A Novel Oncogenic and Drug-Sensitive KIF5B-NTRK1 Fusion in Lung Adenocarcinoma

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Lines and Reagents
2.2. Next-Generation Sequencing (NGS) and Data Processing
2.3. Fluorescence In Situ Hybridization (FISH) Assay
2.4. Immunohistochemistry (IHC)
2.5. Stable Cell Line Generation
2.6. Polymerase Chain Reaction (PCR)
2.7. Cell Proliferation Assay
2.8. Adhesion-Dependent Colony Formation Assay
2.9. Transwell Invasion Assay
2.10. Western Blotting (WB)
2.11. Statistical Analysis
3. Results
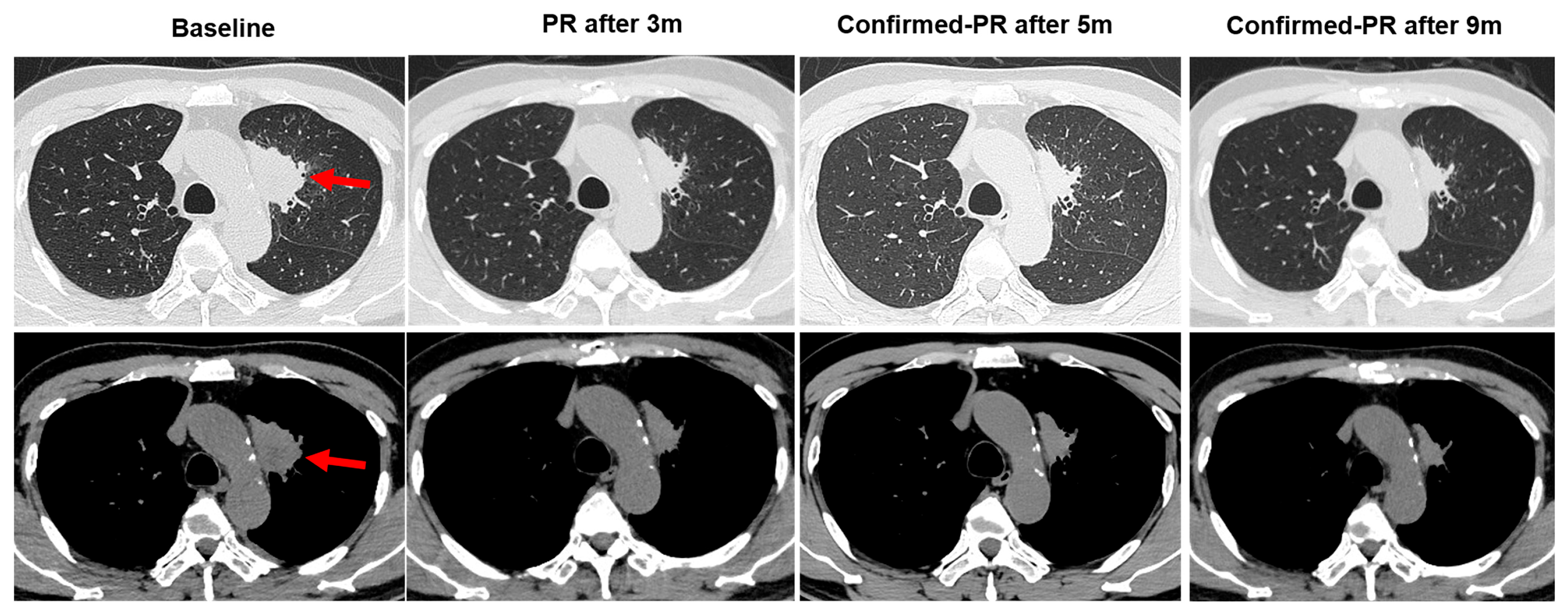
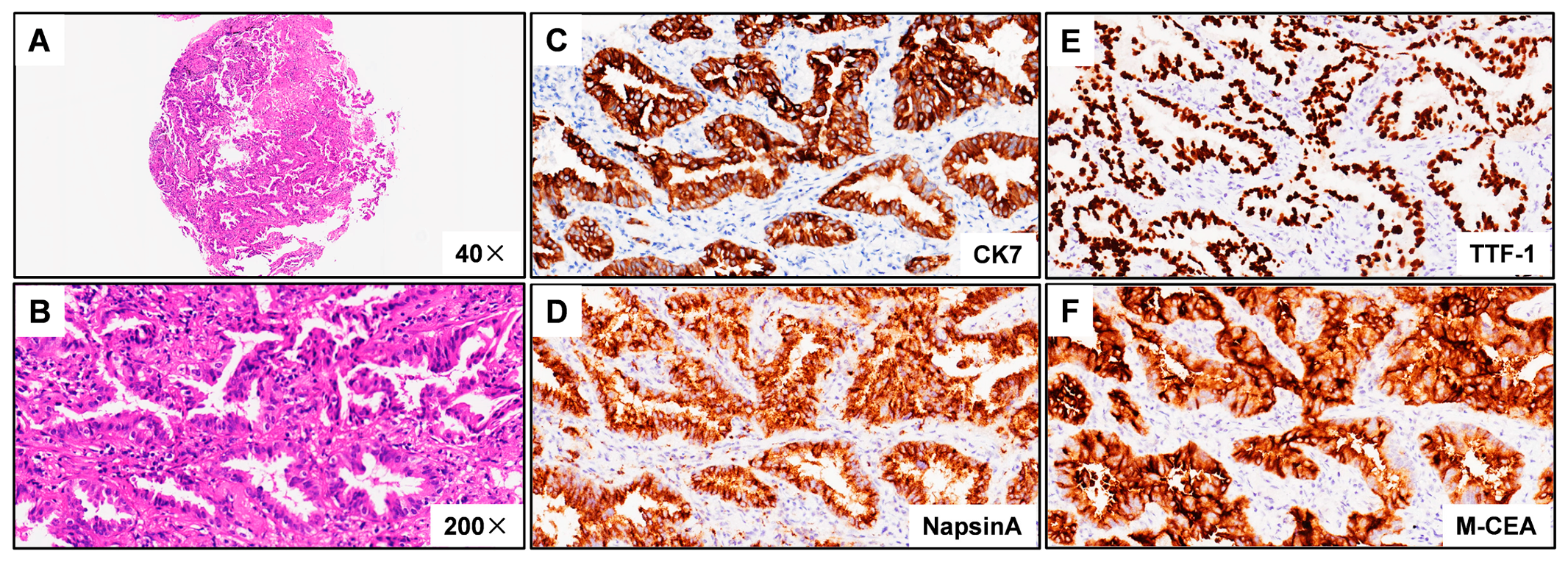
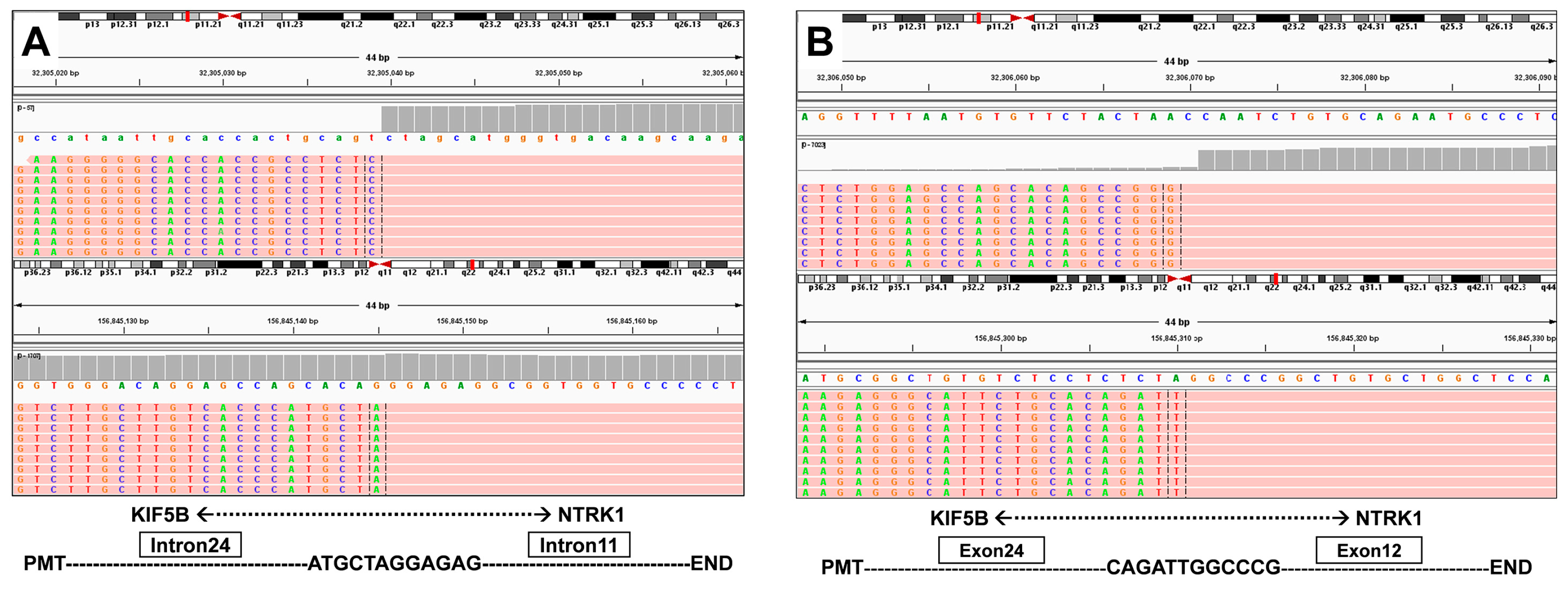
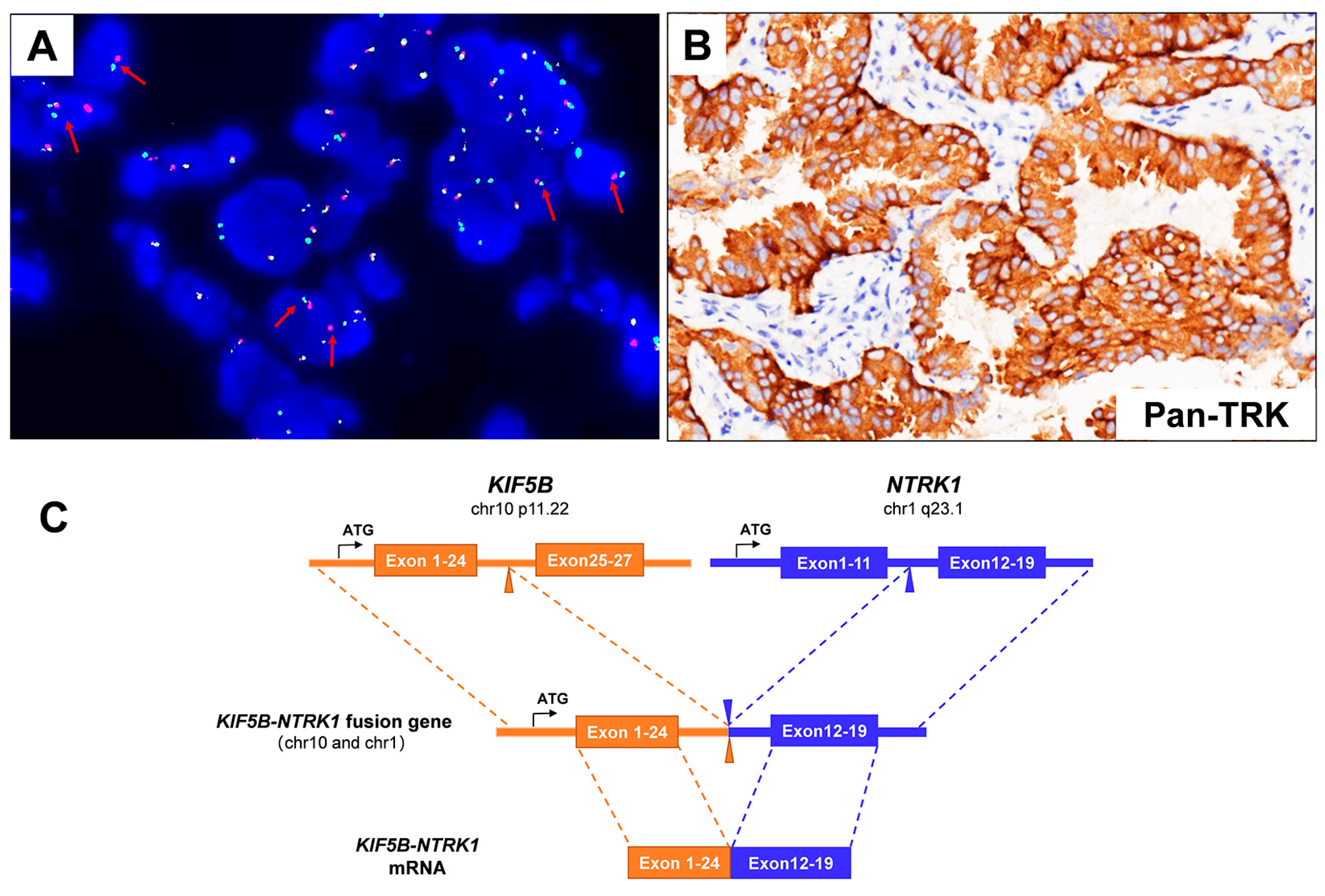
3.1. Case Description
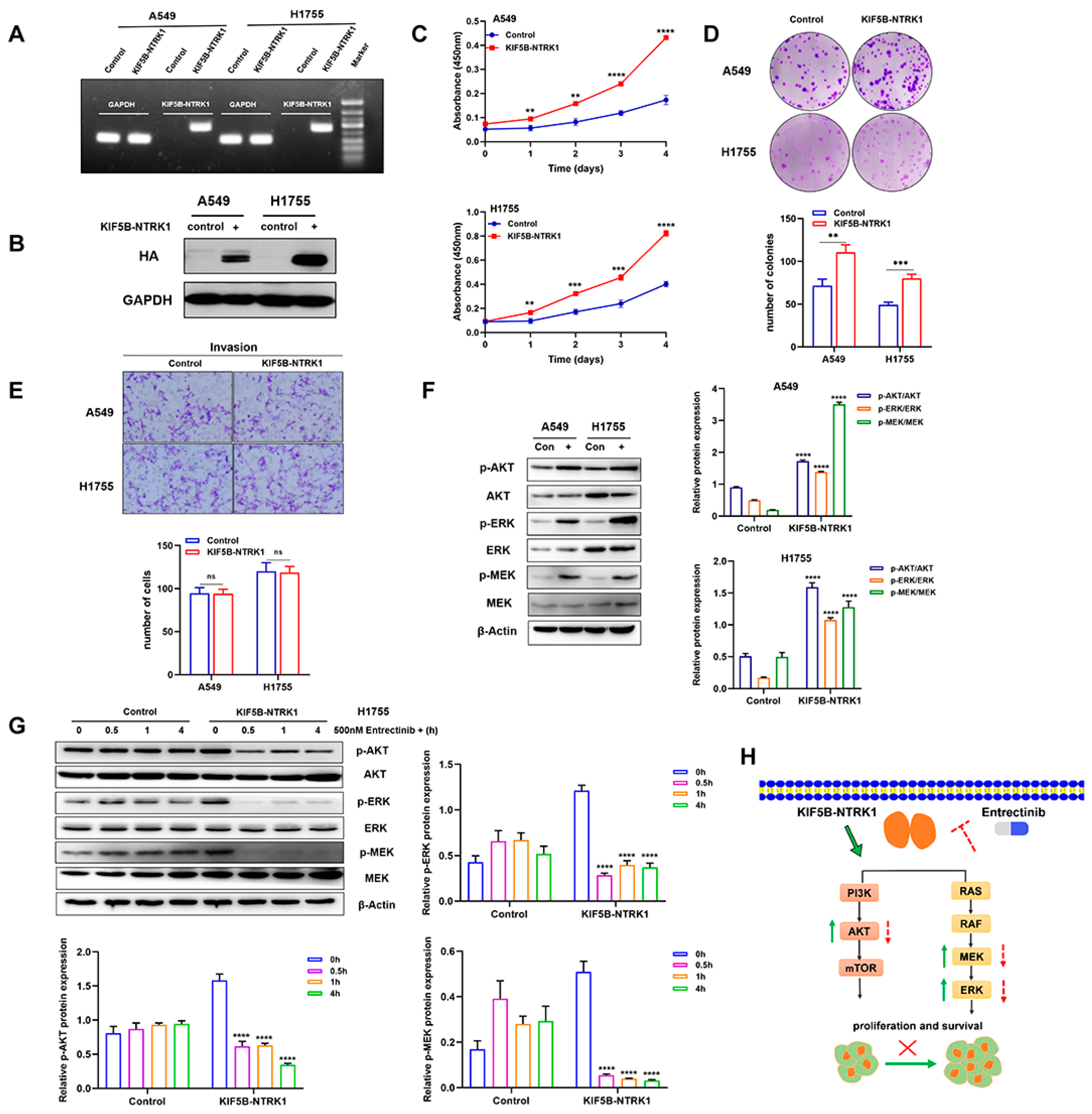
3.2. KIF5B-NTRK1 Promotes Proliferation and Adhesion-Dependent Colony Formation but Not Invasion
3.3. KIF5B-NTRK1 Activates the MAPK and PI3K-AKT Signaling Pathways
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valent, A.; Danglot, G.; Bernheim, A. Mapping of the tyrosine kinase receptors trkA (NTRK1), trkB (NTRK2) and trkC(NTRK3) to human chromosomes 1q22, 9q22 and 15q25 by fluorescence in situ hybridization. Eur. J. Hum. Genet. 1997, 5, 102–104. [Google Scholar] [CrossRef]
- Cunningham, M.E.; Greene, L.A. A function-structure model for NGF-activated TRK. EMBO J. 1998, 17, 7282–7293. [Google Scholar] [CrossRef] [PubMed]
