
Stability Studies of Highly Active Cobalt Catalyst for the Ammonia Synthesis Process

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Catalyst Preparation
- (1)
- Wet impregnation (WI)—the material was impregnated with an appropriate amount of an aqueous solution of barium(II) nitrate. The solvent (water) was removed using a vacuum evaporator. Then, the sample was dried in air at 120 °C for 18 h.
- (2)
- Wet mixing (WM)—the material was mechanically mixed in a mortar with an appropriate amount of an aqueous solution of barium(II) nitrate and dried in air at 120 °C for 18 h.
- (3)
- Deposition–precipitation (DP)—the material was dispersed in an appropriate amount of an aqueous solution of barium(II) nitrate. Then, an aqueous solution of potassium carbonate was introduced dropwise and, maintaining a constant temperature (90 °C) and stirring speed (350 rpm), the process of precipitation of barium carbonate on particles of a mixture of cobalt and cerium oxides was carried out. The precipitate was then filtered, washed, and dried at 120 °C for 18 h.
2.2. Catalyst Characterization Methods
2.3. Catalyst Activity and Stability Tests
3. Results
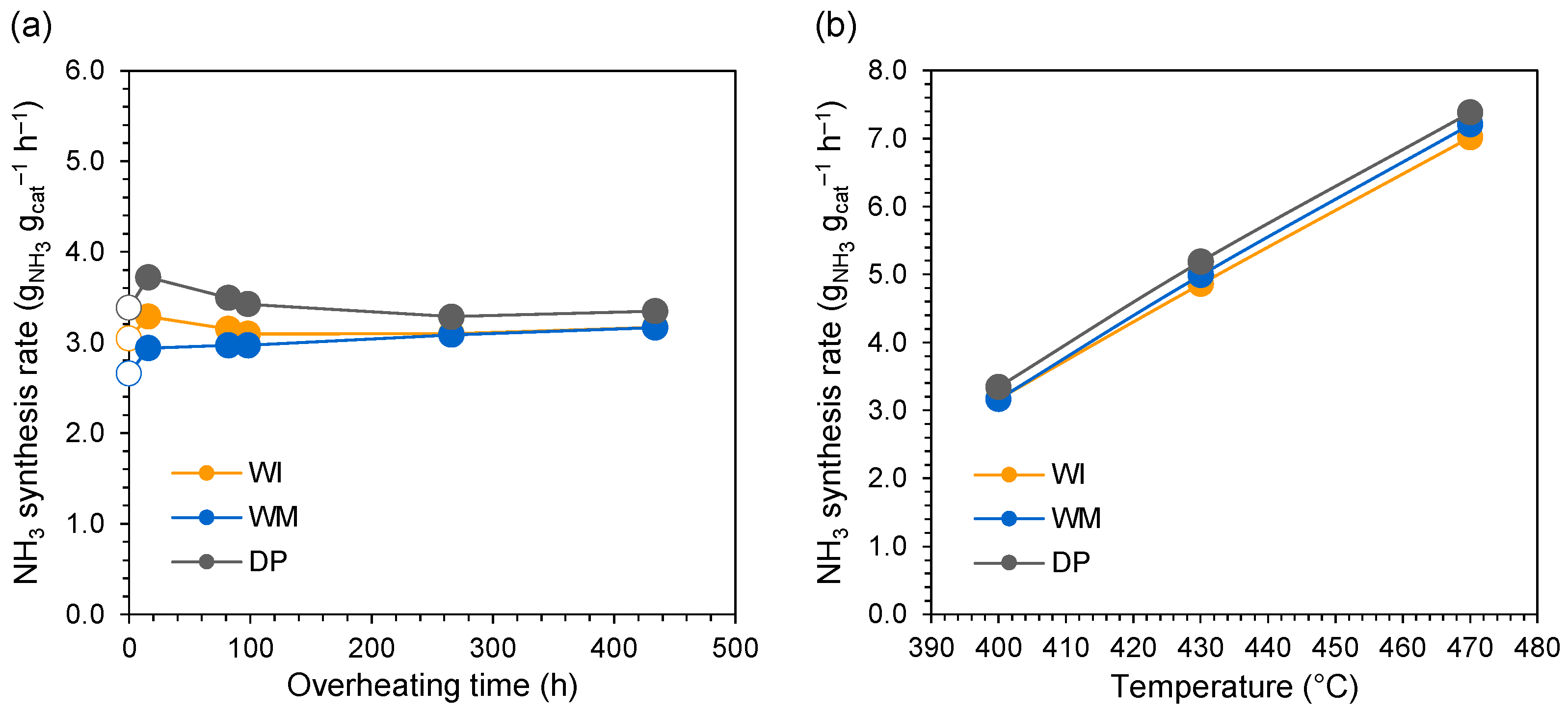
3.1. Evaluation of Catalyst Stability in Ammonia Synthesis Reaction
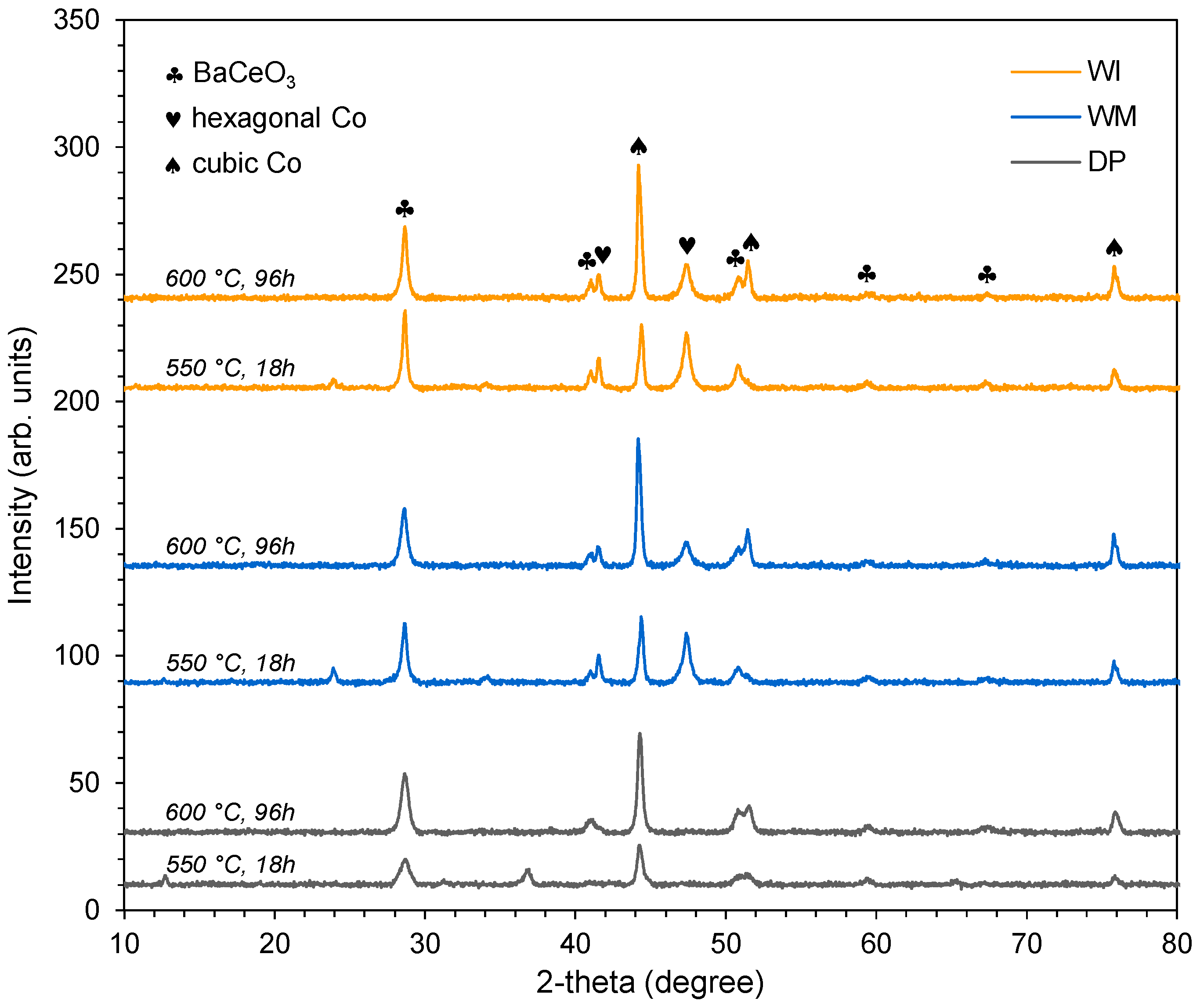
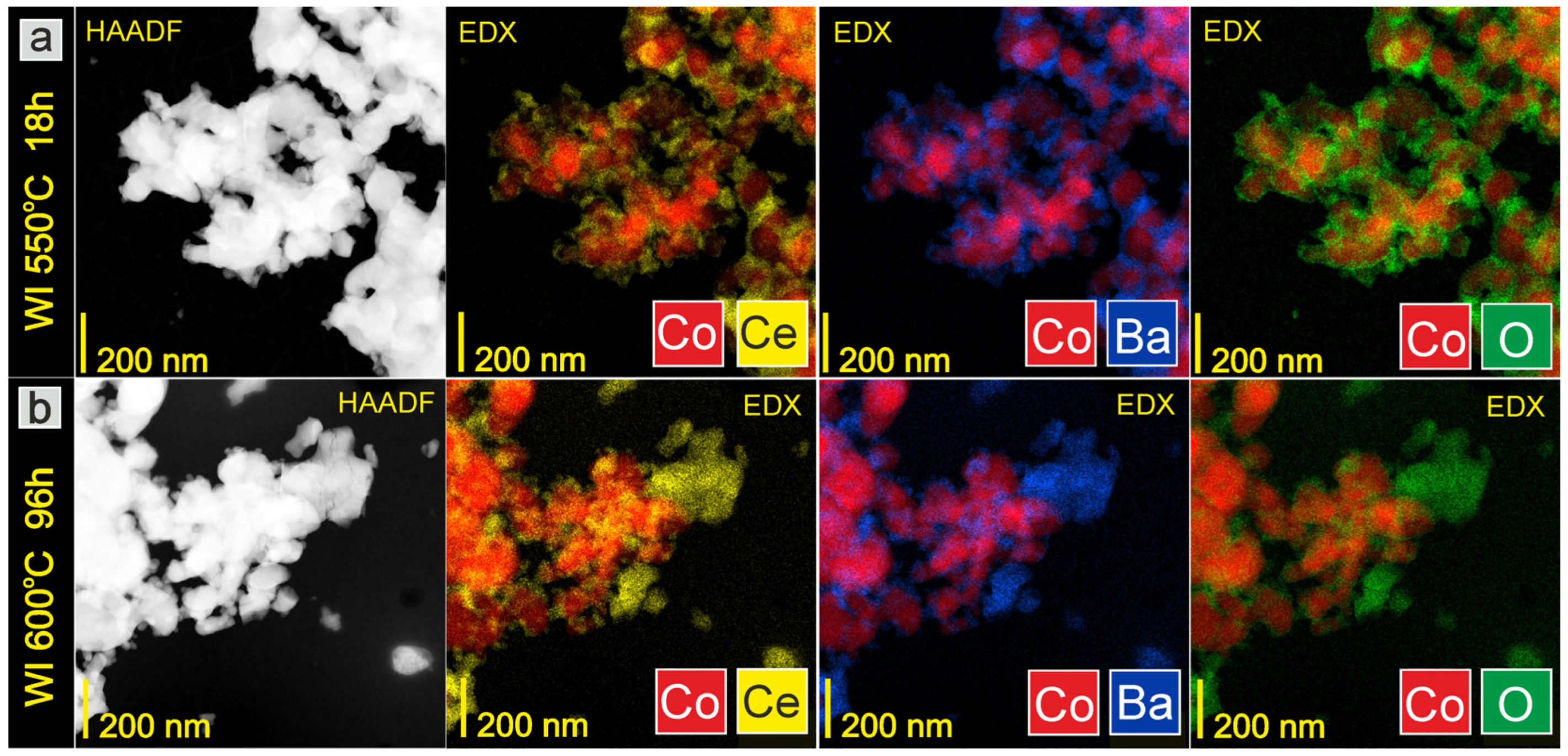
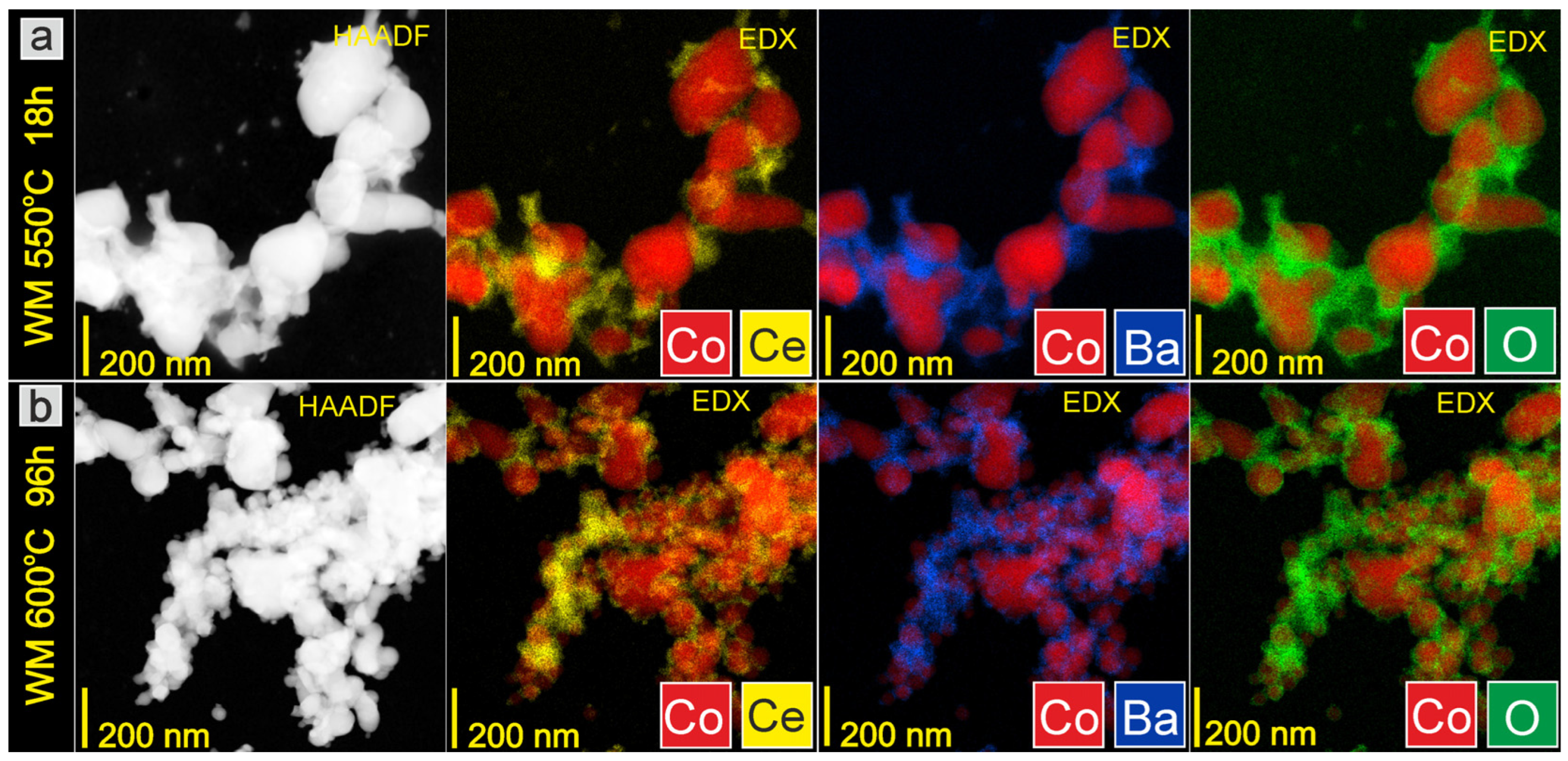
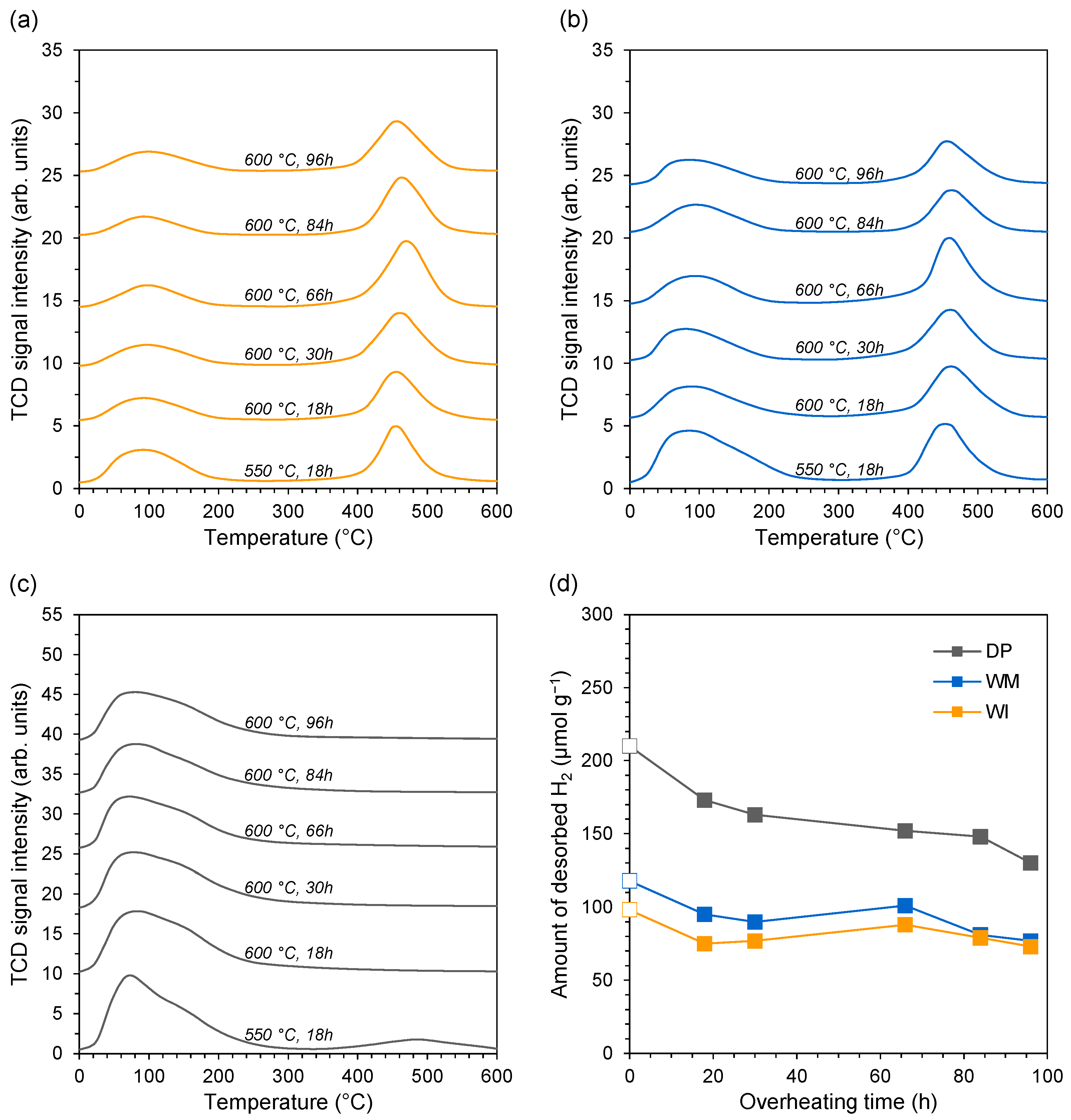
3.2. Evaluation of Catalyst Properties during Stability Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vezirogľu, T.N.; Barbir, F. Hydrogen: The wonder fuel. Int. J. Hydrogen Energy 1992, 17, 391–404. [Google Scholar] [CrossRef]
