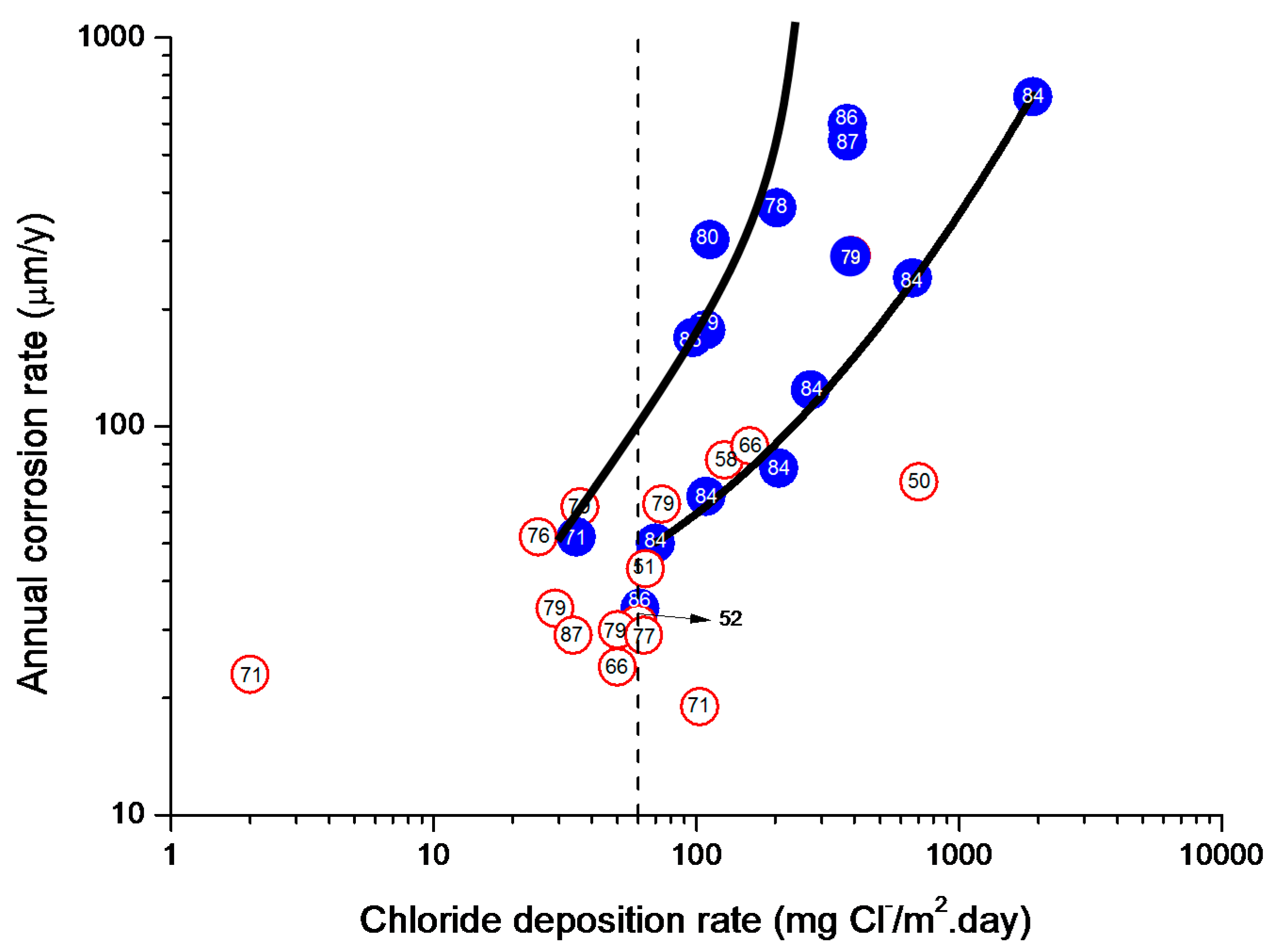
9.5.2. Chloride Deposition Rate
10. Issues Pending
- Experimentation in this field is carried out at atmospheric corrosion testing stations and in the laboratory by means of wet/dry cyclic tests. A great amount of research is under way using both methods, but there are two issues of enormous importance that are not being paid sufficient attention: (a) experimentation in marine atmospheres with high Cl− ion deposition rates (>500 mg/m2·d), where very little information is available; and (b) the standardisation of wet/dry laboratory cyclic tests by means of more specific codes in order to make research results more comparable.It would also be necessary to undertake more research in the case of marine-industrial atmospheres, where there are great discrepancies among researchers. This is a particularly important issue in developing countries where factories are often located in coastal regions.
- One currently unresolved question is concerned with the presence of the amorphous phase in the rust layer and the evolution of its amount with exposure time. With regard to the role played in corrosion mechanisms by the less crystalline phases of rust (ferrihydrite, feroxyhyte, etc.), despite the enormous research effort that has been carried out in recent years by a number of research teams using highly sophisticated analytical techniques, there are still numerous gaps in knowledge.Another matter that generates a great deal of uncertainty is the differentiation of magnetite and maghemite phases, both of which are very similar in many of their characteristics, but which can play a different role in the MAC process.
- In the rust layers formed, aspects such as decoupling of the anodic and cathodic corrosion reactions, localisation, connectivity and reactivity of the different rust phases inside the corrosion layers, as well as characteristics such as porosity, tortuosity, etc., are also of primary importance in the determination of corrosion mechanisms.At high Cl− ion deposition rates, rust layers can exfoliate and become detached from the steel substrate. Although great advances have recently been made in this field, there are still a number of basic aspects that remain to be clarified in order for a complete comprehension of rust exfoliation phenomena.
- Finally, a matter of enormous technical importance for engineers and political policy-makers is to be able to predict steel corrosion rate well into the future (20, 50, 100 years). Data mining and modelling tools can help to improve forecasts and anti-corrosive designs, but despite great progress in the development of damage functions (dose-response) in wide-scale international cooperative research programmes, there is still a long way to go for such long-term modelling of atmospheric corrosion processes.In this sense, better scientific knowledge is needed towards the desirable goal of being able to estimate atmospheric salinity in a given geographic area without the need for measurements involving marine aerosol capturing techniques.
Conflicts of Interest
References
- Evans, U.R. The Corrosion and Oxidation of Metals: Scientific Principles and Practical Applications; Edward Arnold Ltd.: London, UK, 1960. [Google Scholar]
- Tomashov, N.D. Theory of Corrosion and Protection of Metals. The Science of Corrosion; The MacMillan Co.: New York, NY, USA, 1966. [Google Scholar]
- Rozenfeld, I.L. Atmospheric Corrosion of Metals; NACE: Houston, TX, USA, 1972. [Google Scholar]
- Barton, K. Protection against Atmospheric Corrosion; John Wiley and Sons: New York, NY, USA, 1976. [Google Scholar]
- Feliu, S.; Morcillo, M. Corrosión y Protección de los Metales en la Atmósfera; Bellaterra: Barcelona, Spain, 1982. [Google Scholar]
- Graedel, T.E.; Mc Gill, R. Degradation of materials in the atmosphere. Environ. Sci. Technol. 1986, 20, 1093–1100. [Google Scholar] [CrossRef]
- Kucera, V.; Mattsson, E. Atmospheric corrosion. In Corrosion Mechanisms; Mansfeld, F., Ed.; Marcel Dekker: New York, NY, USA, 1987; pp. 211–284. [Google Scholar]
- Leygraf, C.; Odnevall Wallinder, I.; Tidblad, J.; Graedel, T. Atmospheric Corrosion, 2nd ed.; The Electrochemical Society Series; John Wiley and Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Ambler, H.R.; Bain, A.A.J. Corrosion of metals on the tropics. J. Appl. Chem. 1955, 5, 437–527. [Google Scholar] [CrossRef]
- Nishimura, T.; Katayama, H.; Noda, K.; Kodama, T. Electrochemical behavior of rust formed on carbon steel in a wet/dry environment containing chloride ions. Corrosion 2000, 56, 935–941. [Google Scholar] [CrossRef]
- Morcillo, M.; Díaz, I.; Chico, B.; Cano, H.; de la Fuente, D. Wethearing steels: From empirical development to scientific desing. A review. Corros. Sci. 2014, 83, 6–31. [Google Scholar] [CrossRef]
- Morcillo, M.; Chico, B.; Alcántara, J.; Díaz, I.; Simancas, J.; de la Fuente, D. Atmospheric corrosion of mild steel in chloride—Rich environments. Questions to be answered. Mater. Corros. 2015, 66, 882–892. [Google Scholar]
- Cole, I.S.; Ganther, W.D.; Sinclair, J.D.; Lau, D.; Paterson, D.A. A study of the wetting of metal surfaces in order to understand the process controlling atmospheric corrosion. J. Electrochem. Soc. 2004, 151, B627–B635. [Google Scholar] [CrossRef]
- Schindelholz, E.; Risteen, B.E.; Kelly, R.G. Effect of relative humidity on corrosion of steel under sea salt aerosol proxies: I. NaCl. J. Electrochem. Soc. 2014, 161, C450–C459. [Google Scholar] [CrossRef]
- Schindelholz, E.; Risteen, B.E.; Kelly, R.G. Effect of relative humidity on corrosion of steel under sea salt aerosol proxies: II. MgCl2, artificial seawater. J. Electrochem. Soc. 2014, 161, C460–C470. [Google Scholar] [CrossRef]
- Schindelholz, E.; Kelly, R.G. Wetting phenomena and time of wetness in atmospheric corrosion: A review. Corros. Rev. 2012, 30, 135–170. [Google Scholar] [CrossRef]
- Costa, J.M.; Morcillo, M.; Feliu, S. Effect of environmental parameters on atmospheric corrosion of metals. In Encyclopedia of Environmental Control Technology: Air Pollution Control; Cheremisinoff, P.N., Ed.; Gulf Publishing Company: Houston, TX, USA, 1989; Volume 2, pp. 197–238. [Google Scholar]
- Rozenfeld, I.L. Atmospheric corrosion of metals. Some questions of theory. In Proceedings of the 1st International Congress on Metallic Corrosion, London, UK, 10–15 April 1961; pp. 243–253. [Google Scholar]
- Tidblad, J.; Kucera, V.; Ferm, M.; Kreislova, K.; Brüggerhoff, S.; Doytchinov, S.; Screpanti, A.; Grøntoft, T.; Yates, T.; de la Fuente, D.; et al. Effects of air pollution on materials and cultural heritage: ICP materials celebrates 25 years of research. Int. J. Corros. 2012, 2012. [Google Scholar] [CrossRef]
- Haagenrud, S.; Ottar, B. Long range transport of air pollutants and corrosion effects. In Proceedings of the 7th Scandinavian Corrosion Congress, Trondheim, Norway, May 1975; p. 102. [Google Scholar]
- Kucera, V. Effects of sulphur dioxide and acid precipitation on metals and anti-rust painted steel. Ambio 1976, 5, 243–248. [Google Scholar]
- Feliu, S.; Morcillo, M. Corrosión atmosférica. In Corrosión y Protección metáLicas; Andrade, M.C., Feliu, S., Eds.; CSIC (Colección Nuevas Tendencias): Madrid, Spain, 1991; Volume II. [Google Scholar]
- Burstein, G.T. Passivity and localised corrosion. In Corrosion. Metal/Environment Reactions, 3rd ed.; Shreir, L.L., Jarman, R.A., Burstein, G.T., Eds.; Butterworth-Heinemann: Oxford, UK, 1994; Volume 1, p. 1:146. [Google Scholar]
- Feitknecht, W. The breakdown of oxide films on metal surfaces in acidic vapors and the mechanism of atmospheric corrosion. Chimia 1952, 6, 3–13. [Google Scholar]
- Askey, A.; Lyon, S.B.; Thompson, G.E.; Johnson, J.B.; Wood, G.C.; Cooke, M.; Sage, P. The corrosion of iron and zinc by atmospheric hydrogen chloride. Corros. Sci. 1993, 34, 233–247. [Google Scholar] [CrossRef]
- Schikorr, G. On the mechanism of atmospheric corrosion of iron. Werkst. Korros. 1963, 14, 63–80. [Google Scholar]
- International Organization for Standardization. ISO 8565, Metals and Alloys—Atmospheric Corrosion Testing—General Requirements; International Organization for Standardization: Geneva, Switzerland, 2011. [Google Scholar]
- American Society for Testing and Materials. ASTM G50, Conducting Atmospheric Corrosion Tests on Metals; American Society for Testing and Materials: Philadelphia, PA, USA, 1991. [Google Scholar]
- Knotkova, D.; Kreislova, K.; Dean, S.W.J. ISOCORRAG. International Atmospheric Exposure Program: Summary of Results; ASTM: West Conshohocken, PA, USA, 2010. [Google Scholar]
- European Committee for Standardization. EN ISO 9223, Corrosion of Metals and Alloys—Corrosivity of Atmospheres—Classification, Determination and Estimation; European Committee for Standardization: Brussels, Belgium, 2012. [Google Scholar]
- Compton, K.G.; Mendizza, A.; Bradley, W.W. Atmospheric galvanic couple corrosion. Corrosion 1955, 11, 35–44. [Google Scholar] [CrossRef]
- Doyle, D.P.; Godard, H.P. Rapid determination of corrosivity of an atmosphere to aluminium. In Proceedings of the 3rd International Congress on Metallic Corrosion; MIR Publishers: Moscow, Russia, 1969; Volume IV, pp. 429–437. [Google Scholar]
- Doyle, D.P.; Godard, H.P. A rapid method for determining the corrosivity of the atmosphere at any location. Nature 1963, 200, 1167–1168. [Google Scholar] [CrossRef]