- Barbacid, M. The Trk family of neurotrophin receptors. J. Neurobiol. 1994, 25, 1386–1403. [Google Scholar] [CrossRef]
- Martin-Zanca, D.; Hughes, S.H.; Barbacid, M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 1986, 319, 743–748. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Martin-Zanca, D.; Parada, L.F. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature 1991, 350, 158–160. [Google Scholar] [CrossRef]
- Knezevich, S.R.; McFadden, D.E.; Tao, W.; Lim, J.F.; Sorensen, P.H. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat. Genet. 1998, 18, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Knezevich, S.R.; Garnett, M.J.; Pysher, T.J.; Beckwith, J.B.; Grundy, P.E.; Sorensen, P.H. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 1998, 58, 5046–5048. [Google Scholar]
- Rubin, B.P.; Chen, C.-J.; Morgan, T.W.; Xiao, S.; Grier, H.E.; Kozakewich, H.P.; Perez-Atayde, A.R.; Fletcher, J.A. Congenital Mesoblastic Nephroma t(12;15) Is Associated withETV6-NTRK3 Gene Fusion. Am. J. Pathol. 1998, 153, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, I.; Pierotti, M.A.; Monzini, N.; Mondellini, P.; Manenti, G.; Donghi, R.; Pilotti, S.; Grieco, M.; Santoro, M.; Fusco, A.; et al. High frequency of activation of tyrosine kinase oncogenes in human papillary thyroid carcinoma. Oncogene 1989, 4, 1457–1462. [Google