Nanostructured Carbon Material Effect on the Synthesis of Carbon-Supported Molybdenum Carbide Catalysts for Guaiacol Hydrodeoxygenation




Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis of Nanostructured Carbon Materials
2.2. Mo Precursor Impregnation and Carbothermal Hydrogen Reduction
2.3. Catalytic Hydrodeoxygenation of Guaiacol Using Mo2C/NCM
2.4. Characterization of NCM and Mo2C/NCM Catalysts
3. Results and Discussion
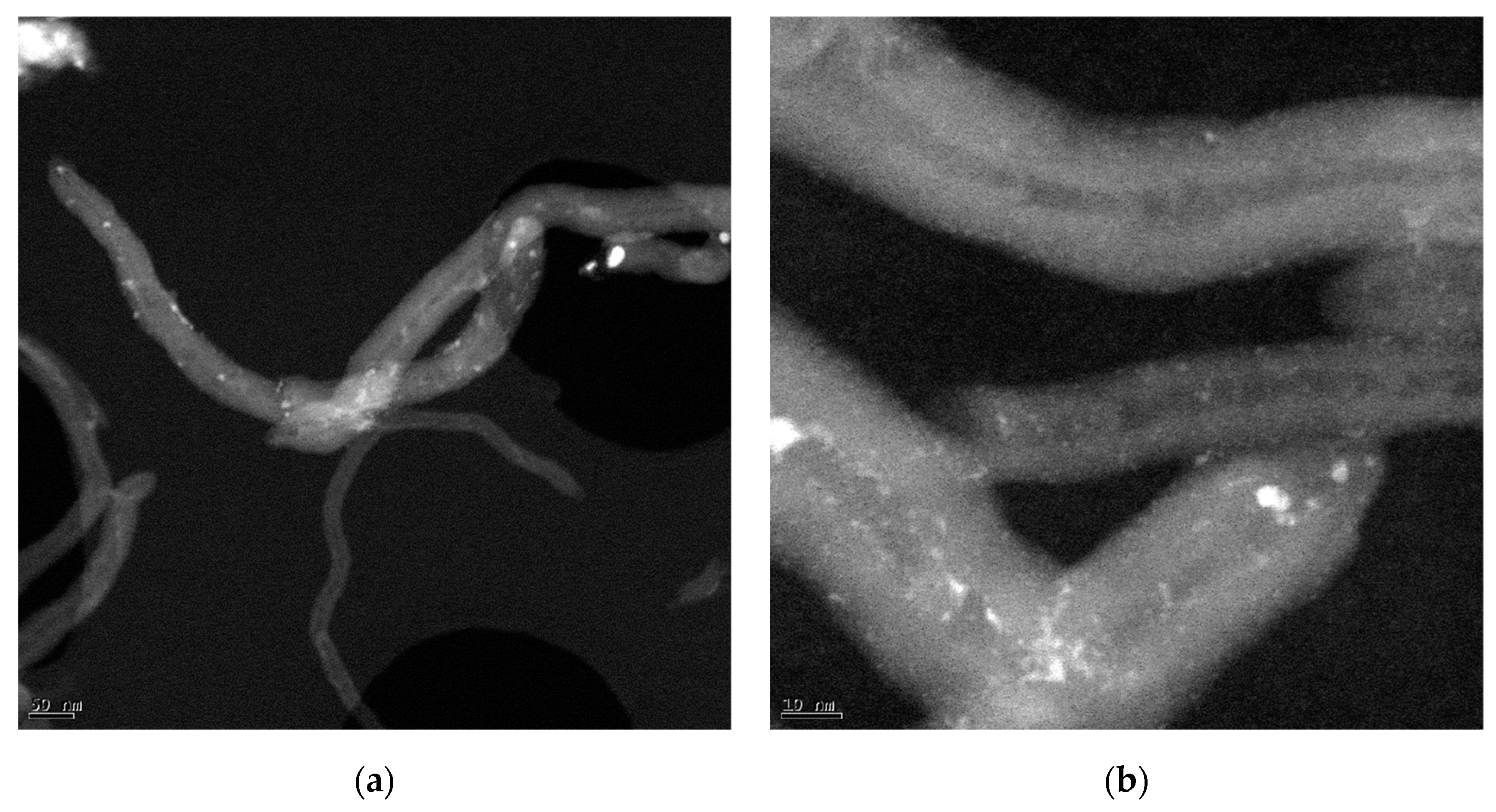
3.1. Characterization of NCM
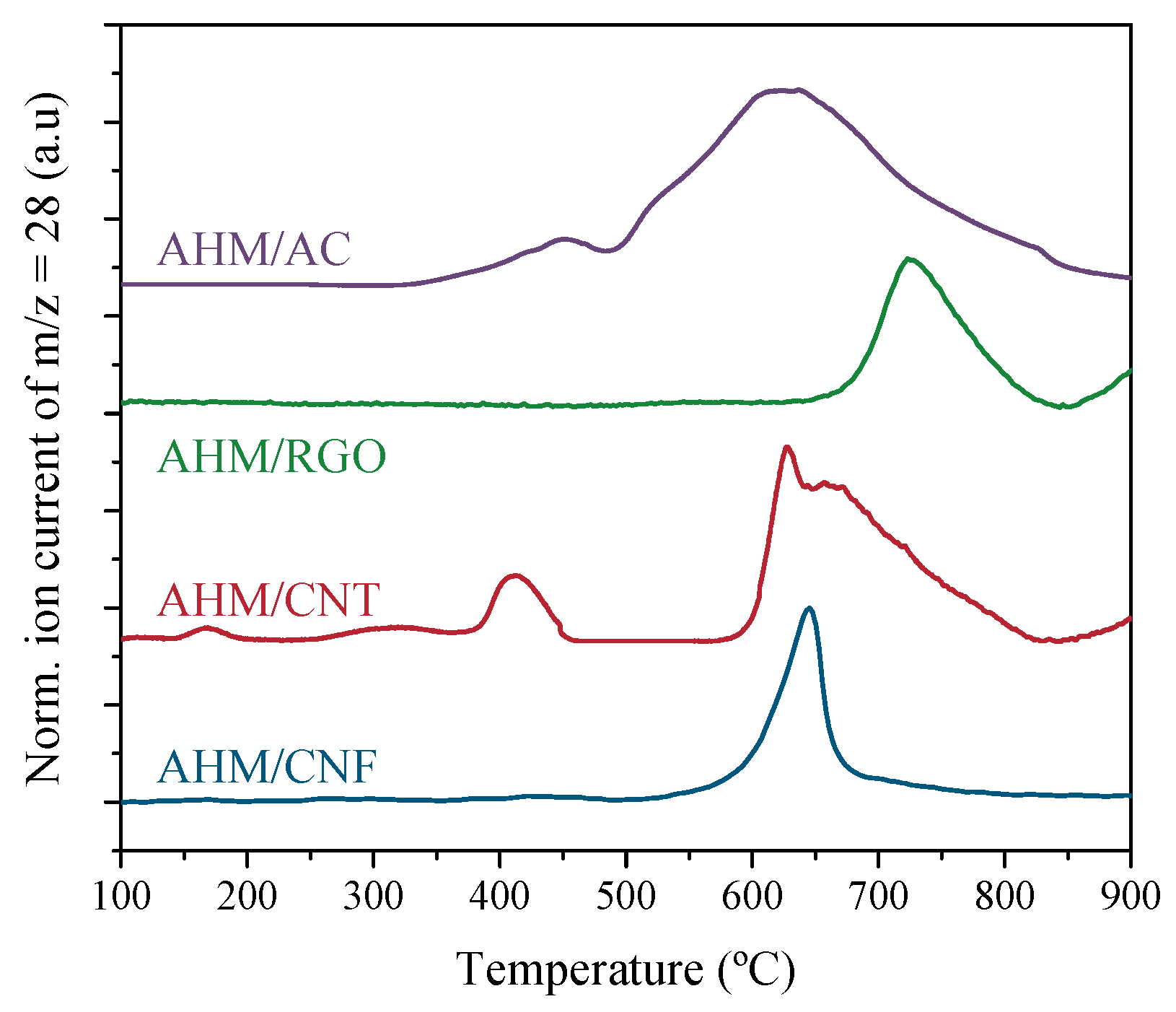
3.2. Carburization Study in Thermobalance
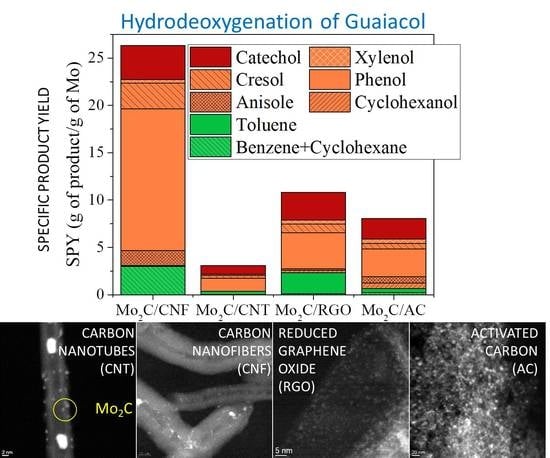
3.3. Characterization of Catalysts Using Different NCM
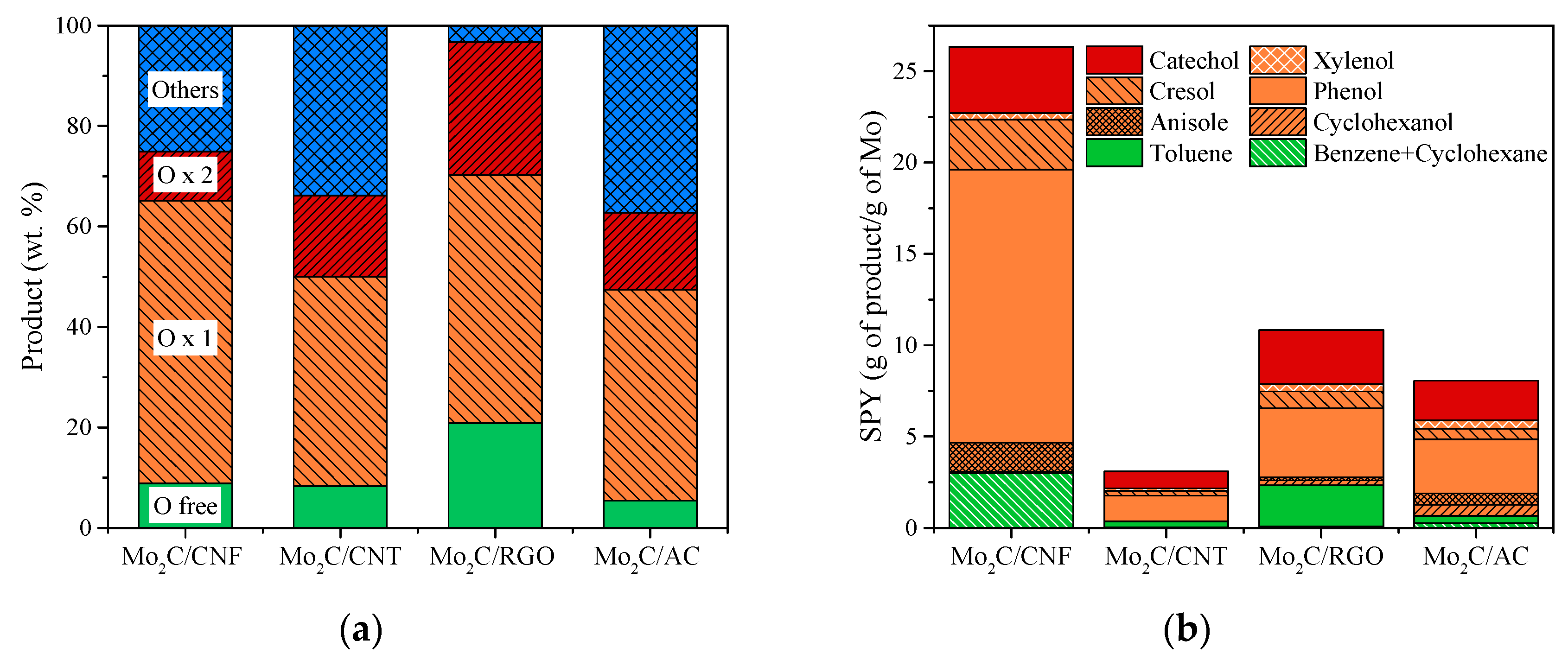
3.4. Catalytic HDO of Guaiacol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Volpe, L.; Boudart, M. Compounds of molybdenum and tungsten with high specific surface area: II. Carbides. J. Solid State Chem. 1985, 59, 348–356. [Google Scholar] [CrossRef]
- Dhandapani, B.; Clair, T.S.; Oyama, S.T. Simultaneous hydrodesulfurization, hydrodeoxygenation, and hydrogenation with molybdenum carbide. Appl. Catal. A: Gen. 1998, 168, 219–228. [Google Scholar] [CrossRef]
- Claridge, J.B.; York, A.P.E.; Brungs, A.J.; Green, M.L.H. Study of the Temperature-Programmed Reaction Synthesis of Early Transition Metal Carbide and Nitride Catalyst Materials from Oxide Precursors. Chem. Mater. 2000, 12, 132–142. [Google Scholar] [CrossRef]
- Xiao, T.-C.; York, A.P.E.; Williams, V.C.; Al-Megren, H.; Hanif, A.; Zhou, X.-Y.; Green, M.L.H. Preparation of Molybdenum Carbides Using Butane and Their Catalytic Performance. Chem. Mater. 2000, 12, 3896–3905. [Google Scholar] [CrossRef]
- Brungs, A.J.; York, A.P.E.; Claridge, J.B.; Márquez-Alvarez, C.; Green, M.L.H. Dry reforming of methane to synthesis gas over supported molybdenum carbide catalysts. Catal. Lett. 2000, 70, 117–122. [Google Scholar] [CrossRef]