- Haynes, G. Cabinet. In Corrosion Tests and Standards. Application and Interpretation; Baboian, R., Ed.; American Society for Testing and Materials: Philadelphia, PA, USA, 1995; pp. 91–97. [Google Scholar]
- American Society for Testing and Materials. ASTM B117, Test Method of Salt Spray (Fog) Testing; American Society for Testing and Materials: Philadelphia, PA, USA, 2011. [Google Scholar]
- International Organization for Standardization. ISO 11130, Corrosion of Metals and Alloys—Alternate Immersion Test in Salt Solution; International Organization for Standardization: Geneve, Switzerland, 1999. [Google Scholar]
- González, J.A. Control de la Corrosión. Estudio y Medida por téCnicas Electroquímicas; CSIC: Madrid, Spain, 1989. [Google Scholar]
- Stratmann, M. The atmospheric corrosion of iron steel. Metal Odlew 1990, 16, 46–52. [Google Scholar]
- Nishimura, T.; Tanaka, K.; Shimizu, Y. Effect of NaCl on rusting of steel in wet and dry corrosion cycle. J. Iron Steel Inst. Jpn. 1995, 81, 1079–1084. [Google Scholar]
- Lair, V.; Antony, H.; Legrand, L.; Chaussé, A. Electrochemical reduction of ferric corrosion products and evaluation of galvanic coupling with iron. Corros. Sci. 2006, 48, 2050–2063. [Google Scholar] [CrossRef]
- Hœrlé, S.; Mazaudier, F.; Dillmann, P.; Santarini, G. Advances in understanding atmospheric corrosion of iron. II. Mechanistic modelling of wet–dry cycles. Corros. Sci. 2004, 46, 1431–1465. [Google Scholar]
- Cook, D.C. Spectroscopic identification of protective and non-protective corrosion coatings on steel structures in marine environments. Corros. Sci. 2005, 47, 2550–2570. [Google Scholar] [CrossRef]
- De Faria, D.L.A.; Venâncio Silva, S.; de Oliveira, M.T. Raman microspectroscopy of some iron oxides and oxyhydroxides. J. Raman Spectrosc. 1997, 28, 873–878. [Google Scholar] [CrossRef]
- Monnier, J.; Réguer, S.; Foy, E.; Testemale, D.; Mirambet, F.; Saheb, M.; Dillmann, P.; Guillot, I. XAS and XRD in situ characterisation of reduction and reoxidation processes of iron corrosion products involved in atmospheric corrosion. Corros. Sci. 2014, 78, 293–303. [Google Scholar] [CrossRef]
- Kimura, M.; Mizoguchi, T.; Kihira, H.; Kaneko, M. Various scale analyses to create functioning corrosion products. In Characterization of Corrosion Products on Steel Surfaces; Waseda, Y., Suzuki, S., Eds.; Advances in Materials Research; Springer: Heildelberg, Germany, 2006; pp. 245–272. [Google Scholar]
- Konishi, H.; Yamashita, M.; Uchida, H.; Mizuki, J. Characterization of rust layer formed on Fe, Fe-Ni and Fe-Cr alloys exposed to Cl-rich environment by Cl and Fe k-edge XANES measurements. Mater. Trans. 2005, 46, 329–336. [Google Scholar] [CrossRef]
- Dillmann, P.; Mazaudier, F.; Hoerlé, S. Advances in understanding atmospheric corrosion of iron. I. Rust characterization of ancient ferrous artefacts exposed to indoor atmospheric corrosion. Corros. Sci. 2004, 46, 1401–1429. [Google Scholar]
- Ishikawa, T. Asessment of rust layers formed on WS in saline environment by gas adsorption. Mater. Corros. 2015, 66, 1460–1466. [Google Scholar] [CrossRef]
- Cole, I.S.; Lau, D.; Paterson, D.A. Holistic model for atmospheric corrosion part 6—From wet aerosol to salt deposit. Corros. Eng. Sci. Technol. 2004, 39, 209–218. [Google Scholar] [CrossRef]
- Johnson, E.; Stanners, J.F. The Characterisation of Corrosion Test Sites in the Community; Commission of the European Communities: Luxembourg, 1981. [Google Scholar]
- Blanchard, D.C.; Woodcock, A.H. The production, concentration and vertical distribution of the sea-salt aerosol. Ann. N. Y. Acad. Sci. 1980, 338, 330–347. [Google Scholar] [CrossRef]
- Meira, G.R.; Pinto, W.T.A.; Lima, E.E.P.; Andrade, C. Vertical distribution of marine aerosol salinity in a brazilian coastal area—The influence of wind speed and the impact on chloride accumulation into concrete. Constr. Build. Mater. 2017, 135, 287–296. [Google Scholar] [CrossRef]
- Zezza, F.; Macrì, F. Marine aerosol and stone decay. Sci. Total Environ. 1995, 167, 123–143. [Google Scholar] [CrossRef]
- Whitby, K.T. The physical characteristics of sulfate aerosols. Atmos. Environ. 1978, 12, 135–159. [Google Scholar] [CrossRef]
- Li, S.; Hihara, L.H. Aerosol salt particle deposition of metals exposed to marine environments: A study related to marine atmospheric corrosion. J. Electrochem. Soc 2014, 161, C268–C275. [Google Scholar] [CrossRef]
- Gustafsson, M.E.R.; Franzén, L.G. Dry deposition and concentration of marine aerosols in a coastal area. Atmos. Environ. 1996, 30, 977–989. [Google Scholar] [CrossRef]
- Cole, I.S.; Azmat, N.S.; Kanta, A.; Venkatraman, M. What really controls the atmospheric corrosion of zinc? Effect of marine aerosols on atmospheric corrosion of zinc. Int. Mater. Rev. 2009, 54, 117–133. [Google Scholar]
- Fitzgerald, J.W. Marine aerosol: A review. Atmos. Environ. 1991, 25A, 533–545. [Google Scholar] [CrossRef]
- Wu, J. Evidence of sea spray produced by bursting bubbles. Science 1981, 212, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Feliu, S.; Morcillo, M.; Chico, B. Effect of distance from sea on atmospheric corrosion rate. Corrosion 1999, 55, 883–891. [Google Scholar] [CrossRef]
- Ohba, R.; Okabayashi, K.; Yamamoto, M.; Tsuru, T. A method for predicting the content of sea salt particles in the atmosphere. Atmos. Environ. 1990, 24A, 925–935. [Google Scholar] [CrossRef]
- Ten Harkel, M.J. The effects of particle-size distribution and chloride depletion of sea-salt aerosols on estimating atmospheric deposition at a coastal site. Atmos. Environ. 1997, 31, 417–427. [Google Scholar] [CrossRef]
- McKay, W.A.; Garland, J.A.; Livesley, D.; Halliwell, C.M.; Walker, M.I. The characteristics of the shore-line sea spray aerosol and the landward transfer of radionuclides discharged to coastal sea water. Atmos. Environ. 1994, 28, 3299–3309. [Google Scholar] [CrossRef]
- Alcántara, J.; Chico, B.; Díaz, I.; de la Fuente, D.; Morcillo, M. Airborne chloride deposit and its effect on marine atmospheric corrosion of mild steel. Corros. Sci. 2015, 97, 74–88. [Google Scholar] [CrossRef]
- Strekalov, P.V.; Panchenko, Y.M. The role of marine aerosols in atmospheric corrosion of metals. Prot. Met. 1994, 30, 254–263. [Google Scholar]
- Morcillo, M.; Chico, B.; Mariaca, L.; Otero, E. Salinity in marine atmospheric corrosion: Its dependence on the wind regime existing in the site. Corros. Sci. 2000, 42, 91–104. [Google Scholar] [CrossRef]
- Meira, G.R.; Andrade, C.; Alonso, C.; Padaratz, I.J.; Borba, J.C., Jr. Salinity of marine aerosols in a brazilian coastal area—Influence of wind regime. Atmos. Environ. 2007, 41, 8431–8441. [Google Scholar] [CrossRef]
- Strekalov, P.V. Wind regimes, chloride aerosol particle sedimentation and atmospheric corrosion of steel and copper. Prot. Met. 1988, 24, 630–641. [Google Scholar]
- Preston, R.S.J.; Sanyal, B. Atmospheric corrosion by nuclei. J. Appl. Chem. 1956, 6, 26–44. [Google Scholar] [CrossRef]
- Evans, U.R.; Taylor, C.A.J. Mechanism of atmospheric rusting. Corros. Sci. 1972, 12, 227–246. [Google Scholar] [CrossRef]
- Morcillo, M.; Chico, B.; Otero, E.; Mariaca, L. Effect of marine aerosol on atmospheric corrosion. Mater. Perform. 1999, 38, 72–77. [Google Scholar]
- Pascual Marqui, R.D. Influencia de la concentración de ion cloruro sobre la corrosión atmosférica de un acero al carbono bajo capa de fase de humedad. Rev. Corros. Prot. 1980, XI, 37–40. [Google Scholar]
- Espada, L.; González, A.M.; Sánchez, A.; Merino, P. Estudio de la velocidad de corrosión de aceros de bajo contenido de carbono en nieblas salinas de distinta concentración. Rev. Iberoam. Corros. Prot. 1988, 19, 227–229. [Google Scholar]
- Hache, A. Contribution à l’étude de la corrosion de l’acier en solutions salines. Rev. Metall. 1956, 53, 76–80. [Google Scholar] [CrossRef]
- Strekalov, P.V. Estimation of atmospheric salinity from analysis of dry and wet chloride precipitates. Prot. Met. 1994, 30, 59–64. [Google Scholar]
- State System for Standardization of Russian Federation. GOST 9.039–74, Unified System of Corrosion and Ageing Protection. Corrosive Aggressiveness of atMosphere; State System for Standardization of Russian Federation: Moscow, Russia, 1974. [Google Scholar]
- Beruksitis, G.K.; Klark, G.B. Corrosion Fatigue of Metals and Metals Coatings under Atmospheric Conditions; Nauka: Moscow, Russia, 1971. [Google Scholar]
- European Committee for Standardization. EN ISO 9225, Corrosion of Metals and Alloys—Corrosivity of Atmospheres—Measurement of Environmental Parameters Affecting Corrosivity of Atmospheres; European Committee for Standardization: Brussels, Belgium, 2012. [Google Scholar]
- Foran, M.R.; Gibbons, E.V.; Wellington, J.R. The measurement of atmospheric sulfur dioxide and chlorides. Chem. Can. 1958, 10, 33–41. [Google Scholar]
- Corvo, F. Atmospheric corrosion of steel in humid tropical climate. Influence of pollution, humidity, temperature, rainfall and sun radiation. Corrosion 1984, 40, 170–175. [Google Scholar]
- Dean, S.W. Atmospheric. In Corrosion Tests and Standards. Application and Interpretation; Baboian, R., Ed.; ASTM: Philadelphia, PA, USA, 1995; pp. 116–125. [Google Scholar]
- Industrial Galvanizers (INGALV). Corrosion Mapping System. Available online: http://www.valmontcoatings.com/locations/asia-pacific (accessed on 12 April 2017).