Scholar]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, S.; Ko, S.; In, Y.; Moon, H.; Ahn, S.K.; Kim, M.K.; Lee, M.; Hwang, J.; Ju, Y.S.; et al. Recurrent fusion transcripts detected by whole-transcriptome sequencing of 120 primary breast cancer samples. Genes Chromosom. Cancer 2015, 54, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Farago, A.F.; Taylor, M.S.; Doebele, R.C.; Zhu, V.W.; Kummar, S.; Spira, A.I.; Boyle, T.A.; Haura, E.B.; Arcila, M.E.; Benayed, R.; et al. Clinicopathologic Features of Non–Small-Cell Lung Cancer Harboring an NTRK Gene Fusion. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Cho, B.C.; Chiu, C.-H.; Massarelli, E.; Buchschacher, G.L.; Goto, K.; Overbeck, T.R.; Loong, H.H.; Chee, C.E.; Garrido, P.; Dong, X.; et al. Updated efficacy and safety of entrectinib in NTRK fusion-positive non-small cell lung cancer. Lung Cancer 2024, 188, 107442. [Google Scholar] [CrossRef]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.; Tosi, F.; Siena, S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann. Oncol. 2019, 30, viii5–viii15. [Google Scholar] [CrossRef]
- Doebele, R.C.; Davis, L.E.; Vaishnavi, A.; Le, A.T.; Estrada-Bernal, A.; Keysar, S.; Jimeno, A.; Varella-Garcia, M.; Aisner, D.L.; Li, Y.; et al. An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov. 2015, 5, 1049–1057. [Google Scholar] [CrossRef]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; de Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a