- Amin, M.; Shah, H.H.; Fareed, A.G.; Khan, W.U.; Chung, E.; Zia, A.; Farooqi, Z.U.; Lee, C. Hydrogen production through renewable and non-renewable energy processes and their impact on climate change. Int. J. Hydrogen Energy 2022, 47, 33112–33134. [Google Scholar] [CrossRef]
- Singh, S.; Jain, S.; Ps, V.; Tiwari, A.K.; Nouni, M.R.; Pandey, J.K.; Goel, S. Hydrogen: A sustainable fuel for future of the transport sector. Renew. Sustain. Energy Rev. 2015, 51, 623–633. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Jiang, L.-W.; Huang, Y.; Zou, Y.; Meng, C.; Xiao, Y.; Liu, H.; Wang, J.-J. Boosting the Stability of Oxygen Vacancies in α-Co(OH)2 Nanosheets with Coordination Polyhedrons as Rivets for High-Performance Alkaline Hydrogen Evolution Electrocatalyst. Adv. Energy Mater. 2022, 12, 2202351. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Huang, Y.; Jiang, L.-W.; Meng, C.; Yin, Z.-H.; Liu, H.; Wang, J.-J. Modulating the electronic structure of CoS2 by Sn doping boosting urea oxidation for efficient alkaline hydrogen production. J. Colloid Interface Sci. 2023, 642, 574–583. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, L.-W.; Liu, H.; Wang, J.-J. Electronic structure regulation and polysulfide bonding of Co-doped (Ni, Fe)1+xS enable highly efficient and stable electrocatalytic overall water splitting. Chem. Eng. J. 2022, 441, 136121. [Google Scholar] [CrossRef]
- Acar, C.; Dincer, I. Review and evaluation of hydrogen production options for better environment. J. Clean. Prod. 2019, 218, 835–849. [Google Scholar] [CrossRef]
- Lucentini, I.; Garcia, X.; Vendrell, X.; Llorca, J. Review of the Decomposition of Ammonia to Generate Hydrogen. Ind. Eng. Chem. Res. 2021, 60, 18560–18611. [Google Scholar] [CrossRef]
