Acute Myeloid Leukemia in Older Patients: From New Biological Insights to Targeted Therapies

 ,
,  and
and
Abstract
1. Introduction
2. Search Strategy and Selection Criteria
3. AML Profiling for Diagnosis and Prognosis
3.1. Nucleic Acids
3.2. Proteomic Profiling
3.3. Metabolic Profiling
3.4. Ambience Profiling
3.5. Extensive Data Analysis, Machine Learning, and AI Tools
3.5.1. Diagnostic and Classification Challenges
3.5.2. Older Patients: New Approaches in Risk Assessment and Monitoring of AML
3.5.3. A Comprehensive Approach in Older AML Patients
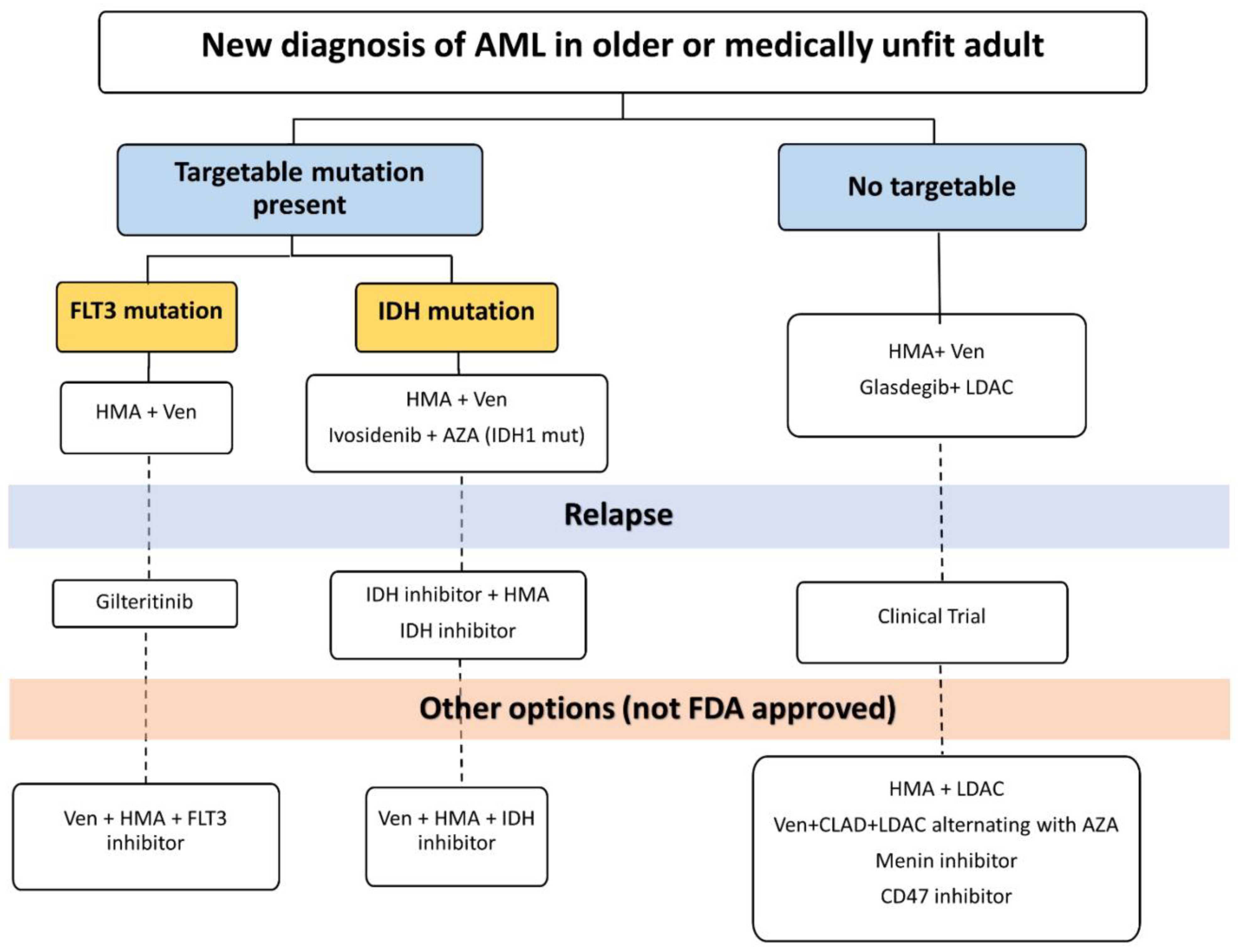
4. Clinical Management of AML in Older Patients in the Current Era
4.1. The AZA/VEN “Revolution”
4.2. Hedgehog Pathway Inhibition
4.3. IDH1/2 Inhibition
4.4. FLT3 Inhibition
4.5. More Therapies for Elder AML Patients
4.6. Intensive Chemotherapy and Allogeneic SCT
4.7. The Holistic Approach to Managing AML in Older Patients: Prioritizing Toxicities Management Alongside Quality of Life and Early Palliative
5. Summary
6. Key Points
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Venugopal, S.; Sekeres, M.A. Contemporary Management of Acute Myeloid Leukemia: A Review. JAMA Oncol. 2024, 8, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Wachter, F.; Pikman, Y. Pathophysiology of Acute Myeloid Leukemia. Acta Haematol. 2024, 147, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.; Abdul-Hay, M.; Choi, J.H. Molecular Features and Treatment Paradigms of Acute Myeloid Leukemia. Biomedicines 2024, 12, 1768. [Google Scholar] [CrossRef]
- Bataller, A.; DiNardo, C.D.; Bazinet, A.; Daver, N.G.; Maiti, A.; Borthakur, G.; Short, N.; Sasaki, K.; Jabbour, E.J.; Issa, G.C.; et al. Targetable genetic abnormalities in patients with acute myeloblastic leukemia across age groups. Am. J. Hematol. 2024, 99, 792–796. [Google Scholar] [CrossRef]
- Aung, M.M.K.; Mills, M.L.; Bittencourt-Silvestre, J.; Keeshan, K. Insights into the molecular profiles of adult and paediatric acute myeloid leukaemia. Mol. Oncol. 2021, 15, 2253–2272. [Google Scholar] [CrossRef]
- Han, H.J.; Choi, K.; Suh, H.S. Impact of aging on acute myeloid leukemia epidemiology and survival outcomes: A real-world, population-based longitudinal cohort study. PLoS ONE 2024, 19, e0300637. [Google Scholar] [CrossRef]
- Snaith, O.; Poveda-Rogers, C.; Laczko, D.; Yang, G.; Morrissette, J.J.D. Cytogenetics and genomics of acute myeloid leukemia. Best Pract. Res. Clin. Haematol. 2024, 37, 101533. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, F.; Garrote, M.; Villamor, N.; Colomer, D.; Esteve, J.; López-Guerra, M. Novel Tools for Diagnosis and Monitoring of AML. Curr. Oncol. 2023, 30, 5201–5213. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef]
- Mishra, S.K.; Millman, S.E.; Zhang, L. Metabolism in acute myeloid leukemia: Mechanistic insights and therapeutic targets. Blood 2023, 141, 1119–1135. [Google Scholar] [CrossRef]
- Jones, C.L.; Inguva, A.; Jordan, C.T. Targeting Energy Metabolism in Cancer Stem Cells: Progress and Challenges in Leukemia and Solid Tumors. Cell Stem Cell 2021, 28, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Appelbaum, F.R. WHO, what, when, where, and why: New classification systems for acute myeloid leukemia and their impact on clinical practice. Best Pract. Res. Clin. Haematol. 2023, 36, 101518. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P. Comparison of the International Consensus and 5th WHO edition classifications of adult myelodysplastic syndromes and acute myeloid leukemia. Am. J. Hematol. 2023, 98, 481–492. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Altman, J.K.; Assi, R.; Bixby, D.; Fathi, A.T.; Foran, J.M.; Gojo, I.; Hall, A.C.; Jonas, B.A.; Kishtagari, A.; et al. Acute Myeloid Leukemia, Version 3.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2023, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Catalano, G.; Zaza, A.; Banella, C.; Pelosi, E.; Castelli, G.; de Marinis, E.; Smigliani, A.; Travaglini, S.; Ottone, T.; Divona, M.; et al. MCL1 regulates AML cells metabolism via direct interaction with HK2. Metabolic signature at onset predicts overall survival in AMLs’ patients. Leukemia 2023, 37, 1600–1610. [Google Scholar] [CrossRef]
- Wang, Y.H.; Orgueira, A.M.; Lin, C.C.; Yao, C.Y.; Lo, M.Y.; Tsai, C.H.; de la Fuente Burguera, A.; Hou, H.A.; Chou, W.C.; Tiene, H.F. Stellae-123 gene expression signature improved risk stratification in taiwanese acute myeloid leukemia patients. Sci. Rep. 2024, 14, 11064. [Google Scholar] [CrossRef]
- Lee, Y.; Baughn, L.B.; Myers, C.L.; Sachs, Z. Machine learning analysis of gene expression reveals TP53 Mutant-like AML with wild type TP53 and poor prognosis. Blood Cancer J. 2024, 14, 80. [Google Scholar] [CrossRef]
- Alhajahjeh, A.; Nazha, A. Unlocking the Potential of Artificial Intelligence in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Curr. Hematol. Malig. Rep. 2024, 19, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Didi, I.; Alliot, J.M.; Dumas, P.Y.; Vergez, F.; Tavitian, S.; Largeaud, L.; Bidet, A.; Rieu, J.B.; Luquet, I.; Lechevalier, N.; et al. Artificial intelligence-based prediction models for acute myeloid leukemia using real-life data: A DATAML registry study. Leuk. Res. 2024, 136, 107437. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, T.Y.; Cho, B.S.; Kwag, D.; Lee, J.M.; Kim, M.; Kim, Y.; Koo, J.; Raman, A.; Kim, T.K.; et al. Prognostic value of European LeukemiaNet 2022 criteria and genomic clusters using machine learning in older adults with acute myeloid leukemia. Haematologica 2024, 109, 1095–1106. [Google Scholar] [PubMed]
- Wysota, M.; Konopleva, M.; Mitchell, S. Novel Therapeutic Targets in Acute Myeloid Leukemia (AML). Curr. Oncol. Rep. 2024, 26, 409–420. [Google Scholar] [CrossRef]
- Alsouqi, A.; Geramita, E.; Im, A. Treatment of Acute Myeloid Leukemia in Older Adults. Cancers 2023, 15, 5409. [Google Scholar] [CrossRef]
- Choi, J.H.; Shukla, M.; Abdul-Hay, M. Acute Myeloid Leukemia Treatment in the Elderly: A Comprehensive Review of the Present and Future. Acta Haematol. 2023, 146, 431–457. [Google Scholar] [CrossRef]
- Roman Diaz, J.L.; Vazquez Martinez, M.; Khimani, F. New Approaches for the Treatment of AML beyond the 7+3 Regimen: Current Concepts and New Approaches. Cancers 2024, 16, 677. [Google Scholar] [CrossRef]
- Abaza, Y.; McMahon, C.; Garcia, J.S. Advancements and Challenges in the Treatment of AML. Am. Soc. Clin. Oncol. Educ. Book 2024, 44, e438662. [Google Scholar] [CrossRef]
- Bhansali, R.S.; Pratz, K.W.; Lai, C. Recent advances in targeted therapies in acute myeloid leukemia. J. Hematol. Oncol. 2023, 16, 29. [Google Scholar] [CrossRef]
- Zimmer, M.; Kadia, T. Approach to the Older Patient with Acute Myeloid Leukemia. Curr. Oncol. Rep. 2023, 25, 1203–1211. [Google Scholar] [CrossRef]