Pan–TRK, ROS1, and ALK Inhibitor with Activity in Multiple Molecularly Defined Cancer Indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Tan, D.S.W.; Lassen, U.N.; Leyvraz, S.; Liu, Y.; Patel, J.D.; Rosen, L.; Solomon, B.; Norenberg, R.; Dima, L.; et al. Efficacy and Safety of Larotrectinib in Patients with Tropomyosin Receptor Kinase Fusion–Positive Lung Cancers. JCO Precis. Oncol. 2022, 6, e2100418. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Liu, Y.; Li, H.; Chen, S.; Guo, Y. Emergence of NOTCH2-NTRK1 After Osimertinib in a Patient with Lung Adenocarcinoma with Neuroendocrine Differentiation. J. Thorac. Oncol. 2021, 22, e157–e159. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pizzutilo, E.G.; Marrapese, G.; Tosi, F.; Cerea, G.; Siena, S. Entrectinib for the treatment of metastatic NSCLC: Safety and efficacy. Expert Rev. Anticancer. Ther. 2020, 20, 333–341. [Google Scholar] [CrossRef]
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Molecular characterization of cancers with NTRK gene fusions. Mod. Pathol. 2019, 32, 147–153. [Google Scholar] [CrossRef]
- Takeuchi, K.; Choi, Y.L.; Togashi, Y.; Soda, M.; Hatano, S.; Inamura, K.; Takada, S.; Ueno, T.; Yamashita, Y.; Satoh, Y.; et al. KIF5B-ALK, a Novel Fusion Oncokinase Identified by an Immunohistochemistry-based Diagnostic System for ALK-positive Lung Cancer. Clin. Cancer Res. 2009, 15, 3143–3149. [Google Scholar] [CrossRef]