- Lan, R.; Irvine, J.T.S.; Tao, S. Ammonia and related chemicals as potential indirect hydrogen storage materials. Int. J. Hydrogen Energy 2012, 37, 1482–1494. [Google Scholar] [CrossRef]
- Klerke, A.; Christensen, C.H.; Nørskov, J.K.; Vegge, T. Ammonia for hydrogen storage: Challenges and opportunities. J. Mater. Chem. 2008, 18, 2304–2310. [Google Scholar] [CrossRef]
- Berwal, P.; Kumar, S.; Khandelwal, B. A comprehensive review on synthesis, chemical kinetics, and practical application of ammonia as future fuel for combustion. J. Energy Inst. 2021, 99, 273–298. [Google Scholar] [CrossRef]
- Herbinet, O.; Bartocci, P.; Grinberg Dana, A. On the use of ammonia as a fuel—A perspective. Fuel Commun. 2022, 11, 100064. [Google Scholar] [CrossRef]
- Elbaz, A.M.; Wang, S.; Guiberti, T.F.; Roberts, W.L. Review on the recent advances on ammonia combustion from the fundamentals to the applications. Fuel Commun. 2022, 10, 100053. [Google Scholar] [CrossRef]
- Valera-Medina, A.; Amer-Hatem, F.; Azad, A.K.; Dedoussi, I.C.; de Joannon, M.; Fernandes, R.X.; Glarborg, P.; Hashemi, H.; He, X.; Mashruk, S.; et al. Review on Ammonia as a Potential Fuel: From Synthesis to Economics. Energy Fuels 2021, 35, 6964–7029. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, J.; Wang, Y.; Mou, T.; Lin, Y.; Yue, L.; Li, T.; Liu, Q.; Luo, Y.; Li, N.; et al. High-Performance Electrochemical NO Reduction into NH3 by MoS2 Nanosheet. Angew. Chem. Int. Ed. 2021, 60, 25263. [Google Scholar] [CrossRef] [PubMed]
- Liu, H. Ammonia Synthesis Catalyst: Innovations and Practice; World Scientific Publishing Co. Pte. Ltd.: Singapore; Chemical Industry Press: Beijing, China, 2013; pp. 686–705. [Google Scholar]