- Bhatia, K.; Sandhu, V.; Wong, M.H.; Iyer, P.; Bhatt, S. Therapeutic biomarkers in acute myeloid leukemia: Functional and genomic approaches. Front. Oncol. 2024, 14, 1275251. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, S.; Puka, B.; Golla, U.; Chachoua, I. Recent Advances towards the Understanding of Secondary Acute Myeloid Leukemia Progression. Life 2024, 14, 309. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Gurbuxani, S.; Crispino, J.D. Malignant progression of pre-leukemic disorders. Blood 2024, 143, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Niscola, P.; Gianfelici, V.; Giovannini, M.; Piccioni, D.; Mazzone, C.; de Fabritiis, P. Latest Insights and Therapeutic Advances in Myelodysplastic Neoplasms. Cancers 2024, 16, 1563. [Google Scholar] [CrossRef]
- Fabre, M.A.; Vassiliou, G.S. The lifelong natural history of clonal hematopoiesis and its links to myeloid neoplasia. Blood 2024, 143, 573–581. [Google Scholar] [CrossRef]
- Molica, M.; Mazzone, C.; Niscola, P.; de Fabritiis, P. TP53 Mutations in Acute Myeloid Leukemia: Still a Daunting Challenge? Front. Oncol. 2021, 10, 610820. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, W.; Yu, J.; Pei, S.; Zhang, Q.; Shi, J.; Huang, H.; Zhao, Y. TP53 in MDS and AML: Biological and clinical advances. Cancer Lett. 2024, 588, 216767. [Google Scholar] [CrossRef] [PubMed]
- Santini, V.; Stahl, M.; Sallman, D.A. TP53 Mutations in Acute Leukemias and Myelodysplastic Syndromes: Insights and Treatment Updates. Am. Soc. Clin. Oncol. Educ. Book 2024, 44, e432650. [Google Scholar] [CrossRef]
- Kim, N.; Hahn, S.; Choi, Y.J.; Cho, H.; Chung, H.; Jang, J.E.; Lyu, C.J.; Lee, S.T.; Choi, J.R.; Cheong, J.W.; et al. Comprehensive insights into AML relapse: Genetic mutations, clonal evolution, and clinical outcomes. Cancer Cell Int. 2024, 24, 174. [Google Scholar] [CrossRef]
- Luque Paz, D.; Jouanneau-Courville, R.; Riou, J.; Ianotto, J.C.; Boyer, F.; Chauveau, A.; Renard, M.; Chomel, J.C.; Cayssials, E.; Gallego-Hernanz, M.P.; et al. Leukemic evolution of polycythemia vera and essential thrombocythemia: Genomic profiles predict time to transformation. Blood Adv. 2020, 4, 4887–4897. [Google Scholar] [CrossRef]
- Zhang, A.; Liu, W.; Guo, X.; Jia, H.; Wei, Y.; Can, C.; He, N.; Ji, C.; Ma, D. Genetic variations in DNA excision repair pathway contribute to the chemosensitivity and prognosis of acute myeloid leukemia. Clin. Chim. Acta 2024, 2, 117899. [Google Scholar] [CrossRef]
- Angelini, D.F.; Ottone, T.; Guerrera, G.; Lavorgna, S.; Cittadini, M.; Buccisano, F.; De Bardi, M.; Gargano, F.; Maurillo, L.; Divona, M.; et al. A Leukemia-Associated CD34/CD123/CD25/CD99+ Immunophenotype Identifies FLT3-Mutated Clones in Acute Myeloid Leukemia. Clin. Cancer Res. 2015, 21, 3977–3985. [Google Scholar] [CrossRef]
- Travaglini, S.; Ottone, T.; Angelini, D.F.; Fiori, V.; Dominici, S.; Noguera, N.I.; Śniegocka, M.; Antonelli, S.; Irno Consalvo, M.A.; De Bardi, M.; et al. CD99 as a novel therapeutic target on leukemic progenitor cells in FLT3-ITDmut AML. Leukemia 2022, 36, 1685–1688. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Li, X.; Xu, Y.; Zhang, T.; Zhu, H.; Yao, D. Recent advances in CAR-T cell therapy for acute myeloid leukemia. J. Cell Mol. Med. 2024, 28, e18369. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Macečková, D.; Vaňková, L.; Holubová, M.; Jindra, P.; Klieber, R.; Jandová, E.; Pitule, P. Current knowledge about FLT3 gene mutations, exploring the isoforms, and protein importance in AML. Mol. Biol. Rep. 2024, 51, 521. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, V.E.; Smith, C.C. FLT3 targeting in the modern era: From clonal selection to combination therapies. Int. J. Hematol. 2023, 19, 1–13. [Google Scholar] [CrossRef]
- Grob, T.; Sanders, M.A.; Vonk, C.M.; Kavelaars, F.G.; Rijken, M.; Hanekamp, D.W.; Gradowska, P.L.; Cloos, J.; Fløisand, Y.; van Marwijk Kooy, M.; et al. Prognostic Value of FLT3-Internal Tandem Duplication Residual Disease in Acute Myeloid Leukemia. J. Clin. Oncol. 2023, 41, 756–765. [Google Scholar] [CrossRef]
- Fruchtman, H.; Avigan, Z.M.; Waksal, J.A.; Brennan, N.; Mascarenhas, J.O. Management of isocitrate dehydrogenase 1/2 mutated acute myeloid leukemia. Leukemia 2024, 38, 927–935. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; DiNardo, C.D.; Loghavi, S. Molecularly Targeted Therapy in Acute Myeloid Leukemia: Current Treatment Landscape and Mechanisms of Response and Resistance. Cancers 2023, 15, 1617. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Shimony, S.; Shallis, R.M.; Liu, Y.; Berton, G.; Schaefer, E.J.; Zeidan, A.M.; Goldberg, A.; Stein, E.; Marcucci, G.; et al. Combination therapy with hypomethylating agents and venetoclax versus intensive induction chemotherapy in IDH1- or IDH2-mutant newly diagnosed acute myeloid leukemia-A multicenter cohort study. Am. J. Hematol. 2024, 99, 1640–1643. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Zarka, J.; Sasaki, K.; Qiao, W.; Pak, D.; Ning, J.; Short, N.J.; Haddad, F.; Tang, Z.; Patel, K.P.; et al. Predictors of outcomes in adults with acute myeloid leukemia and KMT2A rearrangements. Blood Cancer J. 2021, 11, 162. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Martelli, M.P.; Brunetti, L.; Gjertsen, B.T.; Andresen, V. The NPM1 mutant defines AML irrespective of blast count. Am. J. Hematol. 2023, 98, E187–E189. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Martelli, M.P.; Brunetti, L. Mutant NPM1: Nuclear export and the mechanism of leukemogenesis. Am. J. Hematol. 2023, 98, 550–552. [Google Scholar] [CrossRef] [PubMed]
- Thomas, X. Small Molecule Menin Inhibitors: Novel Therapeutic Agents Targeting Acute Myeloid Leukemia with KMT2A Rearrangement or NPM1 Mutation. Oncol. Ther. 2024, 12, 57–72. [Google Scholar] [CrossRef]
- Candoni, A.; Coppola, G. A 2024 Update on Menin Inhibitors. A New Class of Target Agents against KMT2A-Rearranged and NPM1-Mutated Acute Myeloid Leukemia. Hematol. Rep. 2024, 16, 244–254. [Google Scholar] [CrossRef]
- Kühn, M.W.M.; Ganser, A. The Menin story in acute myeloid leukaemia-The road to success. Br. J. Haematol. 2024, 205, 812–815. [Google Scholar] [CrossRef]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature 2023, 615, 920–924. [Google Scholar] [CrossRef]
- Rasouli, M.; Blair, H.; Troester, S.; Szoltysek, K.; Cameron, R.; Ashtiani, M.; Krippner-Heidenreich, A.; Grebien, F.; McGeehan, G.; Zwaan, C.M.; et al. The MLL-Menin Interaction is a Therapeutic Vulnerability in NUP98-rearranged AML. Hemasphere 2023, 7, e935. [Google Scholar] [CrossRef]
- Heikamp, E.B.; Henrich, J.A.; Perner, F.; Wong, E.M.; Hatton, C.; Wen, Y.; Barwe, S.P.; Gopalakrishnapillai, A.; Xu, H.; Uckelmann, H.J.; et al. The menin-MLL1 interaction is a molecular dependency in NUP98-rearranged AML. Blood 2022, 139, 894–906. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature 2023, 615, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Othman, J.; Meggendorfer, M.; Tiacci, E.; Thiede, C.; Schlenk, R.; Dillon, R.; Stasik, S.; Venanzi, A.; Bertoli, S.; Delabesse, E.; et al. Overlapping features of therapy-related and de novo NPM1-mutated AML. Blood 2023, 141, 1846–1857. [Google Scholar] [CrossRef]