- Nagasaka, M.; Brazel, D.; Baca, Y.; Xiu, J.; Al-Hallak, M.N.; Kim, C.; Nieva, J.; Swensen, J.J.; Spetzler, D.; Korn, W.M.; et al. Pan-tumor survey of RET fusions as detected by next-generation RNA sequencing identified RET fusion positive colorectal carcinoma as a unique molecular subset. Transl. Oncol. 2023, 36, 101744. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-Kinase Pathway in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef]
- Dhakar, R.; Dakal, T.C.; Sharma, A. Genetic determinants of lung cancer: Understanding the oncogenic potential of somatic missense mutations. Genomics 2022, 114, 110401. [Google Scholar] [CrossRef]
- Sciot, R. MDM2 Amplified Sarcomas: A Literature Review. Diagnostics 2021, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The Role of MDM2 Amplification and Overexpression in Tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6, a026336. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Zou, Y.; Patel, A.S.; Yoo, S.; Jiang, F.; Sato, T.; Kong, R.; Watanabe, H.; Zhu, J.; Massion, P.P.; et al. Early-Stage Lung Adenocarcinoma MDM2 Genomic Amplification Predicts Clinical Outcome and Response to Targeted Therapy. Cancers 2022, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mori, M.; Taira, M.; Yoshida, T.; Matsukawa, S.; Shimizu, K.; Sekiguchi, M.; Terada, M.; Sugimura, T. Transforming Gene from Human Stomach Cancers and a Noncancerous Portion of Stomach Mucosa. Proc. Natl. Acad. Sci. USA 1986, 83, 3997–4001. [Google Scholar] [CrossRef]