- Hughes, R. Deactivation of Catalysts; Academic Press: London, UK, 1984. [Google Scholar]
- Moulijn, J.A.; van Diepen, A.E.; Kapteijn, F. Catalyst deactivation: Is it predictable? What to do? Appl. Catal. A Gen. 2001, 212, 3–16. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Bartholomew, C.H.; Farrauto, R.J. Fundamentals of Industrial Catalytic Processes, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Birtill, J.J. But will it last until the shutdown? Deciphering catalyst decay! Catal. Today 2003, 81, 531–545. [Google Scholar] [CrossRef]
- Scott, S.L. A Matter of Life(time) and Death. ACS Catal. 2018, 8, 8597–8599. [Google Scholar] [CrossRef]
- Pernicone, N. Methods for laboratory-scale evaluation of catalyst life in industrial plants. Appl. Catal. 1985, 15, 17–31. [Google Scholar] [CrossRef]
- Liu, H.-Z.; Li, X.-N.; Hu, Z.-N. Development of novel low temperature and low pressure ammonia synthesis catalyst. Appl. Catal. A 1996, 142, 209–222. [Google Scholar] [CrossRef]
- Pernicone, N.; Ferrero, F.; Rossetti, I.; Forni, L.; Canton, P.; Riello, P.; Fagherazzi, G.; Signoretto, M.; Pinna, F. Wustite as a new precursor of industrial ammonia synthesis catalysts. Appl. Catal. A 2003, 251, 121–129. [Google Scholar] [CrossRef]
- Lin, B.; Guo, Y.; Lin, J.; Ni, J.; Lin, J.; Jiang, L.; Wang, Y. Deactivation study of carbon-supported ruthenium catalyst with potassium promoter. Appl. Catal. A 2017, 541, 1–7. [Google Scholar] [CrossRef]
- Kowalczyk, Z.; Jodzis, S.; Raróg, W.; Zieliński, J.; Pielaszek, J. Effect of potassium and barium on the stability of a carbon-supported ruthenium catalyst for the synthesis of ammonia. Appl. Catal. A 1998, 173, 153–160. [Google Scholar] [CrossRef]