- Xiao, T.; Hanif, A.; York, A.P.E.; Sloan, J.; Green, M.L.H. Study on preparation of high surface area tungsten carbides and phase transition during the carburisation. Phys. Chem. Chem. Phys. 2002, 4, 3522–3529. [Google Scholar] [CrossRef]
- Hanif, A.; Xiao, T.; York, A.P.E.; Sloan, J.; Green, M.L.H. Study on the Structure and Formation Mechanism of Molybdenum Carbides. Chem. Mater. 2002, 14, 1009–1015. [Google Scholar] [CrossRef]
- Ramanathan, S.; Oyama, S.T. New Catalysts for Hydroprocessing: Transition Metal Carbides and Nitrides. J. Phys. Chem. 1995, 99, 16365–16372. [Google Scholar] [CrossRef]
- Mordenti, D.; Brodzki, D.; Djéga-Mariadassou, G. New Synthesis of Mo2C 14 nm in Average Size Supported on a High Specific Surface Area Carbon Material. J. Solid State Chem. 1998, 141, 114–120. [Google Scholar] [CrossRef]
- Liang, C.; Ying, P.; Li, C. Nanostructured β-Mo2C Prepared by Carbothermal Hydrogen Reduction on Ultrahigh Surface Area Carbon Material. Chem. Mater. 2002, 14, 3148–3151. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Smith, K.J. Synthesis and Hydrodeoxygenation Activity of Carbon Supported Molybdenum Carbide and Oxycarbide Catalysts. Energy Fuels 2016, 30, 6039–6049. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Liu, B.; Montes, V.; Hill, J.M.; Smith, K.J. Carbon and Mo transformations during the synthesis of mesoporous Mo2C/carbon catalysts by carbothermal hydrogen reduction. J. Solid State Chem. 2018, 258, 818–824. [Google Scholar] [CrossRef]
- Liang, C.; Wei, Z.; Xin, Q.; Li, C. Synthesis and characterization of nanostructured Mo2C on carbon material by carbothermal hydrogen reduction. In Studies in Surface Science and Catalysis; Gaigneaux, E., de Vos, D.E., Grange, P., Jacobs, P.A., Martens, J.A., Ruiz, P., Poncelet, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2000; pp. 975–982. [Google Scholar]
- Wang, H.; Liu, S.; Govindarajan, R.; Smith, K.J. Preparation of Ni-Mo2C/carbon catalysts and their stability in the HDS of dibenzothiophene. Appl. Catal. A: Gen. 2017, 539, 114–127. [Google Scholar] [CrossRef]