- Kühn, M.W.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer Discov. 2016, 6, 1166–1181. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Bouhlel, M.A.; Depauw, S.; Vrevin, J.; Blanck, S.; Marot, G.; Figeac, M.; Preudhomme, C.; Quesnel, B.; et al. Induction of AML cell differentiation using HOXA9/DNA binding inhibitors as a potential therapeutic option for HOXA9-dependent AML. Hemasphere 2024, 8, e77. [Google Scholar] [CrossRef]
- Rodríguez-Medina, C.; Stuckey, R.; Bilbao-Sieyro, C.; Gómez-Casares, M.T. Biomarkers of Response to Venetoclax Therapy in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2024, 25, 1421. [Google Scholar] [CrossRef] [PubMed]
- Mestrum, S.G.C.; Roanalis, B.Y.V.; de Wit, N.C.J.; Drent, R.J.M.; Boonen, B.T.; van Hemert, W.L.W.; Hopman, A.H.N.; Ramaekers, F.C.S.; Leers, M.P.G. MDS and AML show elevated fractions of CD34-positive blast cell populations with a high anti-apoptotic versus proliferation ratio. Leuk. Res. 2024, 142, 107520. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D. Toward an improved understanding of hypomethylating agent and venetoclax therapies. Am. J. Hematol. 2024, 99, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Tajeddine, N.; Kroemer, G. Disruption of the hexokinase-VDAC complex for tumor therapy. Oncogene 2008, 27, 4633–4635. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D′Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Paudel, B.B.; Tan, S.F.; Fox, T.E.; Ung, J.; Golla, U.; Shaw, J.J.P.; Dunton, W.; Lee, I.; Fares, W.A.; Patel, S.; et al. Acute myeloid leukemia stratifies as 2 clinically relevant sphingolipidomic subtypes. Blood Adv. 2024, 8, 1137–1142. [Google Scholar] [CrossRef]
- Pino, J.C.; Posso, C.; Joshi, S.K.; Nestor, M.; Moon, J.; Hansen, J.R.; Hutchinson-Bunch, C.; Gritsenko, M.A.; Weitz, K.K.; Watanabe-Smith, K.; et al. Mapping the proteogenomic landscape enables prediction of drug response in acute myeloid leukemia. Cell Rep. Med. 2024, 5, 101359. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Feng, M.; Nakada, D. Metabolic dependencies of acute myeloid leukemia stem cells. Int. J. Hematol. 2024, 120, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Vieira, J.; Weber, D.D.; Silva, S.; Barbosa-Matos, C.; Granja, S.; Reis, R.M.; Queirós, O.; Ko, Y.H.; Kofler, B.; Casal, M.; et al. Glucose Metabolism as a Potential Therapeutic Target in Cytarabine-Resistant Acute Myeloid Leukemia. Pharmaceutics 2024, 16, 442. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D′Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2019, 35, 333–335. [Google Scholar] [CrossRef]
- Banella, C.; Catalano, G.; Travaglini, S.; Pelosi, E.; Ottone, T.; Zaza, A.; Guerrera, G.; Angelini, D.F.; Niscola, P.; Divona, M.; et al. Ascorbate Plus Buformin in AML: A Metabolic Targeted Treatment. Cancers 2022, 14, 2565. [Google Scholar] [CrossRef]
- Panuzzo, C.; Jovanovski, A.; Pergolizzi, B.; Pironi, L.; Stanga, S.; Fava, C.; Cilloni, D. Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. Int. J. Mol. Sci. 2020, 21, 3928. [Google Scholar] [CrossRef]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 13125. [Google Scholar] [CrossRef]
- Mattes, K.; Vellenga, E.; Schepers, H. Differential redox-regulation and mitochondrial dynamics in normal and leukemic hematopoietic stem cells: A potential window for leukemia therapy. Crit. Rev. Oncol. Hematol. 2019, 144, 102814. [Google Scholar] [CrossRef]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Han, Y.; Liu, L.; Zhang, H.; Liu, D.; Wen, J.; Huang, M.; Zhao, L. Targeting Myeloid Leukemia-1 in Cancer Therapy: Advances and Directions. J. Med. Chem. 2024, 67, 5963–5998. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiyari, M.; Liaghat, M.; Aziziyan, F.; Shapourian, H.; Yahyazadeh, S.; Alipour, M.; Shahveh, S.; Maleki-Sheikhabadi, F.; Halimi, H.; Forghaniesfidvajani, R.; et al. The role of bone marrow microenvironment (BMM) cells in acute myeloid leukemia (AML) progression: Immune checkpoints, metabolic checkpoints, and signaling pathways. Cell Commun. Signal. 2023, 21, 252. [Google Scholar] [CrossRef] [PubMed]
- Minciacchi, V.R.; Karantanou, C.; Bravo, J.; Pereira, R.S.; Zanetti, C.; Krack, T.; Kumar, R.; Bankov, K.; Hartmann, S.; Huntly, B.J.; et al. Differential inflammatory conditioning of the bone marrow by acute myeloid leukemia and its impact on progression. Blood Adv. 2024, 8, 4983–4996. [Google Scholar] [CrossRef]
- Filipek-Gorzała, J.; Kwiecińska, P.; Szade, A.; Szade, K. The dark side of stemness—The role of hematopoietic stem cells in development of blood malignancies. Front. Oncol. 2024, 14, 1308709. [Google Scholar] [CrossRef]
- Haouas, H. Angiogenesis and acute myeloid leukemia. Hematology 2014, 19, 311–323. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, C.; Sun, Q.; Li, Y.; Zhou, C.; Sun, C. Susceptibility of acute myeloid leukemia cells to ferroptosis and evasion strategies. Front. Mol. Biosci. 2023, 10, 1275774. [Google Scholar] [CrossRef]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef]
- Fan, C.; Yang, X.; Yan, L.; Shi, Z. Oxidative stress is two-sided in the treatment of acute myeloid leukemia. Cancer Med. 2024, 13, e6806. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Fueling the Revolution: Targeting Metabolism to Enhance Immunotherapy. Cancer Immunol. Res. 2021, 9, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; DeAngelo, D.J.; Lozier, J.N.; Fisher, D.M.; Jonas, B.A.; Magnani, J.L.; Becker, P.S.; Lazarus, H.M.; Winkler, I.G. Targeting hematologic malignancies by inhibiting E-selectin: A sweet spot for AML therapy. Blood Rev. 2024, 65, 101184. [Google Scholar] [CrossRef] [PubMed]
- Vegivinti, C.T.R.; Keesari, P.R.; Veeraballi, S.; Martins Maia, C.M.P.; Mehta, A.K.; Lavu, R.R.; Thakur, R.K.; Tella, S.H.; Patel, R.; Kakumani, V.K.; et al. Role of innate immunological/inflammatory pathways in myelodysplastic syndromes and AML: A narrative review. Exp. Hematol. Oncol. 2023, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cui, J. Targeting the lactic acid metabolic pathway for antitumor therapy. Mol. Ther. Oncolytics 2023, 31, 100740. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, Y.; Li, J.; Hu, Z. Genetic mutations and immune microenvironment: Unveiling the connection to AML prognosis. Hematology 2024, 29, 2346965. [Google Scholar] [CrossRef]
- Chen, Y.; Qiu, X.; Liu, R. Comprehensive characterization of immunogenic cell death in acute myeloid leukemia revealing the association with prognosis and tumor immune microenvironment. BMC Med. Genom. 2024, 17, 107. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Nadorp, B.; Fornerod, M.; Nicolet, D.; Wu, H.; Walker, C.J.; Sun, Z.; Witkowski, M.T.; Tikhonova, A.N.; Guillamot-Ruano, M.; et al. An inflammatory state remodels the immune microenvironment and improves risk stratification in acute myeloid leukemia. Nat. Cancer 2023, 4, 27–42. [Google Scholar] [CrossRef]