- Raróg-Pilecka, W.; Karolewska, M.; Truszkiewicz, E.; Iwanek, E.; Mierzwa, B. Cobalt catalyst doped with cerium and barium obtained by Co-precipitation method for ammonia synthesis process. Catal. Lett. 2011, 141, 678–684. [Google Scholar] [CrossRef]
- Karolewska, M.; Truszkiewicz, E.; Mierzwa, B.; Kępiński, L.; Raróg-Pilecka, W. Ammonia synthesis over cobalt catalysts doped with cerium and barium. Effect of the ceria loading. Appl. Catal. A 2012, 445–446, 280–286. [Google Scholar] [CrossRef]
- Tarka, A.; Patkowski, W.; Zybert, M.; Ronduda, H.; Wieciński, P.; Adamski, P.; Sarnecki, A.; Moszyński, D.; Raróg-Pilecka, W. Synergistic Interaction of Cerium and Barium-New Insight into the Promotion Effect in Cobalt Systems for Ammonia Synthesis. Catalysts 2020, 10, 658. [Google Scholar] [CrossRef]
- Kowalczyk, Z. Effect of potassium on the high-pressure kinetics of ammonia synthesis over fused iron catalysts. Catal. Lett. 1996, 37, 173–179. [Google Scholar] [CrossRef]
- Lin, B.; Liu, Y.; Heng, L.; Wang, X.; Ni, J.; Lin, J.; Jiang, L. Morphology Effect of Ceria on the Catalytic Performances of Ru/CeO2 Catalysts for Ammonia Synthesis. Ind. Eng. Chem. Res. 2018, 57, 9127–9135. [Google Scholar] [CrossRef]
- Han, W.; Li, Z.; Liu, H. La2Ce2O7 supported ruthenium as a robust catalyst for ammonia synthesis. J. Rare Earths 2019, 37, 492–499. [Google Scholar] [CrossRef]
- Jafari, A.; Ebadi, A.; Sahebdelfar, S. Effect of iron oxide precursor on the properties and ammonia synthesis activity of fused iron catalysts. Reac. Kinet. Mech. Cat. 2019, 126, 307–325. [Google Scholar] [CrossRef]