- Souza Macedo, L.; Stellwagen, D.R.; Teixeira da Silva, V.; Bitter, J.H. Stability of Transition-metal Carbides in Liquid Phase Reactions Relevant for Biomass-Based Conversion. ChemCatChem 2015, 7, 2816–2823. [Google Scholar] [CrossRef]
- Lee, J.S.; Oyama, S.T.; Boudart, M. Molybdenum carbide catalysts. J. Catal. 1987, 106, 125–133. [Google Scholar] [CrossRef]
- Guil-López, R.; Nieto, E.; Botas, J.A.; Fierro, J.L.G. On the genesis of molybdenum carbide phases during reduction-carburization reactions. J. Solid State Chem. 2012, 190, 285–295. [Google Scholar] [CrossRef]
- Ochoa, E.; Torres, D.; Moreira, R.; Pinilla, J.L.; Suelves, I. Carbon nanofiber supported Mo2C catalysts for hydrodeoxygenation of guaiacol: The importance of the carburization process. Appl. Catal. B 2018, 239, 463–474. [Google Scholar] [CrossRef]
- Ochoa, E.; Torres, D.; Pinilla, J.L.; Suelves, I. Influence of carburization time on the activity of Mo2C/CNF catalysts for the HDO of guaiacol. Catal. Today 2019. [Google Scholar] [CrossRef]
- Jongerius, A.L.; Gosselink, R.W.; Dijkstra, J.; Bitter, J.H.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Carbon Nanofiber Supported Transition-Metal Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. ChemCatChem 2013, 5, 2964–2972. [Google Scholar] [CrossRef]
- Qin, Y.; Chen, P.; Duan, J.; Han, J.; Lou, H.; Zheng, X.; Hong, H. Carbon nanofibers supported molybdenum carbide catalysts for hydrodeoxygenation of vegetable oils. RSC Adv. 2013, 3, 17485–17491. [Google Scholar] [CrossRef]
- Han, J.; Duan, J.; Chen, P.; Lou, H.; Zheng, X. Molybdenum Carbide-Catalyzed Conversion of Renewable Oils into Diesel-like Hydrocarbons. Adv. Synth. Catal. 2011, 353, 2577–2583. [Google Scholar] [CrossRef]
- Hollak, S.A.W.; Gosselink, R.W.; van Es, D.S.; Bitter, J.H. Comparison of Tungsten and Molybdenum Carbide Catalysts for the Hydrodeoxygenation of Oleic Acid. ACS Catal. 2013, 3, 2837–2844. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Perdu, M.; Pace, R.; Morgan, T.; Crocker, M. Activated Carbon, Carbon Nanofiber and Carbon Nanotube Supported Molybdenum Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. Catalysts 2015, 5, 424. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Smith, K.J.; Kim, C.S. Hydrodeoxygenation of 2-Methoxyphenol over Ru, Pd, and Mo2C Catalysts Supported on Carbon. Energy Fuels 2017, 31, 6378–6388. [Google Scholar] [CrossRef]
- Pielaszek, J.; Mierzwa, B.; Medjahdi, G.; Marêché, J.F.; Puricelli, S.; Celzard, A.; Furdin, G. Molybdenum carbide catalyst formation from precursors deposited on active carbons: XRD studies. Appl. Catal. A: Gen. 2005, 296, 232–237. [Google Scholar] [CrossRef]
- He, L.; Qin, Y.; Lou, H.; Chen, P. Highly dispersed molybdenum carbide nanoparticles supported on activated carbon as an efficient catalyst for the hydrodeoxygenation of vanillin. RSC Adv. 2015, 5, 43141–43147. [Google Scholar] [CrossRef]
- Frank, B.; Friedel, K.; Girgsdies, F.; Huang, X.; Schlögl, R.; Trunschke, A. CNT-Supported MoxC Catalysts: Effect of Loading and Carburization Parameters. ChemCatChem 2013, 5, 2296–2305. [Google Scholar] [CrossRef]
- Frank, B.; Xie, Z.-L.; Ortega, K.F.; Scherzer, M.; Schlogl, R.; Trunschke, A. Modification of the carbide microstructure by N- and S-functionalization of the support in MoxC/CNT catalysts. Catal. Sci. Technol. 2016, 6, 3468–3475. [Google Scholar] [CrossRef]
- Barthos, R.; Széchenyi, A.; Solymosi, F. Efficient H2 Production from Ethanol over Mo2C/C Nanotube Catalyst. Catal. Lett. 2008, 120, 161–165. [Google Scholar] [CrossRef]
- Li, X.; Ma, D.; Chen, L.; Bao, X. Fabrication of molybdenum carbide catalysts over multi-walled carbon nanotubes by carbothermal hydrogen reduction. Catal. Lett. 2007, 116, 63–69. [Google Scholar] [CrossRef]
- Stellwagen, D.R.; Bitter, J.H. Structure-performance relations of molybdenum- and tungsten carbide catalysts for deoxygenation. Green Chem. 2015, 17, 582–593. [Google Scholar] [CrossRef]