- Japan Industrial Standard. JIS-Z-2381, Recommended Practice for Weathering Test; Japan Industrial Standard: Tokyo, Japan, 1987. [Google Scholar]
- Hujioara, M.; Danakka, O. Weathering due to the chloride of ferro-concrete structure. Concr. Eng. 1987, 25, 44–47. [Google Scholar]
- Li, Q.X.; Wang, Z.Y.; Han, W.; Han, E.H. Characterization of the rust formed on weathering steel exposed to Qinghai salt lake atmosphere. Corros. Sci. 2008, 50, 365–371. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.Y.; Ke, W. A study of the evolution of rust on weathering steel submitted to the Qinghai salt lake atmospheric corrosion. Mater. Chem. Phys. 2013, 139, 225–232. [Google Scholar] [CrossRef]
- Morcillo, M.; Chico, B.; Díaz, I.; Cano, H.; de la Fuente, D. Atmospheric corrosion data of weathering steels. A review. Corros. Sci. 2013, 77, 6–24. [Google Scholar] [CrossRef]
- Takebe, M.; Ohya, M.; Ajiki, S.; Furukawa, T.; Adachi, R.; Gan-ei, R.; Kitagawa, N.; Ota, J.; Matsuzaki, Y.; Aso, T. Estimation of quantity of Cl− from deicing salts on weathering steel used for bridges. Int. J. Steel Struct. 2008, 8, 73–81. [Google Scholar]
- Takebe, M.; Matsuzaki, Y.; Ohya, M.; Ajiki, S.; Furukawa, T.; Aso, T. Study of corrosion level and composition of accumulating salt on weathering bridges. J. JSCE 2007, 63, 172–180. [Google Scholar] [CrossRef]
- Hara, S.; Miura, M.; Uchiumi, Y.; Fujiwara, T.; Yamamoto, M. Suppression of deicing salt corrosion of weathering steel bridges by washing. Corros. Sci. 2005, 47, 2419–2430. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, 2nd ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2003. [Google Scholar]
- Gilberg, M.R.; Seeley, N.J. The identity of compounds containing chloride ions in marine iron corrosion products: A critical review. Stud. Conserv. 1981, 26, 50–56. [Google Scholar] [CrossRef]
- Monnier, J.; Neff, D.; Réguer, S.; Dillmann, P.; Bellot-Gurlet, L.; Leroy, E.; Foy, E.; Legrand, L. A corrosion study of the ferrous medieval reinforcement of the amiens cathedral. Phase characterisation and localisation by various microprobes techniques. Corros. Sci. 2010, 52, 695–710. [Google Scholar] [CrossRef]
- Neff, D.; Reguer, S.; Bellot-Gurlet, L.; Dillmann, P. Structural characterization of corrosion products on archaeological iron: An integrated analytical approach to establish corrosion forms. J. Raman Spectrosc. 2004, 35, 739–745. [Google Scholar] [CrossRef]
- Pino, E.; Alcántara, J.; Chico, B.; Díaz, I.; Simancas, J.; de la Fuente, D.; Morcillo, M. Atmospheric corrosion of mild steel in marine atmospheres. Corros. Prot. Mater. 2015, 34, 35–41. [Google Scholar]
- Bernal, J.D.; Dasgupta, D.R.; Mackay, A.L. The oxides and hydroxides of iron and their structural inter-relationships. Clay Miner. Bull. 1959, 4, 15–30. [Google Scholar] [CrossRef]
- Arroyave, C.; Morcillo, M. Atmospheric corrosion products in iron and steels. Trends Corros. 1997, 2, 1–16. [Google Scholar]
- Butler, G.; Beynon, J.G. The corrosion of mild steel in boiling salt solutions. Corros. Sci. 1967, 7, 385–404. [Google Scholar] [CrossRef]
- Refait, P.; Abdelmoula, M.; Génin, J.M.R. Mechanisms of formation and structure of green rust one in aqueous corrosion of iron in the presence of chloride ions. Corros. Sci. 1998, 40, 1547–1560. [Google Scholar] [CrossRef]
- Stampfl, P.P. Ein basisches eisen-II-III-karbonat in rost. Corros. Sci. 1969, 9, 185–187. [Google Scholar] [CrossRef]
- Ståhl, K.; Nielsen, K.; Jiang, J.; Lebech, B.; Hanson, J.C.; Norby, P.; van Lanschot, J. On the akaganéite crystal structure, phase transformations and possible role in post-excavational corrosion of iron artifacts. Corros. Sci. 2003, 45, 2563–2575. [Google Scholar] [CrossRef]
- Watson, J.H.L.; Cardell, R.R.; Heller, W. The internal structure of colloidal crystals of β-Feooh and remarks on their assemblies in schiller layers. J. Phys. Chem. 1962, 66, 1757–1763. [Google Scholar] [CrossRef]
- Keller, P. Occurrence, formation and phase transformation of β-Feooh in rust. Werkst. Korros. 1969, 20, 102–108. [Google Scholar] [CrossRef]
- Rezel, D.; Genin, J.M.R. The substitution of chloride ions to OH−-ions in the akaganeite beta ferric oxyhydroxide studied by Mössbauer effect. Hyperfine Interact. 1990, 57, 2067–2075. [Google Scholar] [CrossRef]
- Gallagher, K.J. The atomic structure of tubular subcrystals of β-iron (III) oxide hydroxide. Nature 1970, 226, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Post, J.E.; Buchwald, V.F. Crystal-structure refinement of akaganeite. Am. Mineral. 1991, 76, 272–277. [Google Scholar]
- Mackay, A.L. β-ferric oxyhydroxide. Mineral. Mag. 1960, 32, 545–557. [Google Scholar] [CrossRef]
- Mackay, A.L. β-ferric oxyhydroxide—Akaganeite. Mineral. Mag. 1962, 33, 270–280. [Google Scholar] [CrossRef]
- Shiotani, K.; Tanimoto, W.; Maeda, C.; Kawabata, F.; Amano, K. Structural analysis of the rust layer on a bare weathering steel bridge exposed in a coastal industrial zone for 27 years. Corros. Eng. 2000, 49, 99–109. [Google Scholar] [CrossRef]
- Morcillo, M.; Chico, B.; Alcántara, J.; Díaz, I.; Wolthuis, R.; de la Fuente, D. SEM/micro-Raman characterization of the morphologies of marine atmospheric corrosion products formed on mild steel. J. Electrochem. Soc. 2016, 163, C426–C439. [Google Scholar] [CrossRef]
- De la Fuente, D.; Alcántara, J.; Chico, B.; Díaz, I.; Jiménez, J.A.; Morcillo, M. Characterisation of rust surfaces formed on mild steel exposed to marine atmospheres using XRD and SEM/micro-Raman techniques. Corros. Sci. 2016, 110, 253–264. [Google Scholar] [CrossRef]
- Rémazeilles, C.; Refait, P. Formation, fast oxidation and thermodynamic data of Fe(II) hydroxychlorides. Corros. Sci. 2008, 50, 856–864. [Google Scholar] [CrossRef]
- Refait, P.; Genin, J.M.R. The mechanism of oxidation of ferrous hydroxychloride β-Fe2(OH)3Cl in aqueous solution: The formation of akaganeite vs goethite. Corros. Sci. 1997, 39, 539–553. [Google Scholar] [CrossRef]
- Rémazeilles, C.; Refait, P. On the formation of β-FeOOH (akaganéite) in chloride-containing environments. Corros. Sci. 2007, 49, 844–857. [Google Scholar] [CrossRef]
- Refait, P.; Génin, J.M.R. The oxidation of ferrous hydroxide in chloride-containing aqueous media and Pourbaix diagrams of green rust one. Corros. Sci. 1993, 34, 797–819. [Google Scholar] [CrossRef]
- Wells, A.F. Structural Inorganic Chemistry, 4th ed.; Oxford University Press: London, UK, 1975. [Google Scholar]
- Fasiska, E.J. Structural aspects of the oxides and oxyhydrates of iron. Corros. Sci. 1967, 7, 833–839. [Google Scholar] [CrossRef]
- Antunes, R.A.; Costa, I.; de Faria, D.L.A. Characterization of corrosion products formed on steels in the first months of atmospheric exposure. Mater. Res. 2003, 6, 403–408. [Google Scholar] [CrossRef]
- Schwarz, H. Über die Wirkung des magnetits beim atmosphärischen Rosten und beim Unterrosten von Anstrichen. Werkst. Korros. 1972, 23, 648–663. [Google Scholar] [CrossRef]
- Singh, A.K. Mössbauer and X-ray diffraction phase analysis of rusts from atmospheric test sites with different environments in Sweden. Corros. Sci. 1985, 25, 931–945. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kondo, Y.; Yasukawa, A.; Kandori, K. Formation of magnetite in the presence of ferric oxyhydroxides. Corros. Sci. 1998, 40, 1239–1251. [Google Scholar] [CrossRef]
- Tanaka, H.; Mishima, R.; Hatanaka, N.; Ishikawa, T.; Nakayama, T. Formation of magnetite rust particles by reacting iron powder with artificial α-, β- and γ-FeOOH in aqueous media. Corros. Sci. 2014, 78, 384–387. [Google Scholar] [CrossRef]
- Hiller, J.E. Phasenumwandlungen im rost. Werkst. Korros. 1966, 17, 943–951. [Google Scholar] [CrossRef]
- Jeffrey, R.J.; Melchers, R.E. The changing composition of the corrosion products of mild steel in severe marine atmospheres. In Proceedings of the Annual Conference of the Australasian Corrosion Association, Melbourne, Australia, 11–14 November 2012. [Google Scholar]
- Haces, C.; Corvo, F.; Furet, N.R. Mecanismo de la corrosión atmosférica del acero en una zona de alta salinidad. In Proceedings of the 3rd Iberoamerican Congress of Corrosion and Protection, ABRACO, Río de Janeiro, Brazil, 26–30 June 1989; Volume 1, pp. 415–419. [Google Scholar]
- Asami, K. Characterization of rust layers on a plain-carbon steel and weathering steels exposed to industrial and coastal atmosphere for years. In Characterization of Corrosion Products on Steel Surfaces; Waseda, Y., Suzuki, S., Eds.; Advances in Materials Research; Springer: Heidelberg, Germany, 2006; pp. 159–197. [Google Scholar]
- Graham, M.J.; Cohen, M. Analysis of iron corrosion products using Mössbauer spectroscopy. Corrosion 1976, 32, 432–438. [Google Scholar] [CrossRef]
- Leidheiser, H., Jr.; Musíc, S. The atmospheric corrosion of iron as studied by Mössbauer spectroscopy. Corros. Sci. 1982, 22, 1089–1096. [Google Scholar] [CrossRef]
- Chico, B.; Alcántara, J.; Pino, E.; Díaz, I.; Simancas, J.; Torres-Pardo, A.; de la Fuente, D.; Jiménez, J.A.; Marco, J.F.; González-Calbet, J.M.; et al. Rust exfoliation on carbon steels in chloride-rich atmopsheres. Corros. Rev. 2015, 33, 263–282. [Google Scholar] [CrossRef]
- Oh, S.J.; Cook, D.C.; Townsend, H.E. Atmospheric corrosion of different steels in marine, rural and industrial environments. Corros. Sci. 1999, 41, 1687–1702. [Google Scholar] [CrossRef]
- Antony, H.; Perrin, S.; Dillmann, P.; Legrand, L.; Chaussé, A. Electrochemical study of indoor atmospheric corrosion layers formed on ancient iron artefacts. Electrochim. Acta 2007, 52, 7754–7759. [Google Scholar] [CrossRef]
- Matsubara, E.; Suzuki, S.; Waseda, Y. Corrosion mechanism of iron from an X-ray structural viewpoint. In Characterization of Corrosion Products on Steel Surfaces; Waseda, Y., Suzuki, S., Eds.; Advances in Materials Research; Springer: Heidelberg, Germany, 2006; pp. 105–129. [Google Scholar]
- Environmental Geochemistry of Ferric Polymers in Aqueous Solutions and Precipitates. Available online: http://geoweb.princeton.edu/research/geochemistry/research/aqueous-polymers.html (accessed on 11 April 2017).
- Waseda, Y.; Suzuki, S.; Saito, M. Structural characterization for a complex system by obtaining middle-range ordering. In Characterization of Corrosion Products on Steel Surfaces; Waseda, Y., Suzuki, S., Eds.; Advances in Materials Research; Springer: Heidelberg, Germany, 2006; pp. 77–104. [Google Scholar]
- Raman, A.; Nasrazadani, S.; Sharma, L. Morphology of rust phases formed on weathering steels in various laboratory corrosion test. Metallography 1989, 22, 79–96. [Google Scholar] [CrossRef]
- Morcillo, M.; Wolthuis, R.; Alcántara, J.; Chico, B.; Díaz, I.; de la Fuente, D. Scanning Electron Microscopy/microRaman: A very useful technique for characterizing the morphologies of rust phases formed on carbon steel in atmospheric exposures. Corrosion 2016, 72, 1044–1054. [Google Scholar]
- Alcántara, J.; Chico, B.; Simancas, J.; Díaz, I.; de la Fuente, D.; Morcillo, M. An attempt to classify the morphologies presented by different rust phases formed during the exposure of carbon steel to marine atmospheres. Mater. Charact. 2016, 118, 65–78. [Google Scholar] [CrossRef]