- Zhou, Y.; Ma, Y.; Lan, G.; Tang, H.; Han, W.; Liu, H.; Li, Y. A highly stable and active mesoporous ruthenium catalyst for ammonia synthesis prepared by a RuCl3/SiO2-templated approach. Chin. J. Catal. 2019, 40, 114–123. [Google Scholar] [CrossRef]
- Zybert, M.; Karasińska, M.; Truszkiewicz, E.; Mierzwa, B.; Raróg-Pilecka, W. Properties and activity of the cobalt catalysts for NH3 synthesis obtained by co-precipitation – the effect of lanthanum addition. Pol. J. Chem. Technol. 2015, 17, 138–143. [Google Scholar] [CrossRef]
- Lin, B.; Liu, Y.; Heng, L.; Ni, J.; Lin, J.; Jiang, L. Effect of barium and potassium promoter on Co/CeO2 catalysts in ammonia synthesis. J. Rare Earths 2018, 36, 703–707. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Sobczak, K.; Moszyński, D.; Albrecht, A.; Sarnecki, A.; Raróg-Pilecka, W. On the effect of metal loading on the performance of Co catalysts supported on mixed MgO–La2O3 oxides for ammonia synthesis. RSC Adv. 2022, 12, 33876–33888. [Google Scholar] [CrossRef]
- Rambeau, G.; Jorti, A.; Amariglio, H. Catalytic activity of a cobalt powder in NH3 synthesis in relation with the allotropic transformation of the metal. J. Catal. 1985, 94, 155–165. [Google Scholar] [CrossRef]
- Ray, A.E.; Smith, S.R.; Scofield, J.D. Study of the phase transformation of cobalt. Phase Equilibr. 1991, 12, 644–647. [Google Scholar] [CrossRef]
- Matsumoto, H. Shift of α→β transformation temperature of cobalt with thermal cycling. J. Mater. Sci. Lett. 1993, 12, 969–970. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Moszyński, D.; Albrecht, A.; Sobczak, K.; Małolepszy, A.; Raróg-Pilecka, W. Co nanoparticles supported on mixed magnesium–lanthanum oxides: Effect of calcium and barium addition on ammonia synthesis catalyst performance. RSC Adv. 2023, 13, 4787–4802. [Google Scholar] [CrossRef] [PubMed]