- Moreira, R.; Ochoa, E.; Pinilla, J.; Portugal, A.; Suelves, I. Liquid-Phase Hydrodeoxygenation of Guaiacol over Mo2C Supported on Commercial CNF. Effects of Operating Conditions on Conversion and Product Selectivity. Catalysts 2018, 8, 127. [Google Scholar] [CrossRef]
- Qin, Y.; He, L.; Duan, J.; Chen, P.; Lou, H.; Zheng, X.; Hong, H. Carbon-Supported Molybdenum-Based Catalysts for the Hydrodeoxygenation of Maize Oil. ChemCatChem 2014, 6, 2698–2705. [Google Scholar] [CrossRef]
- Choi, J.-S.; Bugli, G.; Djéga-Mariadassou, G. Influence of the Degree of Carburization on the Density of Sites and Hydrogenating Activity of Molybdenum Carbides. J. Catal. 2000, 193, 238–247. [Google Scholar] [CrossRef]
- He, Z.; Wang, X. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Energy 2012, 1, 28–52. [Google Scholar] [CrossRef]
- Roldugina, E.A.; Naranov, E.R.; Maximov, A.L.; Karakhanov, E.A. Hydrodeoxygenation of guaiacol as a model compound of bio-oil in methanol over mesoporous noble metal catalysts. Appl. Catal. A: Gen. 2018, 553, 24–35. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Li, D.; Bui, P.; Oyama, S.T. Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts. Appl. Catal. A 2011, 391, 305–310. [Google Scholar] [CrossRef]
- Pinilla, J.L.; Utrilla, R.; Lázaro, M.J.; Suelves, I.; Moliner, R.; Palacios, J.M. A novel rotary reactor configuration for simultaneous production of hydrogen and carbon nanofibers. Int. J. Hydrog. Energy 2009, 34, 8016–8022. [Google Scholar] [CrossRef]
- Torres, D.; Pinilla, J.L.; Suelves, I. Unzipping of multi-wall carbon nanotubes with different diameter distributions: Effect on few-layer graphene oxide obtention. Appl. Surf. Sci. 2017, 424, 101–110. [Google Scholar] [CrossRef]
- Torres, D.; Pinilla, J.L.; Lázaro, M.J.; Moliner, R.; Suelves, I. Hydrogen and multiwall carbon nanotubes production by catalytic decomposition of methane: Thermogravimetric analysis and scaling-up of Fe–Mo catalysts. Int. J. Hydrog. Energy 2014, 39, 3698–3709. [Google Scholar] [CrossRef]
- Torres, D.; Pinilla, J.L.; Moliner, R.; Suelves, I. On the oxidation degree of few-layer graphene oxide sheets obtained from chemically oxidized multiwall carbon nanotubes. Carbon 2015, 81, 405–417. [Google Scholar] [CrossRef]