- Kihira, H. Colloidal aspects of rusting of weathering steel. In Electrical Phenomena at Interfaces, Fundamentals, Measurements and Applications, 2nd ed.; Ohshima, H., Furusawa, K., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1998. [Google Scholar]
- Licheri, G.; Pinna, G. EXAFS and X-ray diffraction in solutions. In EXAFS and Near Edge Structure; Springer: Frascati, Italy, 13–17 September 1982; pp. 240–247. [Google Scholar]
- Jolviet, J.P. Metal Oxide Chemistry and Synthesis; John Wiley & Sons Ltd.: West Sussex, UK, 2000. [Google Scholar]
- Ishikawa, T.; Yoshida, T.; Kandori, K.; Nakayama, T.; Hara, S. Assessment of protective function of steel rust layers by N2 adsorption. Corros. Sci. 2007, 49, 1468–1477. [Google Scholar] [CrossRef]
- Hara, S.; Kamimura, T.; Miyuki, H.; Yamashita, M. Taxonomy for protective ability of rust layer using its composition formed on weathering steel bridge. Corros. Sci. 2007, 49, 1131–1142. [Google Scholar] [CrossRef]
- Calero, J.; Alcántara, J.; Chico, B.; Díaz, I.; Simancas, J.; de la Fuente, D.; Morcillo, M. Wet/dry accelerated laboratory test to simulate the formation of multilayered rust on carbon steel in marine atmospheres. Corros. Eng. Sci. Technol. 2017. [Google Scholar] [CrossRef]
- Ishikawa, T.; Maeda, A.; Kandori, K.; Tahara, A. Characterization of rust on Fe-Cr, Fe-Ni, and Fe-Cu binary alloys by Fourier Transform Infrared and N2 adsorption. Corrosion 2006, 62, 559–567. [Google Scholar] [CrossRef]
- Ishikawa, T.; Katoh, R.; Yasukawa, A.; Kandori, K.; Nakayama, T.; Yuse, F. Influences of metal ions on the formation of β-FeOOH particles. Corros. Sci. 2001, 43, 1727–1738. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kumagai, M.; Yasukawa, A.; Kandori, K. Characterization of rust on weathering steel by gas adsoption. Corrosion 2001, 57, 346–352. [Google Scholar] [CrossRef]
- LaQue, F.L. Corrosion testing. Proc. ASTM 1951, 51, 495–582. [Google Scholar]
- Díaz, I. Corrosión Atmosférica de Aceros Patinables de Nueva Generación. Ph.D. Thesis, Complutense University, Madrid, Spain, October 2012. [Google Scholar]
- Cano, H. Aceros Patinables (Cu, Cr, Ni): Resistencia a la Corrosión Atmosférica y Soldabilidad. Ph.D. Thesis, Complutense University, Madrid, Spain, December 2013. [Google Scholar]
- Díaz, I.; Cano, H.; de la Fuente, D.; Chico, B.; Vega, J.M.; Morcillo, M. Atmospheric corrosion of Ni-advanced weathering steels in marine atmospheres of moderate salinity. Corros. Sci. 2013, 76, 348–360. [Google Scholar] [CrossRef]
- Suzuki, I.; Hisamatsu, Y.; Masuko, N. Nature of atmospheric rust on iron. J. Electrochem. Soc. 1980, 127, 2210–2215. [Google Scholar] [CrossRef]
- De la Fuente, D.; Diaz, I.; Simancas, J.; Chico, B.; Morcillo, M. Long-term atmospheric corrosion of mild steel. Corros. Sci. 2011, 53, 604–617. [Google Scholar] [CrossRef]
- Morcillo, M.; Alcántara, J.; Díaz, I.; Chico, B.; Simancas, J.; de la Fuente, D. Marine atmospheric corrosion of carbon steels. Rev. Metal. Madrid 2015, 51, e045. [Google Scholar] [CrossRef]
- Honzák, J. Schutz von Stahlkonstruktion Gegen Atmospharische Korrosion (Protection of Steel Structures againts Atmospheric Corrosion); 57 Veranstaltung EFK: Prague, Czech Republic, 1971. [Google Scholar]
- Copson, H.R. Atmospheric corrosion of low alloy steels. Proc. ASTM 1952, 52, 1005–1026. [Google Scholar]
- García, K.E.; Morales, A.L.; Barrero, C.A.; Greneche, J.M. On the rusts products formed on weathering and carbon steels exposed to chloride in dry/wet cyclical processes. Hyperfine Interact. 2005, 161, 127–137. [Google Scholar] [CrossRef]
- García, K.E.; Barrero, C.A.; Morales, A.L.; Greneche, J.M. Lost iron and iron converted into rust in steels submitted to dry–wet corrosion process. Corros. Sci. 2008, 50, 763–772. [Google Scholar] [CrossRef]
- Barrero, C.A.; García, K.E.; Morales, A.L.; Greneche, J.M. A proposal to evaluate the amount of corroded iron converted into adherent rust in steels exposed to corrosion. Corros. Sci. 2011, 53, 769–775. [Google Scholar] [CrossRef]
- Cano, H.; Neff, D.; Morcillo, M.; Dillmann, P.; Díaz, I.; de la Fuente, D. Characterization of corrosion products formed on Ni 2.4 wt%–Cu 0.5 wt%–Cr 0.5 wt% weathering steel exposed in marine atmospheres. Corros. Sci. 2014, 87, 438–451. [Google Scholar] [CrossRef]
- Asami, K.; Kikuchi, M. In-depth distribution of rusts on a plain carbon steel and weathering steels exposed to coastal–industrial atmosphere for 17 years. Corros. Sci. 2003, 45, 2671–2688. [Google Scholar] [CrossRef]
- Hara, S. A X-ray diffraction analysis on constituent distribution of heavy rust layer formed on weathering steel using synchrotron radiation. Corros. Eng. Jpn. 2008, 57, 70–75. [Google Scholar] [CrossRef]
- Morcillo, M.; Chico, B.; de la Fuente, D.; Alcántara, J.; Odnevall Wallinder, I.; Leygraf, C. On the mechanism of rust exfoliation in marine environments. J. Electrochem. Soc. 2017, 164, C8–C16. [Google Scholar] [CrossRef]
- Ishikawa, T.; Isa, R.; Kandori, K.; Nakayama, T.; Tsubota, T. Influences of metal chlorides and sulfates on the formation of β-FeOOH particles by aerial oxidation. J. Electrochem. Soc 2004, 151, B586–B594. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Rouguerol, F.; Rouguerol, J.; Sing, K. Adsorption by Powders and Porous Solids; Academic Press: London, UK, 1999. [Google Scholar]
- Yamashita, M.; Misawa, T. Recent progress in the study of protective rust-layer formation on weathering steel. In Proceedings of the Corrosion’ 98, San Diego, CA, USA, 22–27 March 1998; Technical Publication; NACE International: Houston, TX, USA, 31 December 1998; p. 357. [Google Scholar]
- Kamimura, T.; Hara, S.; Miyuki, H.; Yamashita, M.; Uchida, H. Composition and protective ability of rust layer formed on weathering steel exposed to various environments. Corros. Sci. 2006, 48, 2799–2812. [Google Scholar] [CrossRef]
- Henriksen, J.F. Distribution of NaCl on Fe during atmospheric corrosion. Corros. Sci. 1969, 9, 573–577. [Google Scholar] [CrossRef]
- Misawa, T.; Kyuno, Y.; Suëtaka, W.; Shimodaira, S. The mechanism of atmospheric rusting and the effect of Cu and P on the rust formation of low-alloy steels. Corros. Sci. 1971, 11, 35–48. [Google Scholar] [CrossRef]
- Misawa, T.; Hashimoto, K.; Shimodaira, S. The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature. Corros. Sci. 1974, 14, 131–149. [Google Scholar] [CrossRef]
- Misawa, T.; Asami, K.; Hashimoto, K.; Shimodaira, S. Mechanism of atmospheric rusting and protective amorphous rust on low-alloy steel. Corros. Sci. 1974, 14, 279–289. [Google Scholar] [CrossRef]
- Mikhailovskii, N. Atmospheric Corrosion of Metals and Protection Methods; Metallurgiia: Moscow, Russia, 1989. [Google Scholar]
- Worch, H.; Forker, W.; Rahner, D. Rust formation on iron. A model. Werkst. Korros. 1983, 34, 402–410. [Google Scholar] [CrossRef]
- Schikorr, G. Korrosionsverhalten von zink und zinküberzügen and der atmosphäre. Werkst. Korros. 1964, 15, 537–543. [Google Scholar] [CrossRef]
- Lau, T.T.N.; Thoa, N.T.P.; Nishimura, R.; Tsujino, Y.; Yokoi, M.; Maeda, Y. Atmospheric corrosion of carbon steel under field exposure in the southern part of Vietnam. Corros. Sci. 2006, 48, 179–192. [Google Scholar]
- Li, S.; Hihara, L.H. In situ Raman spectroscopy study of NaCl particle-induced marine atmospheric corrosion of carbon steel. J. Electrochem. Soc. 2012, 159, C147–C154. [Google Scholar] [CrossRef]
- Li, S.; Hihara, L.H. Atmospheric corrosion initiation on steel from predeposited NaCl salt particles in high humidity atmospheres. Corros. Eng. Sci. Technol. 2010, 45, 49–56. [Google Scholar] [CrossRef]
- Li, S.; Hihara, L.H. Atmospheric corrosion electrochemistry of NaCl droplets on carbon steel. J. Electrochem. Soc. 2012, 159, C461–C468. [Google Scholar] [CrossRef]
- Risteen, B.E.; Schindelholz, E.; Kelly, R.G. Marine aerosol drop size effects on the corrosion behavior of low carbon steel and high purity iron. J. Electrochem. Soc. 2014, 161, C580–C586. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Tanaka, S. Monitoring the development of rust layers on weathering steel using in situ Raman spectroscopy under wet-and-dry cyclic conditions. J. Solid State Electrochem. 2015, 19, 3559–3566. [Google Scholar] [CrossRef]
- Evans, U.R. Electrochemical mechanism of atmospheric rusting. Nature 1965, 206, 980–982. [Google Scholar] [CrossRef]
- Stratmann, M.; Bohnenkamp, K.; Engell, H.J. An electrochemical study of phase-transitions in rust layers. Corros. Sci. 1983, 23, 969–985. [Google Scholar] [CrossRef]
- Horton, J.B. The Composition, Structure and Growth of the Atmospheric Rust on Various Steels. Ph.D. Thesis, Lehigh University, Bethlehem, PA, USA, May 1964. [Google Scholar]
- Burger, E.; Fenart, M.; Perrin, S.; Neff, D.; Dillmann, P. Use of the gold markers method to predict the mechanisms of iron atmospheric corrosion. Corros. Sci. 2011, 53, 2122–2130. [Google Scholar] [CrossRef]
- Antony, H.; Legrand, L.; Maréchal, L.; Perrin, S.; Dillmann, P.; Chaussé, A. Study of lepidocrocite γ-FeOOH electrochemical reduction in neutral and slightly alkaline solutions at 25 °C. Electrochim. Acta 2005, 51, 745–753. [Google Scholar] [CrossRef]
- Monnier, J.; Burger, E.; Berger, P.; Neff, D.; Guillot, I.; Dillmann, P. Localisation of oxygen reduction sites in the case of iron long term atmospheric corrosion. Corros. Sci. 2011, 53, 2468–2473. [Google Scholar] [CrossRef]
- Nomura, K.; Tasaka, M.; Ujihira, Y. Conversion electron Mössbauer spectrometric study of corrosion products of iron immersed in sodium chloride solution. Corrosion 1988, 44, 131–135. [Google Scholar] [CrossRef]
- Ma, Y.; Li, Y.; Wang, F. The effect of β-FeOOH on the corrosion behavior of low carbon steel exposed in tropic marine environment. Mater. Chem. Phys. 2008, 112, 844–852. [Google Scholar] [CrossRef]
- Ma, Y.; Li, Y.; Wang, F. Corrosion of low carbon steel in atmospheric environments of different chloride content. Corros. Sci. 2009, 51, 997–1006. [Google Scholar] [CrossRef]
- Schwertmann, V.; Taylor, R.M. The transformation of lepidocrocite to goethite. Clays Clay Miner. 1972, 20, 151–158. [Google Scholar] [CrossRef]
- Saha, J.K. Corrosion of Constructional Steels in Marine and Industrial Environment; Springer: New Delhi, India, 2013. [Google Scholar]
- Morcillo, M.; González-Calbet, J.M.; Jiménez, J.A.; Díaz, I.; Alcántara, J.; Chico, B.; Mazarío-Fernández, A.; Gómez-Herrero, A.; Llorente, I.; de la Fuente, D. Environmental conditions for akaganeite formation in marine atmosphere mild steel corrosion products and its characterisation. Corrosion 2015, 71, 872–886. [Google Scholar] [CrossRef]
- De la Fuente, D.; Díaz, I.; Alcántara, J.; Chico, B.; Simancas, J.; Llorente, I.; García-Delgado, A.; Jiménez, J.A.; Adeva, P.; Morcillo, M. Corrosion mechanisms of mild steel in chloride-rich atmospheres. Mater. Corros. 2015, 67, 227–238. [Google Scholar] [CrossRef]
- Institute of Experimental Mineralogy. Crystallographic and Crystallochemical Database for Minerals and Their Structural Analogues; www.Mincryst; Institute of Experimental Mineralogy, Russian Academy of Sciences: Chernogolovka, Russia, 2016; Available online: http://database.iem.ac.ru/mincryst/index.php (accessed on 11 April 2017).