- Cheng, F.M.; Lo, S.C.; Lin, C.C.; Lo, W.J.; Chien, S.Y.; Sun, T.H.; Hsu, K.C. Deep learning assists in acute leukemia detection and cell classification via flow cytometry using the acute leukemia orientation tube. Sci. Rep. 2024, 14, 8350. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef]
- Warnat-Herresthal, S.; Perrakis, K.; Taschler, B.; Becker, M.; Baßler, K.; Beyer, M.; Günther, P.; Schulte-Schrepping, J.; Seep, L.; Klee, K.; et al. Scalable Prediction of Acute Myeloid Leukemia Using High-Dimensional Machine Learning and Blood Transcriptomics. iScience 2020, 23, 100780. [Google Scholar] [CrossRef]
- Cheng, Y.; Yang, X.; Wang, Y.; Li, Q.; Chen, W.; Dai, R.; Zhang, C. Multiple machine-learning tools identifying prognostic biomarkers for acute Myeloid Leukemia. BMC Med. Inform. Decis. Mak. 2024, 24, 2. [Google Scholar] [CrossRef] [PubMed]
- Van Galen, P.; Hovestadt, V.; Wadsworth, M.H., II; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Borsi, E.; Patuelli, A.; Bandini, L.; Mancini, M.; Forte, D.; Nanni, J.; Barone, M.; Grassi, A.; Cristiano, G.; et al. Tracking Response and Resistance in Acute Myeloid Leukemia through Single-Cell DNA Sequencing Helps Uncover New Therapeutic Targets. Int. J. Mol. Sci. 2024, 25, 10002. [Google Scholar] [CrossRef] [PubMed]
- Lucas, F.; Hergott, C.B. Advances in Acute Myeloid Leukemia Classification, Prognostication and Monitoring by Flow Cytometry. Clin. Lab. Med. 2023, 43, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Huber, S.; Baer, C.; Hutter, S.; Dicker, F.; Meggendorfer, M.; Pohlkamp, C.; Kern, W.; Haferlach, T.; Haferlach, C.; Hoermann, G. AML classification in the year 2023: How to avoid a Babylonian confusion of languages. Leukemia 2023, 37, 1413–1420. [Google Scholar] [CrossRef]
- Turkalj, S.; Radtke, F.A.; Vyas, P. An Overview of Targeted Therapies in Acute Myeloid Leukemia. Hemasphere 2023, 7, e914. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhao, D.; Zarif, M.; Davidson, M.B.; Minden, M.D.; Tierens, A.; Yeung, Y.W.T.; Wei, C.; Chang, H. A real-world analysis of clinical outcomes in AML with myelodysplasia-related changes: A comparison of ICC and WHO-HAEM5 criteria. Blood Adv. 2024, 8, 1760–1771. [Google Scholar] [CrossRef]
- Li, X.; Tong, X. Role of Measurable Residual Disease in Older Adult Acute Myeloid Leukemia. Clin. Interv. Aging 2023, 18, 921–931. [Google Scholar] [CrossRef]
- Tiong, I.S.; Loo, S. Targeting Measurable Residual Disease (MRD) in Acute Myeloid Leukemia (AML): Moving beyond Prognostication. Int. J. Mol. Sci. 2023, 24, 4790. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, G.; Zhong, H. Minimal residual disease monitoring in acute myeloid leukemia: Focus on MFC-MRD and treatment guidance for elderly patients. Eur. J. Haematol. 2024, 112, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W.; Jonas, B.A.; Pullarkat, V.; Recher, C.; Schuh, A.C.; Thirman, M.J.; Garcia, J.S.; DiNardo, C.D.; Vorobyev, V.; Fracchiolla, N.S.; et al. Measurable Residual Disease Response and Prognosis in Treatment-Naïve Acute Myeloid Leukemia With Venetoclax and Azacitidine. J. Clin. Oncol. 2022, 40, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Niscola, P.; Gianfelici, V.; Giovannini, M.; Piccioni, D.; Mazzone, C.; Fabritiis, P. Very long-term efficacy of venetoclax combined with hypomethylating agents in two AML elderly: Is it the time for treatment discontinuation strategies? Ann. Hematol. 2024, 103, 1787–1788. [Google Scholar] [CrossRef] [PubMed]
- Garciaz, S.; Dumas, P.Y.; Bertoli, S.; Sallman, D.A.; Decroocq, J.; Belhabri, A.; Orvain, C.; Aspas Requena, G.; Simand, C.; Laribi, K.; et al. Outcomes of acute myeloid leukemia patients who responded to venetoclax and azacitidine and stopped treatment. Am. J. Hematol. 2024, 99, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Boscaro, E.; Urbino, I.; Catania, F.M.; Arrigo, G.; Secreto, C.; Olivi, M.; D′Ardia, S.; Frairia, C.; Giai, V.; Freilone, R.; et al. Modern Risk Stratification of Acute Myeloid Leukemia in 2023: Integrating Established and Emerging Prognostic Factors. Cancers 2023, 15, 3512. [Google Scholar] [CrossRef]
- Song, G.Y.; Kim, H.J.; Kim, T.; Ahn, S.Y.; Jung, S.H.; Kim, M.; Yang, D.H.; Lee, J.J.; Kim, M.Y.; Cheong, J.W.; et al. Validation of the 2022 European LeukemiaNet risk stratification for acute myeloid leukemia. Sci. Rep. 2024, 14, 8517. [Google Scholar] [CrossRef]
- Bazinet, A.; Kantarjian, H.; Arani, N.; Popat, U.; Bataller, A.; Sasaki, K.; DiNardo, C.D.; Daver, N.; Yilmaz, M.; Abbas, H.A.; et al. Evolving trends and outcomes in older patients with acute myeloid leukemia including allogeneic stem cell transplantation. Am. J. Hematol. 2023, 98, 1383–1393. [Google Scholar] [CrossRef]
- Jen, W.Y.; Kantarjian, H.; Kadia, T.M.; DiNardo, C.D.; Issa, G.C.; Short, N.J.; Yilmaz, M.; Borthakur, G.; Ravandi, F.; Daver, N.G. Combination therapy with novel agents for acute myeloid leukaemia: Insights into treatment of a heterogenous disease. Br. J. Haematol. 2024, 205, 30–47. [Google Scholar] [CrossRef]
- Rossi, G.; Borlenghi, E.; Zappasodi, P.; Lussana, F.; Bernardi, M.; Basilico, C.; Molteni, A.; Lotesoriere, I.; Turrini, M.; Frigeni, M.; et al. Adapting the Fitness Criteria for Non-Intensive Treatments in Older Patients with Acute Myeloid Leukemia to the Use of Venetoclax-Hypomethylating Agents Combination-Practical Considerations from the Real-Life Experience of the Hematologists of the Rete Ematologica Lombarda. Cancers 2024, 16, 386. [Google Scholar] [CrossRef]
- Hoff, F.W.; Blum, W.; Huang, Y.; Welkie, R.L.; Swords, R.; Traer, E.; Stein, E.M.; Lin, T.L.; Archer, K.J.; Patel, P.A.; et al. Beat-AML 2024 ELN-Refined Risk Stratification for Older Adults with Newly Diagnosed AML Given Lower-Intensity Therapy. Blood Adv. 2024, 7, 5297–5305. [Google Scholar] [CrossRef]
- Bataller, A.; Bazinet, A.; DiNardo, C.D.; Maiti, A.; Borthakur, G.; Daver, N.G.; Short, N.J.; Jabbour, E.J.; Issa, G.C.; Pemmaraju, N.; et al. Prognostic risk signature in patients with acute myeloid leukemia treated with hypomethylating agents and venetoclax. Blood Adv. 2024, 8, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; DiNardo, C.D.; Wei, A.H.; Löwenberg, B.; Appelbaum, F.; Craddock, C.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; et al. Genetic risk classification for adults with AML receiving less-intensive therapies: The 2024 ELN recommendations. Blood 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Loo, S.; Daver, N.G. How I Treat patients with AML using azacitidine and venetoclax. Blood 2024, 24. [Google Scholar] [CrossRef] [PubMed]
- Getz, T.M.; Bewersdorf, J.P.; Kewan, T.; Stempel, J.M.; Bidikian, A.; Shallis, R.M.; Stahl, M.; Zeidan, A.M. Beyond HMAs: Novel Targets and Therapeutic Approaches. In Seminars in Hematology; WB Saunders: Philadelphia, PA, USA, 2024; Volume 30. [Google Scholar] [CrossRef]