- Torres, D.; Pinilla, J.L.; Galvez, E.M.; Suelves, I. Graphene quantum dots from fishbone carbon nanofibers. RSC Adv. 2016, 6, 48504–48514. [Google Scholar] [CrossRef]
- Ciembroniewicz, G.; Dziembaj, R.; Kalicki, R. Thermal decomposition of Ammonium paramolybdate (APM). J. Therm. Anal. 1983, 27, 125–138. [Google Scholar] [CrossRef]
- Kundu, S.; Wang, Y.; Xia, W.; Muhler, M. Thermal Stability and Reducibility of Oxygen-Containing Functional Groups on Multiwalled Carbon Nanotube Surfaces: A Quantitative High-Resolution XPS and TPD/TPR Study. J. Phys. Chem. C 2008, 112, 16869–16878. [Google Scholar] [CrossRef]
- Liu, C.; Lin, M.; Fang, K.; Meng, Y.; Sun, Y. Preparation of nanostructured molybdenum carbides for CO hydrogenation. RSC Adv. 2014, 4, 20948–20954. [Google Scholar] [CrossRef]
- Chang, J.; Danuthai, T.; Dewiyanti, S.; Wang, C.; Borgna, A. Hydrodeoxygenation of Guaiacol over Carbon-Supported Metal Catalysts. ChemCatChem 2013, 5, 3041–3049. [Google Scholar] [CrossRef]
- Pinilla, J.L.; García, A.B.; Philippot, K.; Lara, P.; García-Suárez, E.J.; Millan, M. Carbon-supported Pd nanoparticles as catalysts for anthracene hydrogenation. Fuel 2014, 116, 729–735. [Google Scholar] [CrossRef]
- Lu, M.; Lu, F.; Zhu, J.; Li, M.; Zhu, J.; Shan, Y. Hydrodeoxygenation of methyl stearate as a model compound over Mo2C supported on mesoporous carbon. React. Kinet. Mech. Catal. 2015, 115, 251–262. [Google Scholar] [CrossRef]
- Bej, S.K.; Bennett, C.A.; Thompson, L.T. Acid and base characteristics of molybdenum carbide catalysts. Appl. Catal. A: Gen. 2003, 250, 197–208. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCM | XPS (at.%) | N2 Physisorption | |||||
|---|---|---|---|---|---|---|---|
| C | O | S | N | Others | SBET (m2/g) | Vt (cm3/g) | |
| CNF | 93.2 | 4.8 | 0.0 | 0.0 | 2.0 | 99 | 0.463 |
| CNT | 95.0 | 4.3 | 0.0 | 0.7 | 0.0 | 104 | 0.590 |
| RGO | 70.3 | 20.5 | 6.7 | 2.5 | 0.0 | 23 | 0.105 |