- Qi, L.; Song, W.; Li, L.; Cao, L.; Yu, Y.; Song, C.; Wang, Y.; Zhang, F.; Li, Y.; Zhang, B.; et al. FGF4 induces epithelial-mesenchymal transition by inducing store-operated calcium entry in lung adenocarcinoma. Oncotarget 2016, 7, 74015–74030. [Google Scholar] [CrossRef]
- Tan, Q.; Li, F.; Wang, G.; Xia, W.; Li, Z.; Niu, X.; Ji, W.; Yuan, H.; Xu, Q.; Luo, Q.; et al. Identification of FGF19 as a prognostic marker and potential driver gene of lung squamous cell carcinomas in Chinese smoking patients. Oncotarget 2016, 7, 18394–18402. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Zhang, J.; Li, W.; Yuan, P.; Xing, P.; Zhang, Z.; Chuai, S.; Li, J.; Ying, J. Response to crizotinib in advanced ALK -rearranged non-small cell lung cancers with different ALK-fusion variants. Lung Cancer 2018, 118, 128–133. [Google Scholar] [CrossRef]
- Iannantuono, G.M.; Riondino, S.; Sganga, S.; Rosenfeld, R.; Guerriero, S.; Carlucci, M.; Capotondi, B.; Torino, F.; Roselli, M. NTRK Gene Fusions in Solid Tumors and TRK Inhibitors: A Systematic Review of Case Reports and Case Series. J. Pers. Med. 2022, 12, 1819. [Google Scholar] [CrossRef]