- Heuser, M.; Fernandez, C.; Hauch, O.; Klibanov, O.M.; Chaudhary, T.; Rives, V. Therapies for acute myeloid leukemia in patients ineligible for standard induction chemotherapy: A systematic review. Future Oncol. 2023, 19, 789–810. [Google Scholar] [CrossRef]
- Barosi, G.; Venditti, A.; Angelucci, E.; Gobbi, M.; Pane, F.; Tosi, P.; Zinzani, P.; Tura, S. Consensus-based definition of unfitness to intensive and non-intensive chemotherapy in acute myeloid leukemia: A project of SIE, SIES and GITMO group on a new tool for therapy decision making. Leukemia 2013, 27, 997–999. [Google Scholar]
- Apolito, V.; Arrigo, G.; Vasseur, L.; Olivi, M.; Perrone, S.; Giai, V.; Secreto, C.; Di Biase, F.; De Simone, M.C.; Copia, C.; et al. Validation of SIE/SIES/GITMO consensus criteria for unfitness to predict early mortality and survival in acute myeloid leukemia patients treated with hypomethylating agents and venetoclax. Br. J. Haematol. 2023, 203, e98–e101. [Google Scholar] [CrossRef]
- Pratz, K.W.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Döhner, H.; Récher, C.; Fiedler, W.; Yamamoto, K.; Wang, J.; et al. Long-term follow-up of VIALE-A: Venetoclax and azacitidine in chemotherapy-ineligible untreated acute myeloid leukemia. Am. J. Hematol. 2024, 99, 615–624. [Google Scholar] [CrossRef]
- He, H.; Wen, X.; Zheng, H. Efficacy and safety of venetoclax-based combination therapy for previously untreated acute myeloid leukemia: A meta-analysis. Hematology 2024, 29, 2343604. [Google Scholar] [CrossRef]
- Lai, C.; Bhansali, R.S.; Kuo, E.J.; Mannis, G.; Lin, R.J. Older Adults with Newly Diagnosed AML: Hot Topics for the Practicing Clinician. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390018. [Google Scholar] [CrossRef]
- Wang, E.S.; Baron, J. Management of toxicities associated with targeted therapies for acute myeloid leukemia: When to push through and when to stop. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 57–66. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Shimony, S.; Shallis, R.M.; Liu, Y.; Berton, G.; Schaefer, E.J.; Zeidan, A.M.; Goldberg, A.D.; Stein, E.M.; Marcucci, G.; et al. Intensive Induction Chemotherapy versus Hypomethylating Agents in Combination with Venetoclax in NPM1-mutant AML. Blood Adv. 2024, 28, 4845–4855. [Google Scholar] [CrossRef] [PubMed]
- Sartor, C.; Brunetti, L.; Audisio, E.; Cignetti, A.; Zannoni, L.; Cristiano, G.; Nanni, J.; Ciruolo, R.; Zingarelli, F.; Ottaviani, E.; et al. A venetoclax and azacitidine bridge-to-transplant strategy for NPM1-mutated acute myeloid leukaemia in molecular failure. Br. J. Haematol. 2023, 202, 599–607. [Google Scholar] [CrossRef]
- Niscola, P.; Mazzone, C.; Fratoni, S.; Ardu, N.R.; Cesini, L.; Giovannini, M.; Ottone, T.; Anemona, L.; Voso, M.T.; de Fabritiis, P. Acute Myeloid Leukemia with NPM1 Mutation and Disseminated Leukemia Cutis: Achievement of Molecular Complete Remission by Venetoclax/Azacitidine Combination in a Very Old Patient. Acta Haematol. 2023, 146, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Madarang, E.; Lykon, J.; Zhao, W.; Sekeres, M.A.; Bradley, T.; Chandhok, N.S.; Taylor, J.; Venugopal, S.; Koru-Sengul, T.; Iyer, S.G.; et al. Venetoclax and hypomethylating agents in octogenarians and nonagenarians with acute myeloid leukemia. Blood Neoplasia 2024, 1, 100016. [Google Scholar] [CrossRef]
- Brown, F.C.; Wang, X.; Birkinshaw, R.W.; Chua, C.C.; Morley, T.D.; Kasapgil, S.; Pomilio, G.; Blombery, P.; Huang, D.C.S.; Czabotar, P.E.; et al. Acquired BCL2 variants associated with venetoclax resistance in acute myeloid leukemia. Blood Adv. 2024, 7. [Google Scholar] [CrossRef]
- Lemos, T.; Merchant, A. The hedgehog pathway in hematopoiesis and hematological malignancy. Front. Oncol. 2022, 12, 960943. [Google Scholar] [CrossRef]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Premnath, N.; Madanat, Y.F. Paradigm Shift in the Management of Acute Myeloid Leukemia—Approved Options in 2023. Cancers 2023, 15, 3002. [Google Scholar] [CrossRef]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.P.; et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2022, 386, 1519–1531. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; Loghavi, S.; Zeng, Z.; Tanaka, T.; Kim, Y.J.; Uryu, H.; Turkalj, S.; Jakobsen, N.A.; Luskin, M.R.; Duose, D.Y.; et al. A Phase Ib/II Study of Ivosidenib with Venetoclax ± Azacitidine in IDH1-Mutated Myeloid Malignancies. Blood Cancer Discov. 2023, 4, 276–293. [Google Scholar] [CrossRef]
- Cai, S.F.; Huang, Y.; Lance, J.R.; Mao, H.C.; Dunbar, A.J.; McNulty, S.N.; Druley, T.; Li, Y.; Baer, M.R.; Stock, W.; et al. A study to assess the efficacy of enasidenib and risk-adapted addition of azacitidine in newly diagnosed IDH2-mutant AML. Blood Adv. 2024, 8, 429–440. [Google Scholar] [CrossRef]
- Watts, J.M.; Baer, M.R.; Yang, J.; Prebet, T.; Lee, S.; Schiller, G.J.; Dinner, S.N.; Pigneux, A.; Montesinos, P.; Wang, E.S.; et al. Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: Phase 1 results of a phase 1/2 trial. Lancet Haematol. 2023, 10, e46–e58. [Google Scholar] [CrossRef]
- Bocchia, M.; Carella, A.M.; Mulè, A.; Rizzo, L.; Turrini, M.; Abbenante, M.C.; Cairoli, R.; Calafiore, V.; Defina, M.; Gardellini, A.; et al. Therapeutic Management of Patients with FLT3 + Acute Myeloid Leukemia: Case Reports and Focus on Gilteritinib Monotherapy. Pharmacogenomics Pers. Med. 2022, 15, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Saburi, M.; Sakata, M.; Maruyama, R.; Kodama, Y.; Takata, H.; Miyazaki, Y.; Kawano, K.; Wada, J.; Urabe, S.; Ohtsuka, E. Gilteritinib as Bridging and Posttransplant Maintenance for Relapsed Acute Myeloid Leukemia with FLT3-ITD Mutation Accompanied by Extramedullary Disease in Elderly. Case Rep. Hematol. 2023, 2023, 7164742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.S.; Wang, J.; Xu, M.Z.; Wu, T.M.; Huang, S.M.; Cao, H.Y.; Sun, A.N.; Liu, S.B.; Xue, S.L. Rapid and Efficient Response to Gilteritinib and Venetoclax-Based Therapy in Two AML Patients with FLT3-ITD Mutation Unresponsive to Venetoclax Plus Azacitidine. Onco Targets Ther. 2022, 15, 159–164. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Perl, A.E.; Larson, R.A.; Podoltsev, N.A.; Strickland, S.; Wang, E.S.; Atallah, E.; Schiller, G.J.; Martinelli, G.; Neubauer, A.; Sierra, J.; et al. Follow-up of patients with R/R FLT3-mutation-positive AML treated with gilteritinib in the phase 3 ADMIRAL trial. Blood 2022, 139, 3366–3375. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Shallis, R.M.; Derkach, A.; Goldberg, A.D.; Stein, A.; Stein, E.M.; Marcucci, G.; Zeidan, A.M.; Shimony, S.; DeAngelo, D.J.; et al. Venetoclax-based salvage therapy in patients with relapsed/refractory acute myeloid leukemia previously treated with FLT3 or IDH1/2 inhibitors. Leuk. Lymphoma 2023, 64, 188–196. [Google Scholar] [CrossRef]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; McCloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2022, 40, 4048–4059. [Google Scholar] [CrossRef]