- Copson, H.R. A theory of the mechanism of rusting of low-alloy steels in the atmosphere. Proc. ASTM 1945, 45, 554–580. [Google Scholar]
- Ericsson, R. Influence of sodium-chloride on atmospheric corrosion of steel. Werkst. Korros. 1978, 29, 400–403. [Google Scholar] [CrossRef]
- Allam, I.M.; Arlow, J.S.; Saricimen, H. Initial stages of atmospheric corrosion of steel in the arabian gulf. Corros. Sci. 1991, 32, 417–432. [Google Scholar] [CrossRef]
- Almeida, E.; Morcillo, M.; Rosales, B. Atmospheric corrosion of mild steel part II—Marine atmospheres. Mater. Corros. 2000, 51, 865–874. [Google Scholar] [CrossRef]
- Liang, C.; Hou, W. Sixteen year atmospheric corrosion exposure study of steels. J. Chin. Soc. Corros. Prot. 2005, 25, 1–6. [Google Scholar]
- Wang, Z. Study of the corrosion behaviour of weathering steels in atmospheric environments. Corros. Sci. 2013, 67, 1–10. [Google Scholar] [CrossRef]
- Scully, J.C. The Fundamentals of Corrosion, 3rd ed.; Pergamon Press: Oxford, UK, 1990; p. 106. [Google Scholar]
- Brown, P.W.; Masters, L.W. Factors affecting the corrosion of metals in the atmosphere. In Atmospheric Corrosion; Ailor, W.H., Ed.; Wiley: New York, NY, USA, 1982; pp. 34–49. [Google Scholar]
- Knotkova, D.; Barton, K.; van Tu, B. Atmospheric corrosion in maritime industrial atmospheres: Laboratory research. In Degradation of Metals in the Atmosphere; ASTM STP 965; Dean, S.W., Lee, T.S., Eds.; American Society for Testing and Materials: Philadelphia, PA, USA, 1987; pp. 290–305. [Google Scholar]
- Bastidas, J.M. Corrosión del Al, Cu, Fe y Zn en atmóSferas Controladas. Ph.D. Thesis, Complutense University, Madrid, Spain, 1981. [Google Scholar]
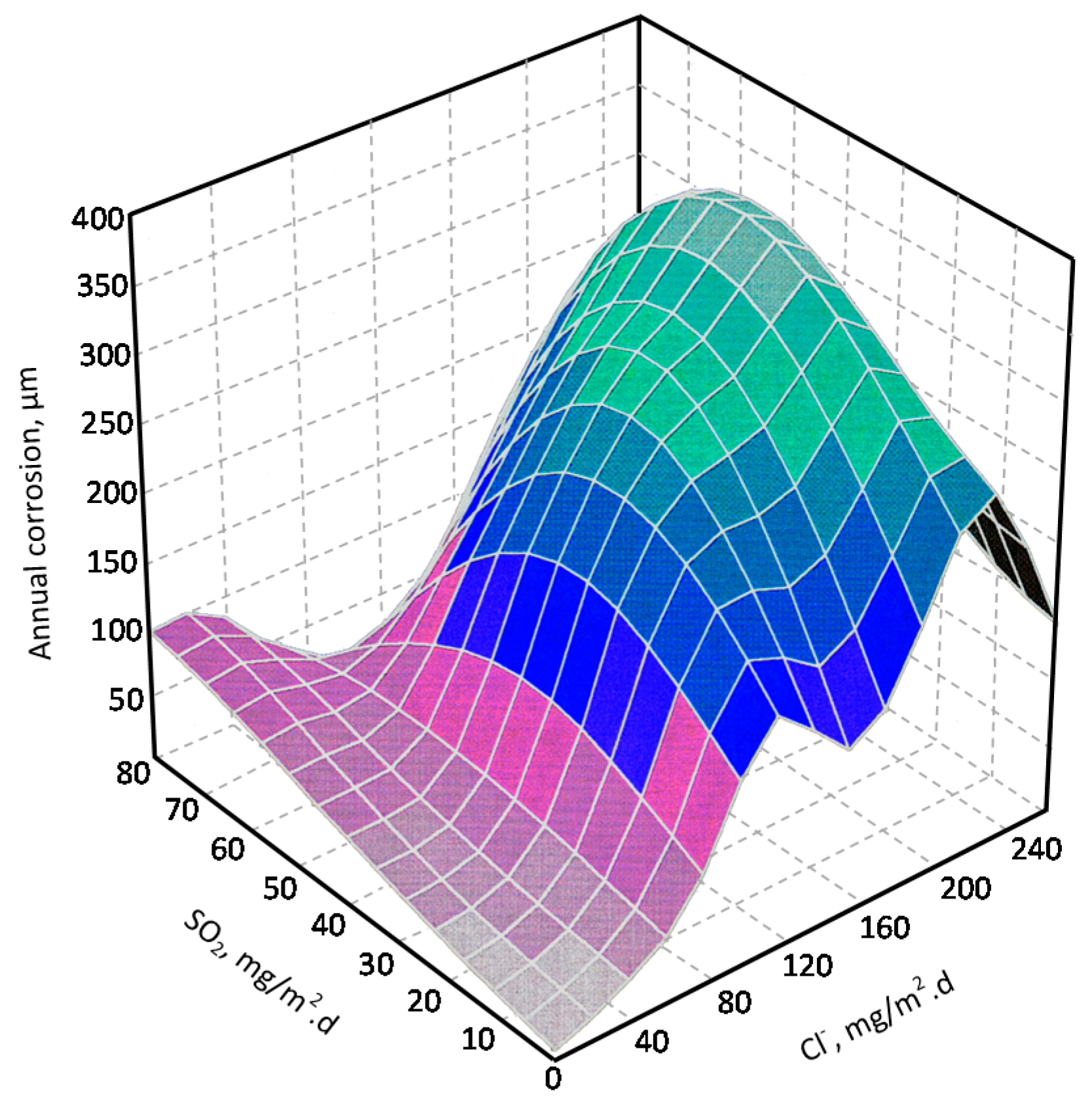
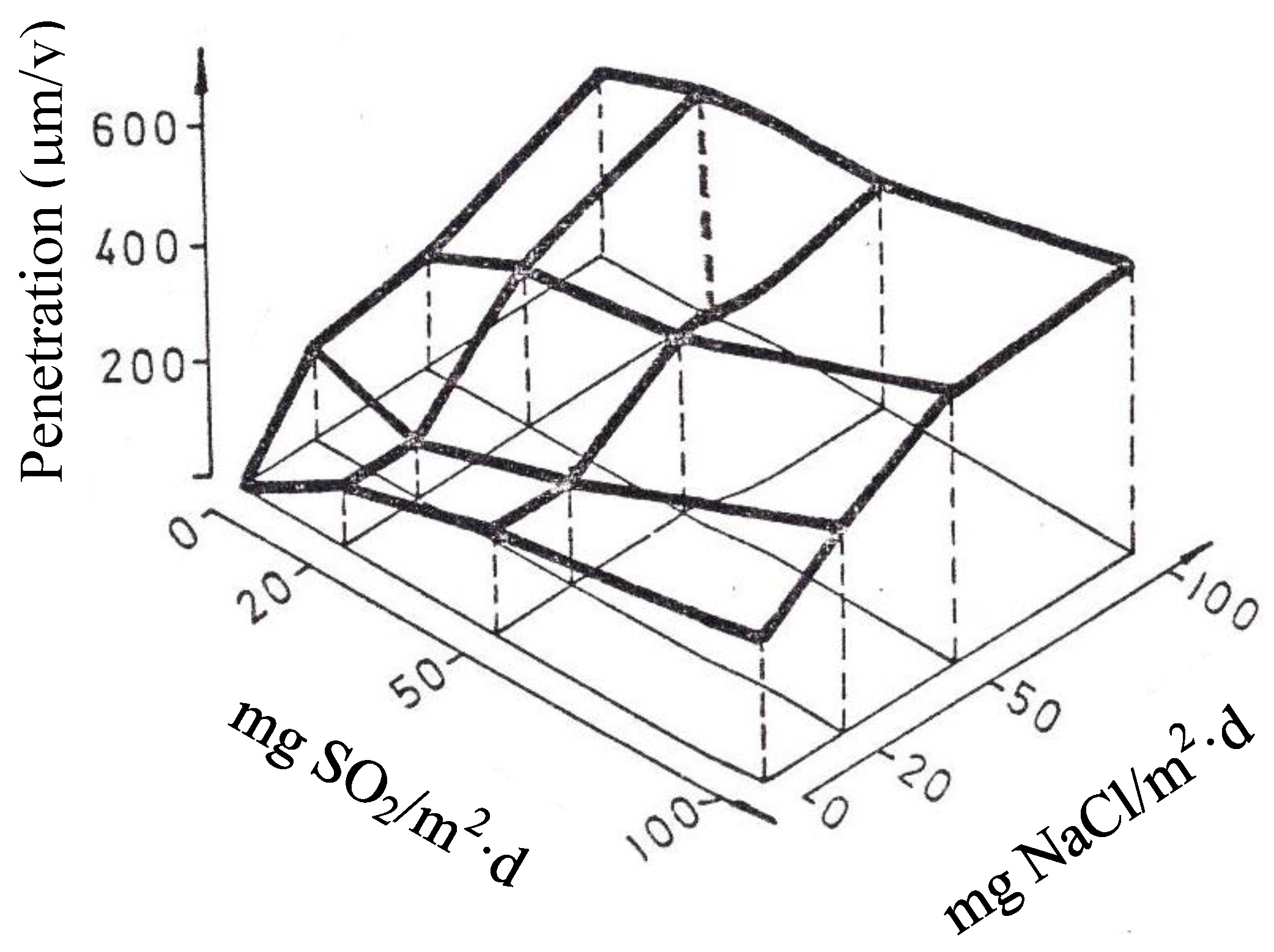
- Chen, W.; Hao, L.; Dong, J.; Ke, W. Effect of sulphur dioxide on the corrosion of a low alloy steel in simulated coastal industrial atmosphere. Corros. Sci. 2014, 83, 155–163. [Google Scholar] [CrossRef]
- UNECE International Cooperative Programme on Effects on Materials Including Historic and Cultural Monuments, Report No. 01: Technical Manual; Swedish Corrosion Institute: Stockholm, Sweden, 1988.