- Russo, M.; Misale, S.; Wei, G.; Siravegna, G.; Crisafulli, G.; Lazzari, L.; Corti, G.; Rospo, G.; Novara, L.; Mussolin, B.; et al. Acquired Resistance to the TRK Inhibitor Entrectinib in Colorectal Cancer. Cancer Discov. 2016, 6, 36–44. [Google Scholar] [CrossRef]
- Drilon, A.; Li, G.; Dogan, S.; Gounder, M.; Shen, R.; Arcila, M.; Wang, L.; Hyman, D.M.; Hechtman, J.; Wei, G.; et al. What hides behind the MASC: Clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann. Oncol. 2016, 27, 920–926. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Liu, H.; Xiao, L.; Gao, H.; Wei, H.; Han, A.; Lin, G. A Novel Oncogenic and Drug-Sensitive KIF5B-NTRK1 Fusion in Lung Adenocarcinoma. Curr. Oncol. 2024, 31, 6621-6631. https://doi.org/10.3390/curroncol31110489
Li H, Liu H, Xiao L, Gao H, Wei H, Han A, Lin G. A Novel Oncogenic and Drug-Sensitive KIF5B-NTRK1 Fusion in Lung Adenocarcinoma. Current Oncology. 2024; 31(11):6621-6631. https://doi.org/10.3390/curroncol31110489
Chicago/Turabian StyleLi, Hui, Huicong Liu, Lisha Xiao, Huabin Gao, Huiting Wei, Anjia Han, and Gengpeng Lin. 2024. "A Novel Oncogenic and Drug-Sensitive KIF5B-NTRK1 Fusion in Lung Adenocarcinoma" Current Oncology 31, no. 11: 6621-6631. https://doi.org/10.3390/curroncol31110489
APA StyleLi, H., Liu, H., Xiao, L., Gao, H., Wei, H., Han, A., & Lin, G. (2024). A Novel Oncogenic and Drug-Sensitive KIF5B-NTRK1 Fusion in Lung Adenocarcinoma. Current Oncology, 31(11), 6621-6631. https://doi.org/10.3390/curroncol31110489