- Venugopal, S.; Watts, J. The future paradigm of HMA + VEN or targeted inhibitor approaches: Sequencing or triplet combinations in AML therapy. Hematol. Am. Soc. Hematol. Educ. Program 2023, 2023, 192–197. [Google Scholar] [CrossRef]
- Short, N.J.; Daver, N.; Dinardo, C.D.; Kadia, T.; Nasr, L.F.; Macaron, W.; Yilmaz, M.; Borthakur, G.; Montalban-Bravo, G.; Garcia-Manero, G.; et al. Azacitidine, Venetoclax, and Gilteritinib in Newly Diagnosed and Relapsed or Refractory FLT3-Mutated AML. J. Clin. Oncol. 2024, 42, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, M.E.; Audemard, É.; Métois, A.; Theret, L.; Lisi, V.; Farah, A.; Spinella, J.F.; Chagraoui, J.; Moujaber, O.; Aubert, L.; et al. Immunotherapeutic targeting of surfaceome heterogeneity in AML. Cell Rep. 2024, 43, 114260. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, E.; Castelli, G.; Testa, U. CD123 a Therapeutic Target for Acute Myeloid Leukemia and Blastic Plasmocytoid Dendritic Neoplasm. Int. J. Mol. Sci. 2023, 24, 2718. [Google Scholar] [CrossRef]
- Martino, G.; Cimino, G.; Caridi, M.; Perta, G.; Cardinali, V.; Sciabolacci, S.; Quintini, M.; Matteucci, C.; Venanzi, A.; Tiacci, E.; et al. One disease, two faces: Clonally-related AML and MPDCP with skin involvement. Ann. Hematol. 2023, 102, 2969–2971. [Google Scholar] [CrossRef]
- Jen, E.Y.; Gao, X.; Li, L.; Zhuang, L.; Simpson, N.E.; Aryal, B.; Wang, R.; Przepiorka, D.; Shen, Y.L.; Leong, R.; et al. FDA Approval Summary: Tagraxofusp-erzs For Treatment of Blastic Plasmacytoid Dendritic Cell Neoplasm. Clin. Cancer Res. 2020, 26, 532–536. [Google Scholar] [CrossRef]
- Zanotta, S.; Galati, D.; De Filippi, R.; Pinto, A. Breakthrough in Blastic Plasmacytoid Dendritic Cell Neoplasm Cancer Therapy Owing to Precision Targeting of CD123. Int. J. Mol. Sci. 2024, 25, 1454. [Google Scholar] [CrossRef]
- Marra, A.; Akarca, A.U.; Martino, G.; Ramsay, A.; Ascani, S.; Perriello, V.M.; O’Nions, J.; Wilson, A.J.; Gupta, R.; Childerhouse, A.; et al. CD47 expression in acute myeloid leukemia varies according to genotype. Haematologica 2023, 108, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.; Piérola, A.A.; Donnellan, W.B.; Yordi, A.M.; Abdul-Hay, M.; Platzbecker, U.; Subklewe, M.; Kadia, T.M.; Alonso-Domínguez, J.M.; McCloskey, J.; et al. First-in-human study of JNJ-67571244, a CD33 × CD3 bispecific antibody, in relapsed/refractory acute myeloid leukemia and myelodysplastic syndrome. Clin. Transl. Sci. 2024, 17, e13742. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Rety, S.; Bonnamy, M.; Aguinaga, L.; Huynh, T.; Parietti, V.; Giraudier, S.; Fenaux, P.; Cassinat, B. Niclosamide combined to Azacitidine to target TP53-mutated MDS/AML cells. Leukemia 2024, 38, 1630–1633. [Google Scholar] [CrossRef]
- Mosna, F.; Borlenghi, E.; Litzow, M.; Byrd, J.C.; Papayannidis, C.; Tecchio, C.; Ferrara, F.; Marcucci, G.; Cairoli, R.; Morgan, E.A.; et al. Long-term survival can be achieved in a significant fraction of older patients with core binding factor acute myeloid leukemia treated with intensive chemotherapy. Haematologica 2024, ahead of print. [CrossRef]
- Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; de Fabritiis, P. CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin. Cancers 2021, 13, 3214. [Google Scholar] [CrossRef] [PubMed]
- Bernal, T.; Moreno, A.F.; de LaIglesia, A.; Benavente, C.; García-Noblejas, A.; Belmonte, D.G.; Riaza, R.; Salamero, O.; Foncillas, M.A.; Roldán, A.; et al. Clinical outcomes after CPX-351 in patients with high-risk acute myeloid leukemia: A comparison with a matched cohort from the Spanish PETHEMA registry. Cancer Med. 2023, 12, 14892–14901. [Google Scholar] [CrossRef] [PubMed]
- Niscola, P.; Tendas, A.; Mazzone, C.; Efficace, F. Pain and related complaints in patients with acute leukemia: Time for simultaneous care in hemato-oncology. Support Care Cancer 2019, 27, 2755–2756. [Google Scholar] [CrossRef]
- El-Jawahri, A.; LeBlanc, T.W.; Kavanaugh, A.; Webb, J.A.; Jackson, V.A.; Campbell, T.C.; O’Connor, N.; Luger, S.M.; Gafford, E.; Gustin, J.; et al. Effectiveness of Integrated Palliative and Oncology Care for Patients with Acute Myeloid Leukemia: A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Zhou, X.; Reeder-Hayes, K.; Jensen, C.E.; Islam, J.; Loh, K.P.; Gupta, A.; Basch, E.; Bennett, A.V.; Bridges, J.F.P.; et al. Home Time Among Older Adults with Acute Myeloid Leukemia Following Chemotherapy. JAMA Oncol. 2024, 13, E241823. [Google Scholar] [CrossRef] [PubMed]
- Le, R.Q.; Przepiorka, D.; Chen, H.; Shen, Y.L.; Pulte, E.D.; Norsworthy, K.; Theoret, M.R.; De Claro, R.A. Complete remission with partial hematological recovery as a palliative endpoint for treatment of acute myeloid leukemia. Blood 2024, 144, 206–215. [Google Scholar] [CrossRef]
- Geissler, K.; Koristek, Z.; Del Castillo, T.B.; Novák, J.; Rodríguez-Macías, G.; Metzelder, S.K.; Illes, A.; Mayer, J.; Arnan, M.; Keating, M.M.; et al. Oral decitabine/cedazuridine versus intravenous decitabine for acute myeloid leukaemia: A randomised, crossover, registration, pharmacokinetics study. Br. J. Haematol. 2024, online ahead of print. [CrossRef]
- De Leeuw, D.C.; Ossenkoppele, G.J.; Janssen, J.J.W.M. Older Patients with Acute Myeloid Leukemia Deserve Individualized Treatment. Curr. Oncol. Rep. 2022, 24, 1387–1400. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Blast Threshold | WHO-5 | ICC | Blast Threshold |
|---|---|---|---|
| No cut-off | AMLs with DGA | APL with t (15; 17) (q24.1; q21.2)/PML: RARA. APL with others RARA rearrangement | 10% |
| APL with PML: RARA fusion gene. | |||
| AML with RUNX1: RUNX1T1 fusion gene. | AML with t (8/21) (q22; q22.1)/RUNX1: RUNX1T1 fusion gene. | ||
| AML with CBFB: MYH11 fusion gene. | AML with inv (16) (p13.1; q22) or t (16; 16) (p13.1; q22)/CBFB: MYH11. | ||
| AML with KMT2A rearrangements. | AML with t (9; 11) (p21.3; q23.3)/MLLT3: KTM2A or other KMT2A rearrangements. | ||
| AML with DEK: NUP214 fusion gene. | AML with t (6; 9) (p22.3; q34.1)/DEK: NUP214. | ||
| AML with MECOM rearrangements | AML with inv (3) (q21.3q; 26.2) or t (3; 3) (q21.3; q26.2)/GATA: MECOM (EV1) or other MECOM rearrangements | ||
| AML with other rare translocations (NUP98; RBM15; MRTF1, DEK: NUP214) | |||
| 20% | AML with BCR: ABL1 fusion gene | AML with t (9; 22) (q34.1; q11.2)/BCR: ABL1 | 20% |
| No cut-off. | AML with NPM1 mutation. | 10% | |
| 20% | AML with CEBPA mutation. | AML with bZIP CEBPA in-frame mutation. | 20% |
| Not classified. | AML with TP53 mutation. | 20% | |
| 20% | AML with MDS-related genetic abnormalities. | AML with MDS-related genetic abnormalities (ASXL1; BCOR, EZH2; RUNX1; SF3B1; SRSF2; STAG2; U2AF1, ZRSR2). AML with MDS-related cytogenetic alterations. | 20% |
| 20% | AMLs defined by differentiation (Table 2). | AML NOS. | 20% |
| 10% | MDS-IB2. | MDS/AML. | 10–19% |
| Myeloid sarcoma | |||
| AML Subtype | Diagnostic Criteria |
|---|---|