- Schlögl, R. Catalytic Synthesis of Ammonia—A “Never-Ending Story”? Angew. Chem. Int. Ed. 2003, 42, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, J.; Lan, R.; Tao, S. Development and Recent Progress on Ammonia Synthesis Catalysts for Haber–Bosch Process. Adv. Energy Sustain. Res. 2021, 2, 2000043. [Google Scholar] [CrossRef]
- Ertl, G. Reactions at Surfaces: From Atoms to Complexity (Nobel Lecture). Angew. Chem. Int. Ed. 2008, 47, 3524–3535. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.; Bukas, V.J.; Shadravan, V.; Wang, Z.; Li, H.; Kibsgaard, J.; Chorkendorff, I.; Nørskov, J.K. A spin promotion effect in catalytic ammonia synthesis. Nat. Commun. 2022, 13, 2382. [Google Scholar] [CrossRef]
- Ertl, G. Elementary steps in ammonia synthesis: The surface approach. In Catalytic Ammonia Synthesis. Fundamentals and Practice, 1st ed.; Jennings, J.R., Ed.; Springer Science+Business Media: New York, NY, USA, 1991; pp. 109–132. [Google Scholar]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Tarka, A.; Jodłowski, P.; Kępiński, L.; Sarnecki, A.; Moszyński, D.; Raróg-Pilecka, W. Tuning the catalytic performance of Co/Mg-La system for ammonia synthesis via the active phase precursor introduction method. Appl. Catal. A 2020, 598, 117553. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Dziewulska, A.; Patkowski, W.; Sobczak, K.; Ostrowski, A.; Raróg-Pilecka, W. Ammonia synthesis using Co catalysts supported on MgO–Nd2O3 mixed oxide systems: Effect of support composition. Surf. Interfaces 2023, 36, 102530. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Element Content (wt%) 1 | SSAred (m2 g−1) 2 | |||