| AC | 90.7 | 7.4 | 0.0 | 0.0 | 1.9 | 1230 | 0.913 |
| CHR | XRD (nm) | ICP-OES (wt.%) | XPS (at.%) | N2 Phys. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | T (°C) | Wt. loss (%) | β-Mo2C | Mo | C | O | Mo | Others | Mo6+ | Mo4+ | Moδ+ | Mo2+ | SBET (m2/g) | Vt (cm3/g) |
| Mo2C/CNF | 750 | 30 | 10.9 | 12.9 | 92.6 | 5.1 | 2.3 | 0.01 | 68.8 | 2.6 | 11.7 | 16.8 | 64 | 0.398 |
| Mo2C/CNT | 650 | 70 | 15.5 | 25.6 | 89.7 | 7.0 | 3.2 | 0.14 | 69.8 | 3.3 | 10.4 | 16.5 | 106 | 0.551 |
| Mo2C/RGO | 750 | 58 | 13.6 | 13.7 | 69.2 | 12.5 | 6.6 | 11.8 | 80.6 | 1.9 | 3.1 | 14.5 | 40 | 0.097 |
| Mo2C/AC | 800 | 70 | 12.5 | 23.6 | 85.8 | 7.9 | 3.1 | 3.13 | 89.6 | 7.9 | 0.04 | 2.5 | 572 | 0.653 |
| Catalyst | Conversion (%) | HDO Ratio (%) | Mass Balance (%) |
|---|---|---|---|
| Mo2C/CNF | 67.0 | 37.20 | 83.28 |
| Mo2C/CNT | 14.23 | 9.09 | 94.34 |
| Mo2C/RGO | 20.68 | 9.59 | 99.99 |
| Mo2C/AC | 42.00 | 24.97 | 83.72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochoa, E.; Torres, D.; Pinilla, J.L.; Suelves, I. Nanostructured Carbon Material Effect on the Synthesis of Carbon-Supported Molybdenum Carbide Catalysts for Guaiacol Hydrodeoxygenation. Energies 2020, 13, 1189. https://doi.org/10.3390/en13051189
Ochoa E, Torres D, Pinilla JL, Suelves I. Nanostructured Carbon Material Effect on the Synthesis of Carbon-Supported Molybdenum Carbide Catalysts for Guaiacol Hydrodeoxygenation. Energies. 2020; 13(5):1189. https://doi.org/10.3390/en13051189
Chicago/Turabian StyleOchoa, Elba, Daniel Torres, José Luis Pinilla, and Isabel Suelves. 2020. "Nanostructured Carbon Material Effect on the Synthesis of Carbon-Supported Molybdenum Carbide Catalysts for Guaiacol Hydrodeoxygenation" Energies 13, no. 5: 1189. https://doi.org/10.3390/en13051189
APA StyleOchoa, E., Torres, D., Pinilla, J. L., & Suelves, I. (2020). Nanostructured Carbon Material Effect on the Synthesis of Carbon-Supported Molybdenum Carbide Catalysts for Guaiacol Hydrodeoxygenation. Energies, 13(5), 1189. https://doi.org/10.3390/en13051189