| AML with minimal differentiation. | Cytochemistry: MPO and SBB negative blasts (<3%). |
| MFC: expression of myeloid antigens (two or more), such as CD13, CD33, and CD117. | |
| AML without maturation. | Morphology: <10% maturing myeloid progenitors of the BM nucleated cells. |
| Cytochemistry: ≥3% blasts positive for MPO or SBB and negative for NSE. | |
| MFC: expression of myeloid antigens (two or more), such as MPO, CD13, CD33, and CD117. | |
| AML with maturation. | Morphology: >10% maturing myeloid progenitors and <20% of the monocytic lineage cells of the BM nucleated cells. |
| Cytochemistry: ≥3% blasts positive for MPO or SBB. | |
| MFC: expression of myeloid antigens (two or more), such as MPO, CD13, CD33, and CD117. | |
| Acute basophilic leukemia. | Morphology: blasts and mature/immature basophils. |
| Cytochemistry. Basophils: metachromasia on toluidine blue staining. Blasts: negative for MPO, SBB, and NSA. | |
| MFC: negative CD117 (to exclude mast cell leukemia). | |
| Acute myelomonocytic leukemia. | Morphology: ≥20% monocytes or their precursors and ≥20% maturing granulocytic cells. |
| Cytochemistry and/or MFC: ≥3% of MPO-positive blasts. | |
| Acute monocytic leukemia. | Morphology: ≥80% of monocytes and/or their precursors (monoblasts and/or promonocytes); ≤20% of maturing granulocytic cells. |
| MFC/cytochemistry: expression of monocytic antigens (two or more), such as CD11c, CD14, CD36, and CD64, on blasts and promonocytes or their NSE positivity. | |
| Acute erythroid leukemia. | Morphology: erythroid predominance in the BM (>80% of BM cellularity); >30% of immature erythroid (proerythroblasts). |
| Acute megakaryoblastic leukemia. | MFC: expression of one or more platelet GPs: CD41(GP IIb), CD61 (GP IIIa), or CD42b (GP Ib). |
| ELN 2022 and ICC 2022 | WHO-5 | |
|---|---|---|
| MDS/AML (without AML defining genetic alterations). | 10–19% blasts | Designated as MDS-IB2 (10–19% BM or 5–19% PB or Auer Roads). |
| AML with antecedent MDS, MDS/MPM, or prior exposure to therapy. | MDS was added as a diagnostic qualifier. | Included as a separate entity, “AML-MR”. |
| AML with NPM1 mutations, KMT2A rearrangement, MECOM rearrangement, and NUP98 rearrangement. | Requires ≥ 10% blasts in BM or PB. | It can be diagnosed irrespective of blast count. |
| AML with CEPA mutation. | Requires ≥ blasts in BM or PB. | Requires ≥ 20% blasts in BM or PB. Includes bi-allelic and bzip mutations. |
| TP53 mutation. | Included separately in the hierarchical classification. | Not included in a separate entity for AML. |
| Therapy-related. | Added as a diagnostic qualifier. | Included as a separate entity, “AML pCT”. |
| Risk category | Favorable | Genetic abnormalities |
| t (8; 21) (q22; q22.1)/RUNX1: RUNX1T1 °*. | ||
| inv (16) (p13.1q22) or t (16; 16) (p13.1; q22)/CBFB: MYH11 °*. | ||
| Mutated NPM1 °, ^ without FLT3-ITD. | ||
| bZIP in-frame mutated CEBPA °°. | ||
| Intermediate | Mutated NPM1 °, * with FLT3-ITD. | |
| Wild-type NPM1 with FLT3-ITD (without ARG). | ||
| t (9; 11) (p21.3; q23.3)/MLLT3: KMT2A ° | ||
| Cytogenetic and/or molecular abnormalities not classified as favorable or adverse. | ||
| Adverse | t (6; 9) (p23.3; q34.1)/DEK: NUP214. | |
| t (v; 11q23.3)/KMT2A-rearranged °°°. | ||
| t (9; 22) (q34.1; q11.2)/BCR: ABL1. | ||
| t (8; 16) (p11.2; p13.3)/KAT6A: CREBBP. | ||
| inv (3) (q21.3q26.2) or t (3; 3) (q21.3; q26.2)/GATA2, MECOM(EVI1). | ||
| t (3q26.2; v)/MECOM(EVI1)-rearranged. | ||
| Monosomy 5 or del(5q); −7; −17/abn(17p). | ||
| Complex karyotype ^^ monosomic karyotype ^^. | ||
| Mutated ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, and/or ZRSR2 ^^^. | ||
| Mutated TP53 ***. |
| Risk Category | Genetic Marker | Median OS (Months) |
|---|---|---|
| Favorable | Mutated NPM1 (FLT3-ITD neg, NRAS wt, KRAS wt, TP53 wt) | 39 |
| Mutated IDH2 (FLT3-ITD neg, NRAS wt, KRAS wt, TP53 wt) | 37 | |
| Mutated IDH1 (TP53 wt) | 29 | |
| Mutated DDX41 | >24 | |
| AML with MDS-related gene mutations (FLT3-ITD neg, NRAS wt, KRA Swt, TP53 wt) | 23 | |
| Intermediate | AML with MDS-related gene mutations (FLT3-ITD pos and/or NRAS mut and/or KRAS mut; TP53 wt) | 13 |
| Other cytogenetic and molecular abnormalities (FLT3-ITD pos and/or NRAS mut and/or KRAS mut; TP53 wt) | 12 | |
| Adverse | Mutated TP53 | 5–8 |
| Therapeutic Mechanisms and Biological Targets | Therapeutic Agent | Indications | |
|---|---|---|---|
| Antiapoptotic by inhibition of BCL2 overexpression | Venetoclax | ND AML in patients > 75 years old or with comorbidities in combination with HMA or LODAC | |
| FLT3 | FLT-3 ITD FLT-3 TKD | Midostaurin, Quizartinib | Frontline, in combination with ICT |
| Gilteritinib | R/R setting | ||
| Sorafenib | Maintenance following consolidation | ||
| IDH1 | IDH1 | Ivosidenib | ND AML in patients > 75 years old or with comorbidities; R/R setting |
| Olutasidenib | R/R setting | ||
| IDH2 | IDH2 | Enasidenib | R/R setting |
| Inhibition of Hedgehog pathway | Glasdegib | Adults older than 75 years who have comorbidities. | |
| ICT with liposomal compounds in s-AML and t-AML | CPX-351 | As induction ICT for ND s-AML and t-AML | |
| Anti-CD33 monoclonal antibodies | GO | During induction, ICT for CD33-positive AML or as a single agent in the R/R setting. | |
| Targeting CD123 membrane receptor, cell death via disruption of intracellular protein synthesis by CD123 binding and internalization of the drug | Tagraxofusp (anti-CD123 conjugate with toxin) | Treatment of BPDCN | |
| Clinical Study | ClinicalTrials.Gov Identifier |
|---|---|
| Investigating The Prognostic Significance Of Malnutrition And Sarcopenia in Older Adults with Acute Myeloid Leukemia. | NCT05458258 |
| A Pilot Randomized Controlled Trial of a Patient-Centered Communication Tool (UR-GOAL) for Older Patients With Acute Myeloid Leukemia, Their Caregivers, and Their Oncologists. | NCT05335369 |
| Allogeneic Hematopoietic Cell Transplantation Versus Best Available Standard of Care Therapy in Elderly Patients With Acute Myeloid Leukemia: a Randomized Phase 3 Trial. | NCT04822766 |
| A Randomized Phase II Study of Venetoclax and HMA-Based Therapies for the Treatment of Older and Unfit Adults With Newly Diagnosed FLT3-Mutated Acute Myeloid Leukemia (AML): A myelomatch Treatment Trial. | NCT06317649 |
| Phase I/II Clinical Trial Assessing the Combination of Sulfasalazine With Standard of Care Induction Therapy in Newly Diagnosed Acute Myeloid Leukemias (AML) Patients 60 Years or Older- the SALMA Study. | NCT05580861 |
| A Phase Ib Trial of Azacitidine, Venetoclax and Allogeneic NK Cells for Acute Myeloid Leukemia (ADVENT-AML). | NCT05834244 |
| Relatlimab With Nivolumab and 5-Azacytidine for the Treatment of AML (AARON). | NCT04913922 |
| Dual Growth Factor (rhtpo + G-CSF) and Chemotherapy Combination Regimen for Elderly Patients with Acute Myeloid Leukemia: A Phase II Single-Arm Multicenter Study. | NCT05258799 |