- Morcillo, M.; Almeida, E.; Rosales, B.; Uruchurtu, J.; Marrocos, M. Corrosion y Protección de Metales en las Atmósferas de Iberoamérica. Parte I—Mapas de Iberoamérica de Corrosividad Atmosférica (Proyecto MICAT, XV.1/CYTED); CYTED: Madrid, Spain, 1998. [Google Scholar]
- Benarie, M.; Lipfert, F.L. A general corrosion function in terms of atmospheric pollutant concentrations and rain pH. Atmos. Environ. 1986, 20, 1947–1958. [Google Scholar] [CrossRef]
- Panchenko, Y.M.; Marshakov, A.I.; Igonin, T.N.; Kovtanyuk, V.V.; Nikolaeva, L.A. Long-term forecast of corrosion mass losses of technically important metals in various world regions using a power function. Corros. Sci. 2014, 88, 306–316. [Google Scholar] [CrossRef]
- McCuen, R.H.; Albrecht, P.; Cheng, J.G. A new approach to power-model regression of corrosion penetration data. In Corrosion Forms and Control for Infrastructure; Chaker, V., Ed.; ASTM STP 1137; American Society for Testing and Materials: Philadelphia, PA, USA, 1992; Volume 1137, pp. 46–76. [Google Scholar]
- Panchenko, Y.M.; Marshakov, A.I. Long-term prediction of metal corrosion losses in atmosphere using a power-linear function. Corros. Sci. 2016, 109, 217–229. [Google Scholar] [CrossRef]
- Albrecht, P.; Hall, T.T. Atmospheric corrosion resistance of structural steels. J. Mater. Civ. Eng. 2003, 15, 2–24. [Google Scholar] [CrossRef]
- European Committee for Standardization. EN ISO 9224, Corrosion of Metals and Alloys—Corrosivity of Atmospheres—Guiding Values for the Corrosivity Categories; European Committee for Standardization: Brussels, Belgium, 2012. [Google Scholar]
- Melchers, R.E. A new interpretation of the corrosion loss processes for weathering steels in marine atmospheres. Corros. Sci. 2008, 50, 3446–3454. [Google Scholar] [CrossRef]
- Melchers, R.E. Long-term corrosion of cast irons and steel in marine and atmospheric environments. Corros. Sci. 2013, 68, 186–194. [Google Scholar] [CrossRef]
- International Organization for Standardization. ISO 9223 1st Edition, Corrosion of Metals and alloys—Corrosivity of Atmospheres—Classification; International Organization for Standardization: Geneve, Switzerland, 1992. [Google Scholar]