|---|---|---|---|---|---|
| Co | Ce | Ba | After Reduction in H2 Flow at 550 °C for 10 h | After Overheating in H2 Flow at 600 °C for 10/20/30 h | |
| WI | 70.1 | 8.4 | 12.6 | 5.6 | 3.9/4.2/3.7 |
| WM | 70.3 | 8.2 | 12.8 | 13.3 | 7.2/8.0/6.0 |
| DP | 70.7 | 8.1 | 12.3 | 11.2 | 11.8/12.3/10.4 |
| Catalyst | After Reduction in H2 Flow at 550 °C for 18 h | After Overheating in H2 Flow at 600 °C for 96 h | ||
|---|---|---|---|---|
| Hexagonal Co (nm) | Cubic Co (nm) | Hexagonal Co (nm) | Cubic Co (nm) | |
| WI | 15 | 26 | 17 | 27 |
| WM | 17 | 25 | 15 | 26 |
| DP | – | 20 | – | 23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zybert, M.; Ronduda, H.; Patkowski, W.; Rybińska, W.; Ostrowski, A.; Sobczak, K.; Raróg-Pilecka, W. Stability Studies of Highly Active Cobalt Catalyst for the Ammonia Synthesis Process. Energies 2023, 16, 7787. https://doi.org/10.3390/en16237787
Zybert M, Ronduda H, Patkowski W, Rybińska W, Ostrowski A, Sobczak K, Raróg-Pilecka W. Stability Studies of Highly Active Cobalt Catalyst for the Ammonia Synthesis Process. Energies. 2023; 16(23):7787. https://doi.org/10.3390/en16237787
Chicago/Turabian StyleZybert, Magdalena, Hubert Ronduda, Wojciech Patkowski, Weronika Rybińska, Andrzej Ostrowski, Kamil Sobczak, and Wioletta Raróg-Pilecka. 2023. "Stability Studies of Highly Active Cobalt Catalyst for the Ammonia Synthesis Process" Energies 16, no. 23: 7787. https://doi.org/10.3390/en16237787
APA StyleZybert, M., Ronduda, H., Patkowski, W., Rybińska, W., Ostrowski, A., Sobczak, K., & Raróg-Pilecka, W. (2023). Stability Studies of Highly Active Cobalt Catalyst for the Ammonia Synthesis Process. Energies, 16(23), 7787. https://doi.org/10.3390/en16237787