| Dual Growth Factor (rhtpo + G-CSF) and Chemotherapy Combination Regimen in Acute Myeloid Leukemia: Study Protocol for a Randomized Controlled Trial. | NCT05382390 |
| A Prospective, Single-arm, Open-label, Non-interventional, Multi-centre, Post Marketing Surveillance (PMS) Study of Mylotarg®. | NCT05189639 |
| Randomized, Sequential, Open-Label Study to Evaluate the Efficacy of IDH Targeted/Non-Targeted Versus Non-targeted/IDH-targeted Approaches in the Treatment of Newly Diagnosed IDH Mutated AML Patients Not Candidates for Intensive Induction Therapy (I-DATA Study). | NCT05401097 |
| Dynamics of Resistance Emergence to Azacitidine-based Therapies in Acute Myeloid Leukemia. | NCT06225128 |
| Phase IA/B Combination Study of ADI-PEG 20, Venetoclax and Azacitidine in Patients with Acute Myeloid Leukemia (AML). | NCT05001828 |
| The Feasibility of Telehealth-Based Palliative Care Intervention and Digital Symptom Monitoring on Patients With AML Receiving Low-Intensity Induction Therapy. | NCT04885127 |
| An Investigator-Sponsored Randomized Phase II Study of Selinexor in Combination With Induction/Consolidation Therapy in Acute Myeloid Leukemia Patients. | NCT02835222 |
| A Prospective Non-interventional Study Documenting the Management and Outcomes of Adult Patients With Acute Myeloid Leukemia (AML). | NCT04777916 |
| Integrative “Omics” Approaches for Leukemia Target Identification and Matched Therapeutic Intervention. | NCT06626893 |
| Maintenance Treatment With Oral Azacitidine for Patients With de Novo AML Including t-AML and AML-MRC in First Remission After CPX-351. | NCT06349239 |
| Phase II Study of Maintenance Ruxolitinib After Allogeneic Stem Cell Transplantation for Older Patients With Acute Myeloid Leukemia (AML) or Myelodysplastic Syndrome (MDS) in Complete Remission. | NCT03286530 |
| Do Decreased Dietary Fat and Increased Fiber Reduce Recurrence of Clostridioides Difficile Infection in Oncology Patients? | NCT04940468 |
| A Telehealth Advance Care Planning Intervention for Older Patients With Myeloid Malignancies: A Pilot Randomized Controlled Trial. | NCT05875805 |
| Phase 1a/1b Study of Itacitinib (INCB039110) for Cytokine Release Syndrome Prevention and Minimization of Immunosuppression Following Nonmyeloablative Related Partially HLA-mismatched Peripheral Blood Stem Cell Transplant (PBSCT) With High-dose Posttransplantation Cyclophosphamide in Older Patients (Age 60 Years). | NCT05823571 |
| Prospective, Observational Study of the Role of Primary Antifungal Prophylaxis to Prevent Invasive Aspergillosis in Elderly Patients With Acute Myeloid Leukemia Undergoing Consolidation Therapy. | NCT06382922 |
| A Master Protocol for Biomarker-Based Treatment of AML (The Beat AML Trial). | NCT03013998 |
| Phase 1 Trial for Patients With Advanced Hematologic Malignancies Undergoing Reduced Intensity Allogeneic HCT With a T-cell Depleted Graft With Infusion of Conventional T-cells and Regulatory T-cells. | NCT05088356 |
| A Phase II Trial of HSCT for the Treatment of Patients With Fanconi Anemia Lacking a Genotypically Identical Donor, Using a Risk-Adjusted Chemotherapy Only Cytoreduction With Busulfan, Cyclophosphamide and Fludarabine. | NCT02143830 |
| A Single Arm Phase II Trial to Assess Cobicistat Boosted Venetoclax in Combination With Azacitidine (sc) in Adult Patients With Newly Diagnosed Acute Myeloid Leukaemia (AML) Who Are Not Considered Candidates for Intensive Treatment Regimens. | NCT06014489 |
| Carolina Senior: UNC Registry for Older Cancer Patients. | NCT01137825 |
| Combined Haploidentical Reduced Intensity Bone Marrow and Kidney Transplantation for Patients With Chronic Kidney Disease and Advanced Hematological Disorders. | NCT01758042 |
| Collection of Blood, Bone Marrow, Skin, Saliva, and Stool Samples From Healthy Volunteers Used for Comparative Analysis of Myeloid Malignancies. | NCT05588154 |
| A Phase I/II Trial of Eltanexor (KPT-8602) With Inqovi (Decitabine-Cedazuridine) in High-Risk Myelodysplastic Syndromes. | NCT05918055 |
| Phase I/II Trial to Determine the Lowest Effective Dose of Post-Transplantation Cyclophosphamide in Combination With Sirolimus and Mycophenolate Mofetil as Graft-Versus-Host Disease Prophylaxis After Reduced Intensity Conditioning and Peripheral Blood Stem Cell Transplantation. | NCT05436418 |
| Source: https://clinicaltrials.gov/ (accessed on 16 October 2024) | |
| Therapeutic Agent | Most Common Toxicities and Comments | |
|---|---|---|
| Venetoclax (ND AML in patients > 75 years old or with comorbidities in combination with HMA or LODAC) | Myelosuppression, notably prolonged neutropenia, could be managed by prolonging treatment intervals and using antimicrobial prophylaxis. G-CSF will be allowed in patients with AML in remission. | |
| FLT-3 ITD FLT-3 TKD | Midostaurin, Quizartinib (Frontline, in combination with ICT) | GI side effects. The survival benefit of quizartinib was limited to patients younger than 60. There is a high risk of early mortality in older patients. |
| Gilteritinib (R/R setting) | Differentiation syndrome, long QT syndrome, and posterior reversible encephalopathy. | |
| IDH1 | Ivosidenib (ND and R/R AML) | Differentiation syndrome, long QT syndrome. |
| Olutasidenib (R/R setting) | Differentiation syndrome, hepatotoxicity. | |
| IDH2 | Enasidenib (R/R setting) | Differentiation syndrome, hyperbilirubinemia. |
| Glasdegib (adults older than 75 years who have comorbidities). | Black, tarry stools, bleeding gums, chest pain, chills, confusion, and cough. | |
| CPX-351 (as induction ICT for ND s-AML and t-AML). | Myelosuppression, prolonged neutropenia. | |
| Tagraxofusp (anti-CD123 conjugate with toxin). Treatment of BPDCN. | CLS, nausea, tiredness (fatigue), fever, swelling in your legs or feet, and weight gain. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niscola, P.; Gianfelici, V.; Catalano, G.; Giovannini, M.; Mazzone, C.; Noguera, N.I.; de Fabritiis, P. Acute Myeloid Leukemia in Older Patients: From New Biological Insights to Targeted Therapies. Curr. Oncol. 2024, 31, 6632-6658. https://doi.org/10.3390/curroncol31110490
Niscola P, Gianfelici V, Catalano G, Giovannini M, Mazzone C, Noguera NI, de Fabritiis P. Acute Myeloid Leukemia in Older Patients: From New Biological Insights to Targeted Therapies. Current Oncology. 2024; 31(11):6632-6658. https://doi.org/10.3390/curroncol31110490
Chicago/Turabian StyleNiscola, Pasquale, Valentina Gianfelici, Gianfranco Catalano, Marco Giovannini, Carla Mazzone, Nelida Ines Noguera, and Paolo de Fabritiis. 2024. "Acute Myeloid Leukemia in Older Patients: From New Biological Insights to Targeted Therapies" Current Oncology 31, no. 11: 6632-6658. https://doi.org/10.3390/curroncol31110490
APA StyleNiscola, P., Gianfelici, V., Catalano, G., Giovannini, M., Mazzone, C., Noguera, N. I., & de Fabritiis, P. (2024). Acute Myeloid Leukemia in Older Patients: From New Biological Insights to Targeted Therapies. Current Oncology, 31(11), 6632-6658. https://doi.org/10.3390/curroncol31110490