- Cole, I.S.; Neufeld, A.K.; Kao, P.; Ganther, W.D.; Chotimongkol, L.; Bharmornsut, C.; Hue, N.V.; Bernardo, S.; Purwadaria, S. Development of performance verification methods for the durability of metallic components in tropical countries. In Proceedings of the 11th Asia-Pacific Corrosion Control Conference, Ho Chi Minh City, Vietnam, 1–5 November 1999; Volume 2, pp. 555–570. [Google Scholar]
- Cole, I.S.; Furman, S.A.; Neufeld, A.K.; Ganther, W.D.; King, G.A. A holistic approach to modelling in atmospheric corrosion. Procededings of the 14th International Corrosion Congress, Cape Town, South Africa, 26 September–1 October 1999. [Google Scholar]
- Cole, I.S.; King, G.A.; Trinidad, G.S.; Chan, W.Y.; Paterson, A. An Australia-wide map of corrosivity: A gis approach. In Proceedings of the 8th International Conference on Durability of Building Materials and Components, Vancouver, BC, Canada, 30 May–3 June 1999. [Google Scholar]
- Nakajima, M. Mapping method for salt attack hazzard using topographic effects analysis. In Proceedings of the 1st Asia/Pacific Conference on Harmonisation of Durability Standards and Performance Tests for Components in the Building Industry, Bangkok, Thailand, 8–10 September 1999. [Google Scholar]
- Klassen, R.D.; Roberge, P.R. The effects of wind on local atmospheric corrosivity. In Corrosion 2001; NACE International: Houston, TX, USA, 2001. [Google Scholar]
- Doyle, D.P.; Wright, T.E. Rapid method for determining atmospheric corrosivity and corrosion resistance. In Atmospheric Corrosion; Ailor, W.H., Ed.; Wiley: New York, NY, USA, 1982; pp. 227–243. [Google Scholar]
- Roberge, P.R.; Klassen, R.D.; Haberecht, P.W. Atmospheric corrosivity modeling—A review. Mater. Des. 2002, 23, 321–330. [Google Scholar] [CrossRef]





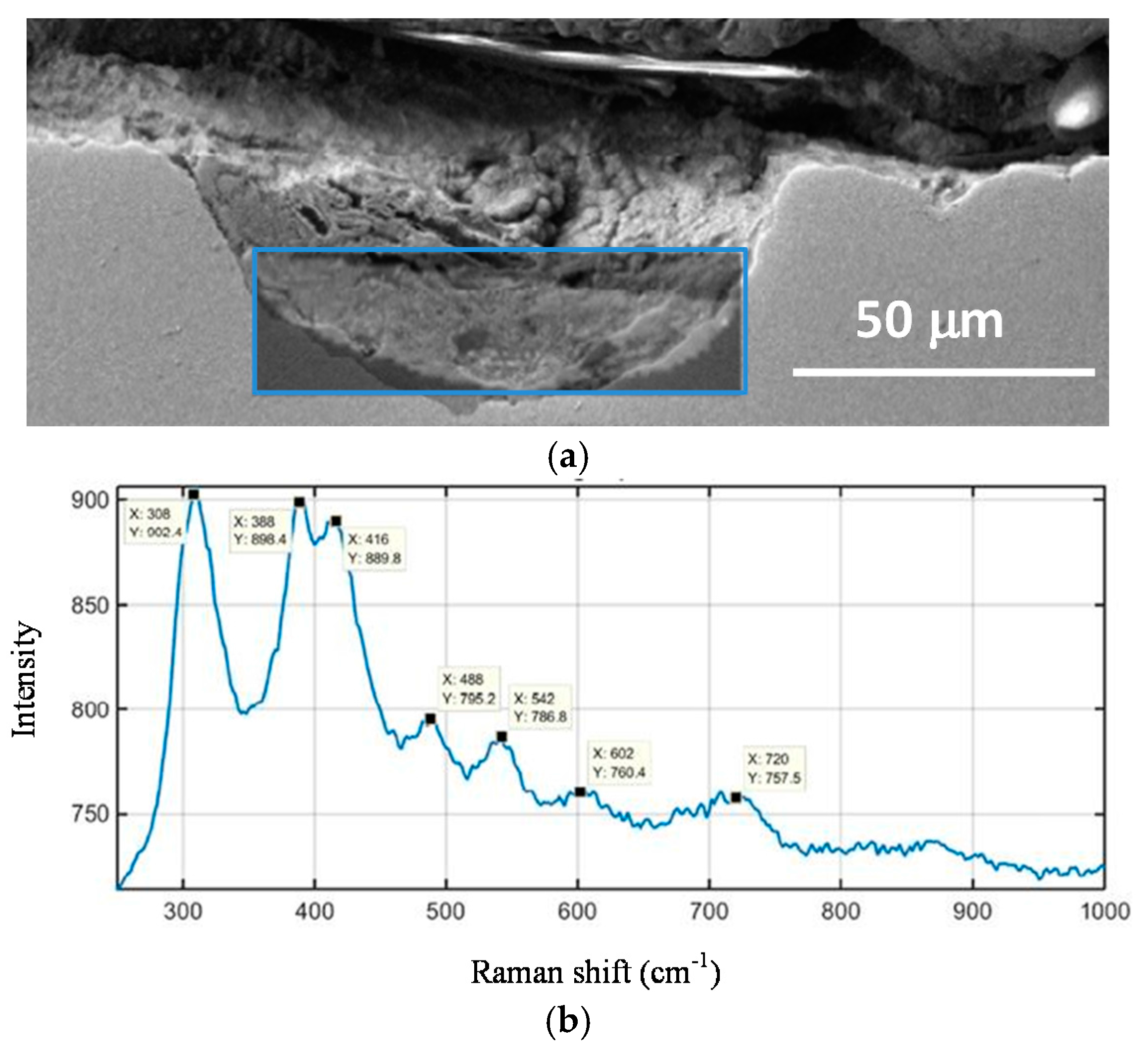
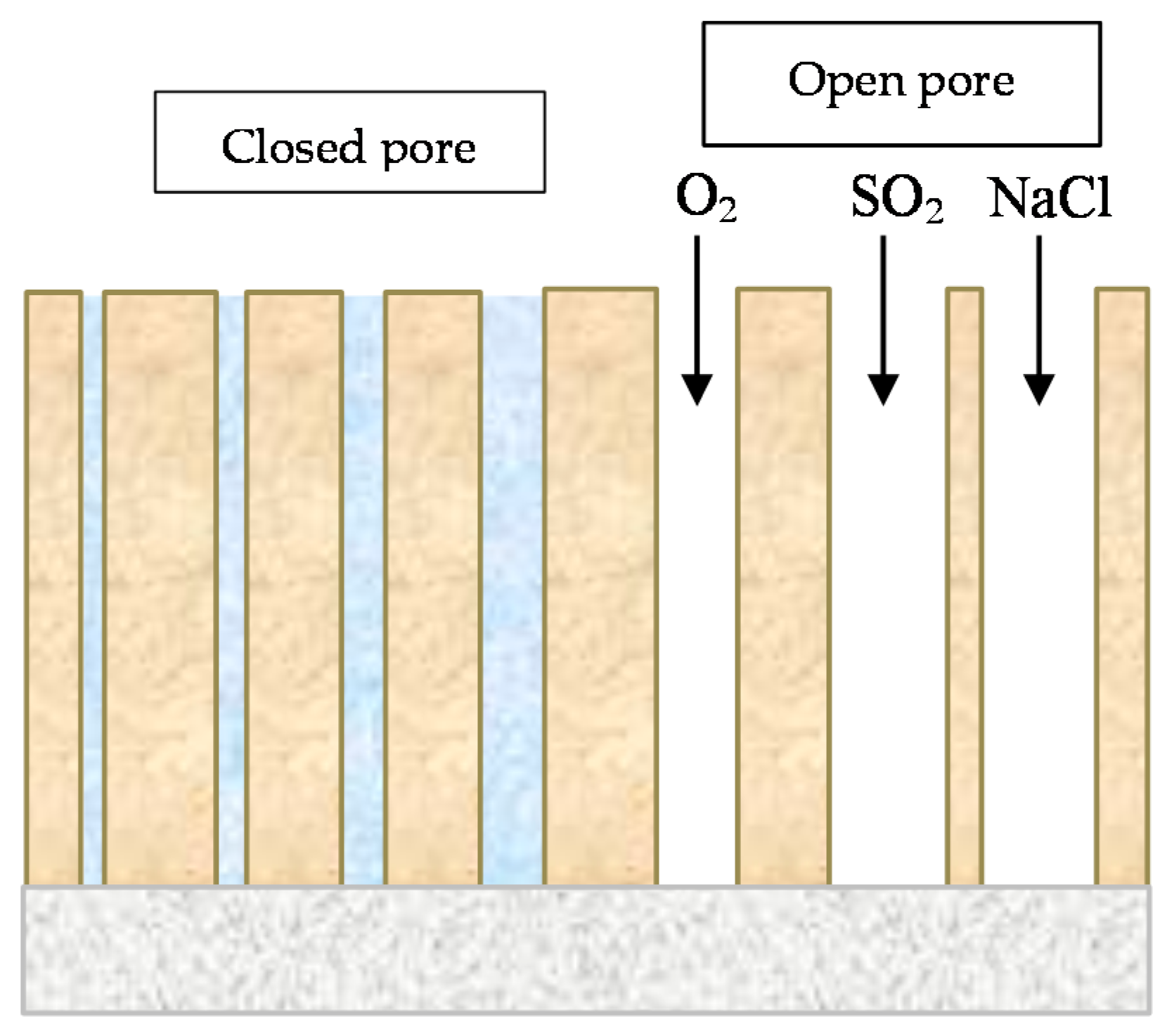
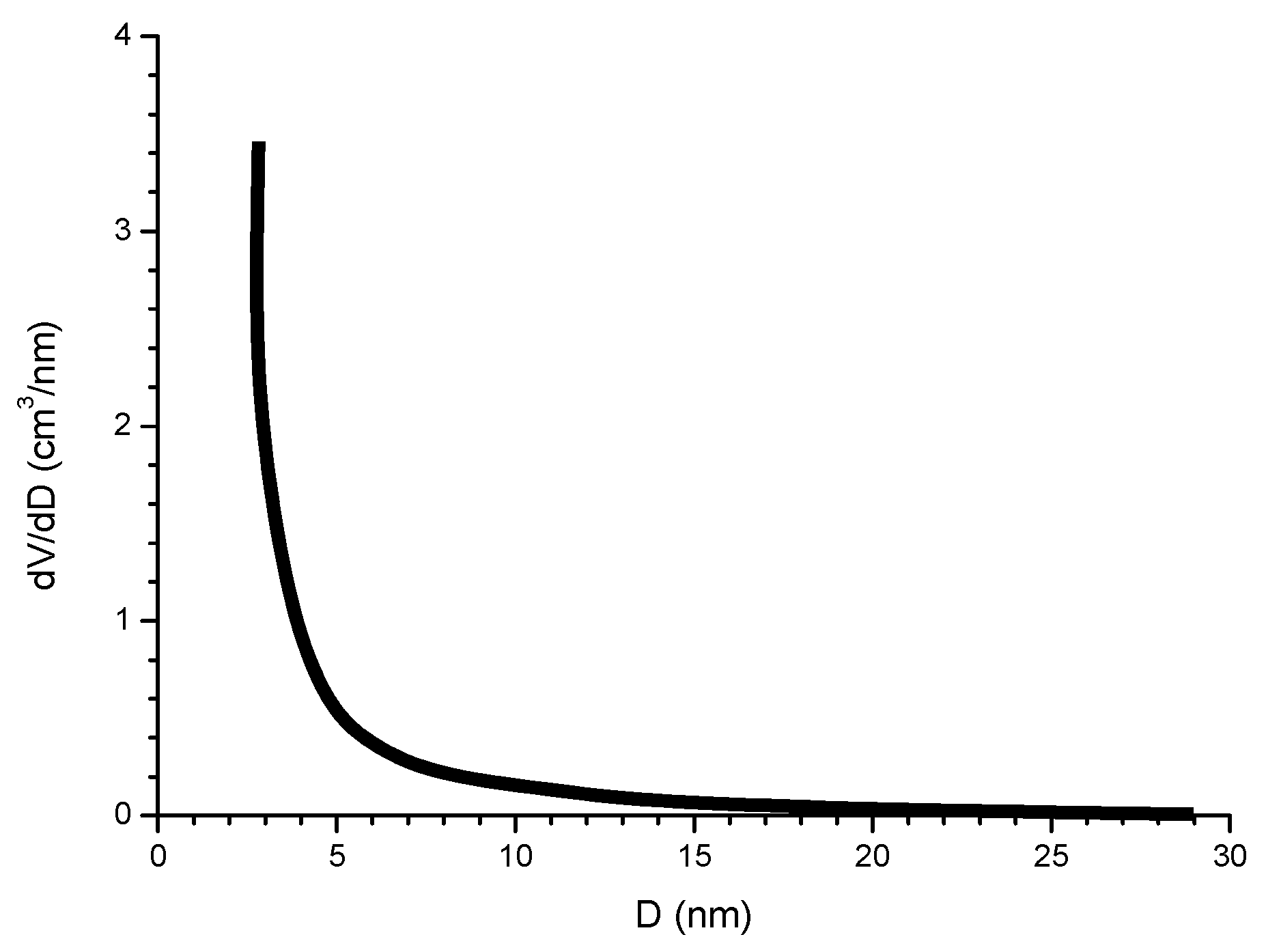
 5.6 mg NaCl/m2; wet period: 56%;
5.6 mg NaCl/m2; wet period: 56%;  10.7 mg NaCl /m2; wet period: 82%; p/po is the relative pressure of water or relative humidity (RH).
5.6 mg NaCl/m2; wet period: 56%; 10.7 mg NaCl /m2; wet period: 82%; p/po is the relative pressure of water or relative humidity (RH).
10.7 mg NaCl /m2; wet period: 82%; p/po is the relative pressure of water or relative humidity (RH).
5.6 mg NaCl/m2; wet period: 56%; 10.7 mg NaCl /m2; wet period: 82%; p/po is the relative pressure of water or relative humidity (RH).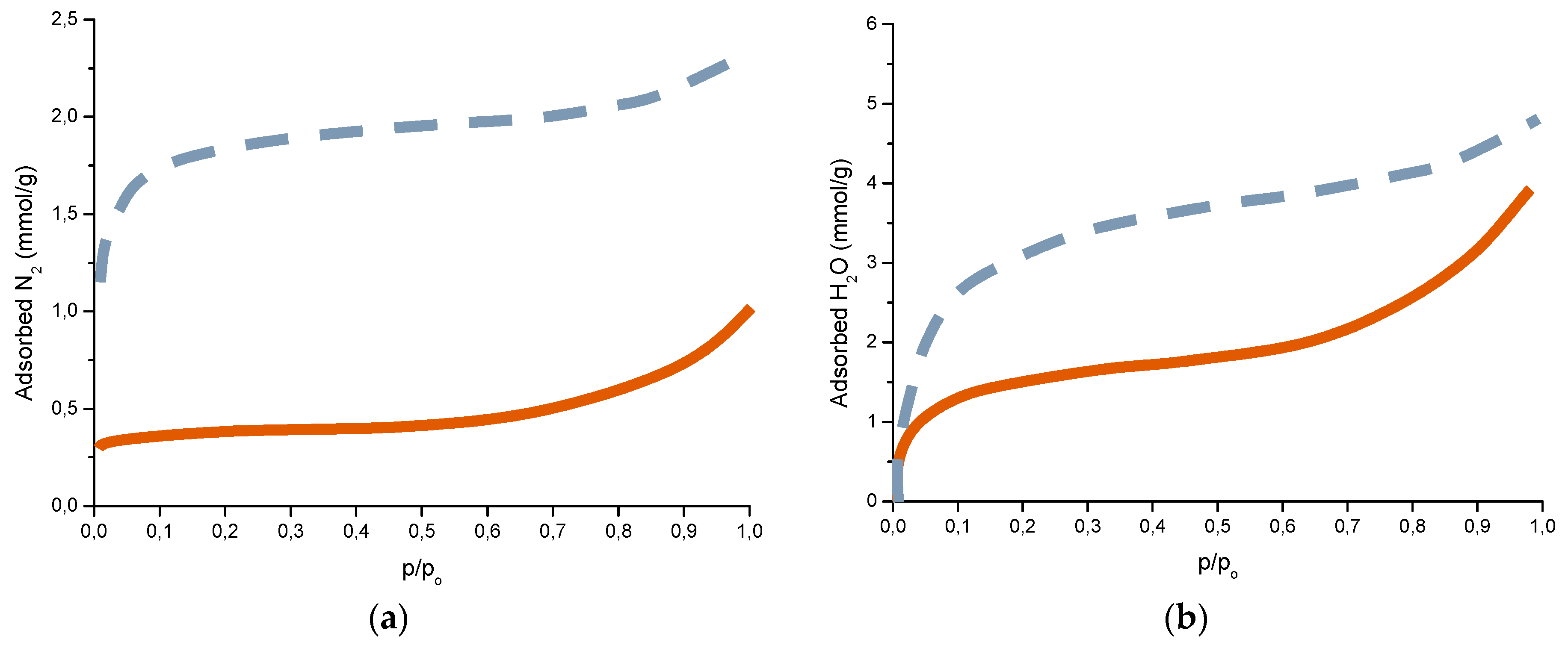


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Name | Formula |
|---|---|---|
| Oxides | Magnetite | Fe3O4 |
| Maghemite | γ-Fe203 | |
| Hematite | α-Fe203 | |
| Hydroxides | - | Fe(OH)2 |
| Bernalite | Fe(OH)3 | |
| Green rusts | FexIII FeyII (OH)3x+2y−z (A−)z where A− = Cl−; ½SO42− | |
| Ferrydrite | Fe5O8H·H2O | |
| Oxyhydroxides | Goethite | α-FeOOH |
| Lepidocrocite | γ-FeOOH | |
| Akaganeite | β-FeOOH | |
| Feroxyhite | δ-FeOOH | |
| Schwertmannite | Fe16O16(OH)y(SO4)z·nH2O |
| Name | Formula |
|---|---|
| Ferrous chloride (lawrencite) | FeCl2 |
| Ferric chloride (molysite) | FeCl3 |
| Ferric oxychloride | FeOCl |
| Ferrous hydroxychloride | β-Fe2(OH)3Cl |
| Green rusts | GR1 (GR Cl) |
| β-oxihydroxide (akaganeite) | β-FeOOH |
| Iron Oxide | Morphology |
|---|---|
| Lepidocrocite (γ-FeOOH) | Laths |
| Goethite (α-FeOOH) | Acicular |
| Akaganeite (β-FeOOH) | Rods, somatoids |
| Feroxyhyte (δ-FeOOH) | Plates |
| Magnetite (Fe3O4) | Octohedra |
| Maghemite (γ-Fe2O3) | Laths or cubes |
| Hematite (α-Fe2O3) | Hexagonal plates, rhombohedra |
| Ferrihydrite (Fe5HO8·4H2O) | Spheres |
| Test Site | Annual Average Chloride Deposition Rate, mg/m2·d | wt % | |||
|---|---|---|---|---|---|
| Lepidocrocite | Goethite | Akaganeite | Spinel | ||
| Ponte do Porto | 4 | 100 | * | 0 | 0 |
| Cabo Vilano-1 | 30 | 80.0 | 16.0 | 0 | 4.0 |
| Cabo Vilano-2 | 70 | 59.6 | 20.0 | 17.6 | 2.7 |
| Cabo Vilano-3 | 665 | 35.8 | 27.0 | 12.5 | 24.7 |
| Phase | Molar Volume (cm3/mol) |
|---|---|
| Goethite (α-FeOOH) | 20.84 |
| Lepidocrocite (γ-FeOOH) | 22.40 |
| Maghemite (γ-Fe2O3) | 43.71 |
| Magnetite (Fe3O4) | 44.56 |
| Akaganeite (β-FeOOH) | 101.62 |
| Non-Marine (Rural-Urban-Industrial) Atmospheres | Marine Atmospheres | ||
|---|---|---|---|
| Av. n | Range of n in Equation (41) | Av. n | Range of n in Equation (41) |
| 0.49 | 0.26–0.76 | 0.73 | 0.37–0.98 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcántara, J.; Fuente, D.d.l.; Chico, B.; Simancas, J.; Díaz, I.; Morcillo, M. Marine Atmospheric Corrosion of Carbon Steel: A Review. Materials 2017, 10, 406. https://doi.org/10.3390/ma10040406
Alcántara J, Fuente Ddl, Chico B, Simancas J, Díaz I, Morcillo M. Marine Atmospheric Corrosion of Carbon Steel: A Review. Materials. 2017; 10(4):406. https://doi.org/10.3390/ma10040406
Chicago/Turabian StyleAlcántara, Jenifer, Daniel de la Fuente, Belén Chico, Joaquín Simancas, Iván Díaz, and Manuel Morcillo. 2017. "Marine Atmospheric Corrosion of Carbon Steel: A Review" Materials 10, no. 4: 406. https://doi.org/10.3390/ma10040406
APA StyleAlcántara, J., Fuente, D. d. l., Chico, B., Simancas, J., Díaz, I., & Morcillo, M. (2017). Marine Atmospheric Corrosion of Carbon Steel: A Review. Materials, 10(4), 406. https://doi.org/10.3390/ma10040406